

Graphical abstract
Keywords: Cytomegalovirus, Antiviral agents, Azetidine-containing peptides, Solid-phase synthesis, γ-Turn conformation
Abstract
SAR studies on an azetidine-containing dipeptide prototype inhibitor of HCMV are described. Three series of structurally modified analogues, involving substitutions at the N- and C-terminus, and at the C-terminal side-chain were synthesized and evaluated for antiviral activity. Aliphatic or no substituents at the C-carboxamide group, an aliphatic C-terminal side-chain, as well as a benzyloxycarbonyl moiety at the N-terminus were absolute requirements for anti-HCMV activity. The conformational restriction induced by the 2-azetidine residue into the dipeptide derivatives, identified by 1H NMR as a γ-type reverse turn, seems to have influence on the activity of these molecules.
1. Introduction
Human cytomegalovirus (HCMV) is a prevalent β-herpesvirus that can cause serious, life-threatening diseases in immunologically immature or immunocompromised individuals, like neonates, AIDS patients, and transplant recipients.1 Moreover, infection with HCMV is suggested to be associated with certain vascular diseases (atherosclerosis, restenosis, etc.).2, 3
The current antiviral agents approved for HCMV infection, ganciclovir, acyclovir, their Val pro-drugs valganciclovir and valacyclovir, cidofovir, foscarnet and the antisense RNA fomivirsen, demonstrate in general suboptimal efficacy and safety profiles.4, 5 Moreover, these drugs suffer from unfavourable pharmacokinetic properties, and for some of them antiviral drug resistance have emerged.6 Therefore, there is still a need for more effective and safe therapeutic agents for HCMV disease.
While most of the anti-HCMV approved drugs, except fomivirsen, are inhibitors of the viral DNA polymerase, compounds with different mechanisms of action have also appeared. Among them, the l-ribose benzimidazole derivative maribavir, targeting to the viral UL97 protein kinase, is currently under clinical trials.7 A series of thiourea small-molecules were identified as potent anti-HCMV agents, inhibiting virion envelope fusion, and some piperidine-derived compounds were described as the first class of small-molecule ligands for the chemokine receptor US28, thus also acting at an early stage of the replication cycle.8 Quite recently, some γ-sultone derivatives with anti-HCMV activity were shown to keep full antiviral sensitivity against a panel of mutant HCMV strains, selected for resistance against approved drugs, and accordingly suggesting a new mechanism of action, which is still unclear.9
The HCMV protease, essential for capsid assembly and viral maturation, is being considered an attractive target for anti-HCMV chemotherapy, and several structurally diverse inhibitors have been described.10 Most of them include an activated carbonyl group or are mechanism-based inhibitors that covalently interact with the Ser residue within the enzyme active site.11, 12 Among the latter, we and others have discovered simple and small β-lactam derivatives with anti-HCMV activity.13, 14 In our case, a simple deletion of the carbonyl group of the β-lactam ring resulted in the corresponding azetidine derivatives, which behave as non-covalent inhibitors of HCMV replication.14b Preliminary structure–activity relationship studies suggested the importance of a certain hydrophobic environment by the carboxylate in position 2 of the azetidine ring. Moreover, within this series, some C-terminal carboxamide derivatives showed restricted rotation around the N1–CO bond, due to the stabilization of a γ-turn-like folded conformation.14b To investigate further the structural and conformational issues within these azetidine derivatives and to determine their interest as antiviral agents, we initiated a program directed to the modification of compound 1, selected as a prototype (Fig. 1 ). To this end, diverse substituents have been built-in the C-terminal carboxamide moiety (A), amino acids with side-chains of different character were selected as C-terminal residues (B), and the N-terminal benzyloxycarbonyl group was replaced by different urethane, acyl and urea moieties containing aromatic groups (C).
Figure 1.
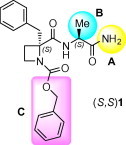
Prototype and points of modification (A–C).
This paper deals with the solution/solid-phase synthesis, conformational analysis by NMR, and biological assays of the compounds resulting from the three indicated series A–C of azetidine-containing dipeptides.
2. Results and discussion
2.1. Synthesis
To increase the hydrophobic character of compound 1, different alkyl groups were incorporated at the C-terminal carboxamide to generate derivatives 4 (Series A). The preparation of these compounds was achieved by coupling the 2-carboxy azetidine 2 with the corresponding l-alanyl carboxamides 3 under standard solution methodologies (Scheme 1 ). All final compounds 4a–e were obtained in good yield as mixtures of two diastereoisomers that could be chromatographically resolved. Since the starting azetidine was a ∼2:1 S:R enantiomeric mixture,15 the major components of these mixtures were assigned as the S,S-diastereoisomers.
Scheme 1.
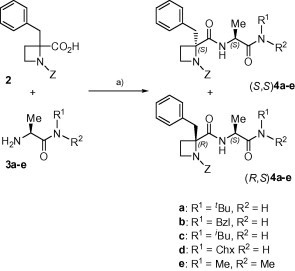
Synthesis of C-terminal amide-modified compounds (A). Reagents: (a) BOP/TEA/THF.
To understand the role of the C-terminal amino acid side-chain, d-Ala-, Phe-, Val-, Leu-, Lys- and Glu-bearing derivatives 7a–e were designed (series B). Solid-phase synthetic methodologies, employing a Rink-amide polystyrene resin as insoluble matrix, were used for the preparation of these compounds (Scheme 2 ). After removal of the Fmoc-protecting group from the commercial resin, the corresponding Fmoc-Xaa-OH amino acids were coupled, using DIC/HOBt, to afford intermediates 5a–f. A similar procedure was followed for the subsequent incorporation of the 1-benzyloxycarbonyl azetidine 2. The resulting resin-bound azetidine-containing dipeptides 6a–f were finally detached from the polymeric support by treatment with a 95:4:1 TFA/H2O/TIPS cleavage cocktail. As for the previous series, compounds 7 were obtained in excellent yields as mixtures of two diastereoisomers that were separated by column chromatography, and configurationally assigned as previously indicated for analogues 4.
Scheme 2.
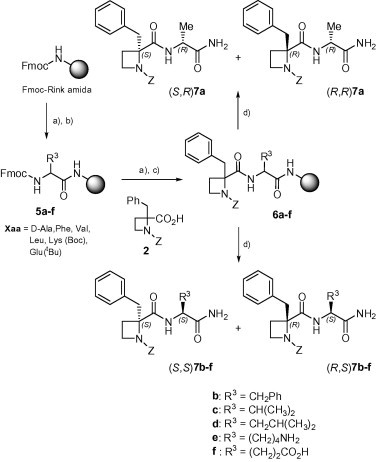
Synthesis of C-terminal side-chain-modified compounds (B). Reagents: (a) 20% pip/DMF; (b) Fmoc-Xaa-OH/DIC/HOBt/DMF; (c) 2/DIC/HOBt/DMF; (d) TFA/H2O/TIPS (95:4:1).
Taking into account the inactivity of the N-methoxycarbonyl-substituted analogue of 1,14b which suggested a role for the aromatic phenyl ring of the Z-group, different Ar-containing urethane, acyl and urea groups were envisaged as substituents at the N1 position of the azetidine moiety (series C). The good results in the synthesis of compounds 7 prompted us to apply a similar solid-phase approach for the preparation of the third series of derivatives (Scheme 3 ). In this case, the Fmoc-l-Ala resin 5g was deprotected and coupled to the Fmoc-azetidine derivative 8 to provide key intermediate 9. Unfortunately, this coupling step was difficult and, despite it was repeated, the Kaiser test indicated an incomplete incorporation. The direct cleavage of 9 afforded after purification the Fmoc-azetidine dipeptide 10a. Removal of the Fmoc-protecing group from 9 followed by reaction with different chloroformates or acyl- or sulfonyl-chlorides in the presence of propylene oxide gave to derivatives 10b–g, after cleavage. Alternatively, the treatment with isocyanates, followed by exposure to the cleavage cocktail, allowed the preparation of urea analogues 10h–i. In all these cases, only the major S,S-diastereoisomer of the mixture was isolated in enough purity for biological assays. The deficient coupling of azetidine derivative 8 could be responsible for the formation of the complex reaction crudes, making the purification step more difficult.
Scheme 3.
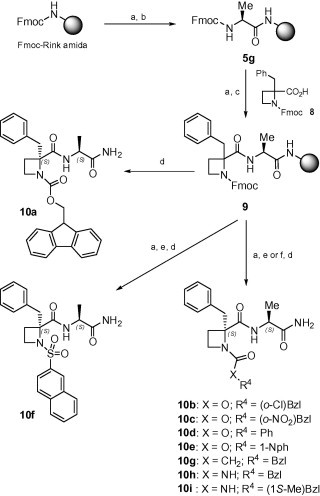
Synthesis of N-terminal-modified compounds (C). Reagents: (a) 20% pip/DMF; (b) Fmoc-Ala-OH/DIC/HOBt/DMF; (c) 8/DIC/HOBt/DMF; (d) TFA/H2O/TIPS (95:4:1); (e) R4XCOCl or R4SO2Cl/propylene oxide/DMF; (f) R4NCO/DMF.
All prepared compounds were characterised by standard analytical and spectroscopic methods. The 1H NMR spectra always showed cis/trans rotamer around the CON1 bond, fluctuating between 10% and 20% of cis conformer in CDCl3 and increasing up to 40% in DMSO-d 6.
2.2. Conformational analysis by 1H NMR
In our previous work on anti-HCMV N-benzyloxycarbonylazetidines, we found that some active 2-carbamoyl derivatives had low conformational flexibility, due to the formation of a reverse γ-turn, stabilized through an intramolecular hydrogen bond between the Z carbonyl group and the NH proton of the 2-carboxamide.14 Recent studies on 2-alkyl-2-carboxy azetidine-containing model peptides confirmed that these non-proteinogenic amino acids are valuable γ-turn inducers.16 To ascertain whether this particular folded conformation could play or not a role in the antiviral activity of the new prepared compounds, their chemical shifts and temperature coefficients for amide protons were evaluated.
In CDCl3, the chemical shift values for the α-NH amide protons in the major trans-rotamer were always >7 ppm, and the variation when changing to DMSO-d 6 was very small (0.01–0.3 ppm in most cases), suggesting the participation of these protons in intramolecular stable hydrogen bonds that protect them from solvent influence.17 On the contrary, the C-terminal CONH2 or CONHR amide protons showed chemical shifts in the range of 5.27–6.82 ppm in CDCl3, while the change of solvent resulted in an important fluctuation of δ values, indicative of solvent exposure (normally >1 ppm, see Table 1S, Supplementary data).
The variation of amide proton chemical shifts with the temperature was measured in DMSO-d 6 for all compounds 4, 7 and 10, and the calculated temperature coefficients are recorded in Table 1 . It is established that, in small peptides, values below 3 ppb/K (in absolute value) are indicative of solvent-protected NH, probably implicated in a hydrogen bond, while values above 4 ppb/K indicated accessibility to solvent or non-hydrogen-bonded states, and those between 3 and 4 ppb/K are in the range of uncertainty.18 As shown in the table, most compounds showed Δδ/ΔT values for the α-NH amide proton <3 ppb/K, while for a small number of them the values were not conclusive. These data point towards the stabilization of a γ-turn structure through the formation of an intramolecular hydrogen bond implicating the α-NH amide proton. In contrast, the temperature coefficients for the C-terminal amide protons are large enough, indicating total accessibility to the solvent. The only exception was compound (R,S)-4b, for which the Δδ/ΔT values for both amide protons suggested either the coexistence of two conformational states, β- and γ-turn or a single double-turned conformation.
Table 1.
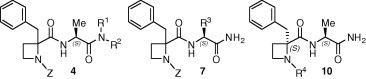
| Compd | R1, R2 | Δδ/ΔT (ppb/K) |
|
|---|---|---|---|
| α-NHa | CONHa,b | ||
| (S,S)-4a | H, C(CH)3 | −2.5 | −3.5 |
| (R,S)-4a | H, C(CH)3 | −2.0 | −4.0 |
| (S,S)-4b | H, CH2Ph | −2.5 | −4.5 |
| (R,S)-4b | H, CH2Ph | −1.6 | −2.9 |
| (S,S)-4c | H, CH2CH(CH3)2 | −3.5 | −4.0 |
| (S,S)-4d | H, Chx | −3.0 | −4.0 |
| (R,S)-4d | H, Chx | −2.5 | −5.5 |
| (S,S)-4e | CH3, CH3 | −2.5 | — |
| (R,S)-4e | CH3, CH3 | −2.5 | — |
| Compd | R3 | ||
| (S,R)-7a | CH3 | −3.3 | −5.1 |
| (R,R)-7a | CH3 | −2.4 | −4.1, −5.4 |
| (S,S)-7b | CH2Ph | −3.6 | −4.5 |
| (R,S)-7b | CH2Ph | −2.2 | −5.3 |
| (S,S)-7c | CH(CH3)2 | −2.5 | −8.1 |
| (R,S)-7c | CH(CH3)2 | −1.8 | −4.8, −5.2 |
| (S,S)-7d | CH2CH(CH3)2 | −3.5 | −5.6 |
| (R,S)-7d | CH2CH(CH3)2 | −2.2 | −4.6 |
| Compd | R4 | ||
| (S,S)-10a | Fmoc | −2.8 | −4.7 |
| (S,S)-10b | CO2CH2C6H4Cl | −3.0 | −4.3, −5.5 |
| (S,S)-10c | CO2CH2C6H4NO2 | −3.0 | −4.7, −5.1 |
| (S,S)-10d | CO2Ph | −2.1 | −4.0 |
| (S,S)-10e | CO2C10H7 | −3.1 | −4.5 |
| (S,S)-10f | SO2C10H7 | −4.0 | −4.6, −5.7 |
| (S,S)-10g | CO2(CH2)2Ph | −2.7 | −4.9, −5.6 |
| (S,S)-10h | CONHCH2Ph | −4.0 | −4.8 |
| (S,S)-10i | CONHCH(Me)Ph | −3.8 | −4.7 |
Determined by least-squares linear regression analysis from measurements over the range 30–60 °C (seven points), in DMSO-d6.
CONH2 for series B and C.
2.3. Antiviral activity
The azetidine-derived dipeptides of the three series, A–C, were evaluated for their ability to inhibit the replication of HCMV in vitro.19 The results were compared to those of the reference compound 1 and the commercial drug ganciclovir (Table 2, Table 3, Table 4 ).
Table 2.
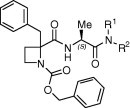
| Compd | R1, R2 | EC50 (μM) | Cytotoxicity (μM) |
|
|---|---|---|---|---|
| HCMVa | MCCb | CC50c | ||
| (S,S)-4a | H, C(CH)3 | 45 | >100 | >100 |
| (R,S)-4a | H, C(CH)3 | 49 | >100 | >100 |
| (S,S)-4b | H, CH2Ph | >20 | 100 | 42 |
| (R,S)-4b | H, CH2Ph | >4 | ⩾20 | 42 |
| (S,S)-4c | H, CH2CH(CH3)2 | 63 | >100 | >100 |
| (R,S)-4c | H, CH2CH(CH3)2 | >100 | >100 | >100 |
| (S,S)-4d | H, Chx | >100 | >100 | >100 |
| (R,S)-4d | H, Chx | >20 | 100 | 46 |
| (S,S)-4e | CH3, CH3 | >100 | >100 | >100 |
| (R,S)-4e | CH3, CH3 | >100 | >100 | >100 |
| (S,S)-1 | H, H | 32 | >50 | 50 |
| 11 | Z-Phe-Ala-NH2 | >100 | >100 | >100 |
| 12 | Z-Phe-Ala-NHtBu | >100 | >100 | >100 |
| GCV | 7.0 | ⩾1575 | 580 | |
Effective concentration required to reduce virus plaque formation by 50% (Davis strain). Virus input was 100 plaque forming units (PFU).
Minimum cytotoxic concentration that causes a microscopically detectable alteration of cell morphology.
Cytotoxic concentration required to reduce cell growth by 50%.
Table 3.
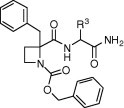
| Compd | R3 | EC50 (μM) | Cytotoxicity (μM) |
|
|---|---|---|---|---|
| HCMVa | MCCb | CC50c | ||
| (S,R)-7a | CH3 | 63 | >100 | 36 |
| (R,R)-7a | CH3 | >100 | >100 | >100 |
| (S,S)-7b | CH2Ph | >20 | 100 | 33 |
| (R,S)-7b | CH2Ph | >20 | 100 | 37 |
| (S,S)-7c | CH(CH3)2 | 76 | >100 | 100 |
| (R,S)-7c | CH(CH3)2 | >100 | >100 | 85 |
| (S,S)-7d | CH2CH(CH3)2 | >20 | 100 | 50 |
| (R,S)-7d | CH2CH(CH3)2 | 45 | >100 | 66 |
| 7e | (CH2)4NH2 | >100 | >100 | >100 |
| 7f | (CH2)2CO2H | >100 | >100 | >100 |
| (S,S)-1 | CH3 | 32 | >50 | 50 |
| GCV | 7.0 | ⩾1575 | 580 | |
a, b and c as defined in Table 2.
Table 4.
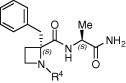
| Compd | R4 | EC50 (μM) | Cytotoxicity (μM) |
|
|---|---|---|---|---|
| HCMVa | MCCb | CC50c | ||
| (S,S)-10a | 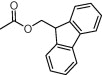 |
>4 | 20 | 14 |
| (S,S)-10b | 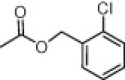 |
>20 | >100 | >100 |
| (S,S)-10c | 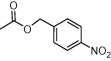 |
>20 | 100 | >100 |
| (S,S)-10d | 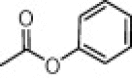 |
>20 | >100 | >100 |
| (S,S)-10e | 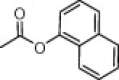 |
45 | >100 | >100 |
| (S,S)-10f | 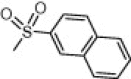 |
>100 | >100 | >100 |
| (S,S)-10g | 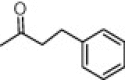 |
>100 | 100 | >100 |
| (S,S)-10h | 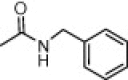 |
>100 | >100 | >100 |
| (S,S)-10i | 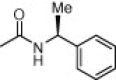 |
>100 | >100 | >100 |
| (S,S)-1 | 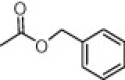 |
32 | >50 | 50 |
| GCV | 5.6 | 1575 | 63 | |
a, b and c as defined in Table 2.
As shown in Table 2, derivatives (S,S)-4a, (R,S)-4a and (S,S)-4c, with tert-butyl and iso-butyl groups at the C-terminal amide, retain the anti-HCMV activity with EC50 values comparable to model dipeptide 1. Benzyl- and cyclohexyl-substituted analogues were inactive, indicating a preference for aliphatic non-cyclic substituents at the C-terminal position. The lack of antiviral activity of the N,N-dimethyl derivatives 4e could suggest the need for a proton at the amide group and its possible implication in the direct interaction with the biological target. This could explain the inactivity of the ester-substituted analogues found in our previous work.14b Although only a few compounds showed significant activity, a potential preference for the S,S-configuration could be deduced from the comparison of the inhibitory potency between the diastereoisomeric pairs of compounds 4a and 4c (and data obtained from the B and C series of compounds).
As indicated in the previous section, the described azetidine dipeptides adopt a γ-turn-like conformation, centred at the azetidine residue. To explore to which extent this structural characteristic could be important for HCMV antiviral activity, dipeptides Z-Phe-Ala-NH2 (11) and Z-Phe-Ala-NHtBu (12), non-constrained analogues of active compounds 1 and 4a, respectively, were synthesized and evaluated. All the amide protons of peptides 11 and 12 showed temperature coefficients higher than 4.5 ppb/K, in absolute value, indicating random coil conformations, and therefore higher flexibility than azetidine-containing analogues. Both dipeptides were unable to inhibit the replication of HCMV in cell culture (Table 2), suggesting a key function of the azetidine ring in the observed antiviral activity. A direct role, in which the methylene groups of the four-member ring interact with the target, or an indirect function to spatially pre-organise the molecule to facilitate the interaction through other functionalities, could be envisaged.
In series B (Table 3), only compounds (S,R)-7a, (S,S)-7c and (R,S)-7d, derived from d-Ala, -Val and -Leu, respectively, maintain measurable anti-HCMV activity, while the Phe, Lys, and Glu analogues were inactive. Again, a preference for aliphatic groups at the C-terminal residue is observed. However, neither an increase of the aliphatic side-chain volume (Val or Leu vs Ala) nor a change in the configuration of the Ala residue succeeded in improving the activity of prototype 1. Active compounds in the B series are—as a rule—also somewhat cytotoxic, specially the d-Ala- and Phe-containing analogues.
As shown in Table 4, most prepared derivatives in compound series C were totally inactive at 100 μM, except for compound (S,S)-10e, bearing a 1-naphtylcarbamate group. This group can be considered as a conformationally restricted analogue of the benzyloxycarbonyl substituent present in prototype 1. These results suggest that the Z group plays a key role in the activity of these azetidine-derived compounds. In fact, small changes, such as the incorporation of different types of substituents at the aromatic phenyl ring (10b, 10c), the shortening of the benzyl group to phenyl (10d) and the replacement of the urethane oxygen atom by CH2 or NH (10g, 10h, 10i), led to inactive analogues. Compound 10a, being inactive against HCMV, was the most cytotoxic/cytostatic compound among the three series of azetidine derivatives (Table 3).
To assess HCMV selectivity, all compounds were also evaluated for antiviral activity against a wide panel of DNA and RNA viruses. Some active derivatives against HCMV also inhibited the replication of varicella-zoster virus (VZV, Table 2S within Supplementary data), while they were inactive against a wide variety of other DNA or RNA viruses at subtoxic concentrations (20–100 μM). Dual anti-VZV and anti-HCMV activity was also described for non-nucleoside 4-benzyloxy-γ-sultones and bicyclic furanopyrimidine-derived nucleosides.(b), 20
3. Conclusion
Structure–activity studies on a new family of azetidine-containing dipeptide inhibitors of HCMV are described. Compounds modified at the C-terminal residue (carboxamide and amino acid side-chain) and at the N-terminus were easily synthesized by application of both solid-phase and solution methodologies. The results of the biological assays indicated quite strict structural requirements for activity. Thus, only derivatives with aliphatic groups at the C-terminal residue, either at the carboxamide moiety or at the side-chain showed significant inhibitory activity against HCMV replication in cell cultures. Additionally, at the C-carboxamide group, the presence of a hydrogen atom is an absolute requirement. Substitutions at N-terminus recommended the initial benzyloxycarbonyl (Z) group as the best choice at this position. In addition, the conformational restriction induced by the reverse turn (γ-type) seems important for activity, since more flexible dipeptide analogues were totally inactive. Further SAR studies are needed to improve the activity of the present series of inhibitors.
4. Experimental
For details on general methods and complete 1H, 13C and MS characterisation of all intermediates and final compounds, see the Supplementary data. Azetidine 2 was synthesized according to the previously reported method.14b
4.1. (2R,S)-2-Benzyl-1-(9-fluorenylmethoxy)carbonyl-2-carboxyazetidine (8)
A solution of (2R,S)-2-benzyl-2-methoxycarbonylazetidine15a (1.57 g, 7.64 mmol) in MeOH (20 mL) was treated with 2 N NaOH (19.1 mL, 9.55 mmol). The reaction was stirred at rt for 24 h. The solvent was removed and the residue dissolved in water and washed with EtOAc. The aqueous layer was lyophilised and the resulting residue was dissolved in a mixture of Na2CO3 (10%)/dioxane (20 mL/12 mL). The reaction was cooled at 0 °C and a solution of Cl-Fmoc (1.97 g, 7.64 mmol) in dioxane (12 mL) was added. After 1 h, the temperature was allowed to raise at rt and the reaction was stirred for 3 h. Then, water was added and the mixture was washed with Et2O. The organic layer was acidified with 1 M HCl up to pH 2 and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4 and the solvent removed under vacuum. The residue was purified by flash chromatography employing a mixture of EtOAc/CH2Cl2 (1:7) to afford 1.89 g (60%) of the title azetidine 8 as a solid. Mp: 88–90 °C. HPLC: R t = 5.78 min (A/B = 35:65). 1H NMR (CDCl3) rotamers ratio: 3.2:1. Major rotamer δ: 7.85–7.18 (m, 13H, Ar), 4.56–4.48 (m, 2H, CH 2-Fmoc), 4.34 (m, 1H, CH-Fmoc), 3.75 (m, 1H, 4-H), 3.56 (d, 1H, J = 14.2, 2-CH 2), 3.18 (m, 1H, 4-H), 3.09 (d, 1H, J = 14.2, 2-CH 2), 2.63 (m, 1H, 3-H), 2.32 (m, 1H, 3-H). Anal. Calcd for C26H23NO4: C, 75.53; H, 5.61; N, 3.39. Found: C, 75.78; H, 5.73; N, 3.42.
4.2. Synthesis of azetidine-derived dipeptides
4.2.1. Series A
General procedure for the coupling reaction: A solution of the corresponding H-Ala-NR1R2 trifluoroacetate (0.36 mmol) in anhydrous THF, was treated with azetidine 2 (58 mg, 0.18 mmol), BOP (0.16 g, 0.36 mmol) and Et3N (0.1 mL, 0.72 mmol). The reaction was stirred overnight at rt. The solvent was evaporated and the resulting residue was dissolved in EtOAc and washed with citric acid (10%), NaHCO3 (10%) and brine. The organic layer was dried over Na2SO4 and evaporated affording a residue, which was purified by flash chromatography (EtOAc/Et2O/hexane, 1:1:1).
4.2.1.1. (2S,1′S)-2-Benzyl-1-benzyloxycarbonyl-2-[(1′-tert-butylcarba-moyl)ethyl]carbamoylazetidine, (S,S)-4a
Syrup. Yield: 58%. [α]D = +7.3 (c 0.49, CHCl3). HPLC: R t = 19.79 min (A/B = 50:50). 1H NMR (DMSO-d 6), rotamers ratio: 5.2:1. Major rotamer δ: 8.20 (br d, 1H, J = 7.7, 1′-NH), 7.79 (br d, 1H, J = 7.3, NHtBu), 7.45–7.13 (m, 10H, Ph), 5.24 (d, 1H, J = 12.4, CH 2-Z), 5.04 (d, 1H, J = 12.4, CH 2-Z), 4.29 (m, 1H, 1′-H), 3.53 (m, 1H, 4-H), 3.31 (d, 1H, J = 13.9, 2-CH 2), 3.03 (d, 1H, J = 13.9, 2-CH 2), 2.88 (dd, 1H, J = 8.1 and 16.5, 4-H), 2.32 (m, 1H, 3-H), 2.10 (m, 1H, 3-H), 1.27 (s, 9H, tBu),1.20 (d, 3H, J = 6.9, 1′-CH 3). MS: 452.2 [M+1]+, 474.2 [M+Na]+. Anal. Calcd for C26H33N3O4: C, 69.16; H, 7.37; N, 9.31. Found: C, 68.87; H, 7.61; N, 9.29.
4.2.1.2. (2R,1′S)-2-Benzyl-1-benzyloxycarbonyl-2-[(1′-tert-butylcarba-moyl)ethyl]carbamoylazetidine, (R,S)-4a
Syrup. Yield: 25%. [α]D = −47.5 (c 0.61, CHCl3). HPLC: R t = 17.60 min (A/B = 50:50). 1H NMR (DMSO-d 6), rotamers ratio: 4.6:1. Major rotamer δ: 8.14 (d, 1H, J = 7.7, 1′-NH), 7.81 (br d, 1H, J = 7.6, NHtBu), 7.46–7.13 (m, 10H, Ph), 5.23 (d, 1H, J = 12.4, CH 2-Z), 5.06 (d, 1H, J = 12.4, CH 2-Z), 4.29 (m, 1H, 1′-H), 3.50 (m, 1H, 4-H), 3.36 (d, 1H, J = 13.9, 2-CH 2), 2.98 (d, 1H, J = 13.9, 2-CH 2), 2.79 (m, 1H, 4-H), 2.30 (m, 1H, 3-H), 2.11 (m, 1H, 3-H), 1.25 (s, 9H, tBu), 1.23 (d, 3H, J = 7.3, 1′-CH 3). MS: 452.2 [M+1]+, 474.2 [M+Na]+. Anal. Calcd for C26H33N3O4: C, 69.16; H, 7.37; N, 9.31. Found: C, 68.94; H, 7.42; N, 9.28.
4.2.1.3. (2S,1′S)-2-Benzyl-1-benzyloxycarbonyl-2-[(1′-isobutylcarba-moyl)ethyl]carbamoylazetidine (S,S)-4c
Foam. Yield: 60%. [α]D = +14.7 (c 0.5, CHCl3). HPLC: R t = 22.29 min (A/B = 47:53). 1H NM (DMSO-d 6), rotamers ratio: 2.8:1. Major rotamer δ: 8.29 (d, 1H, J = 7.3, 1′-NH), 7.85 (m, 1H, NHiBu), 7.44–7.10 (m, 10H, Ph), 5.25 (d, 1H, J = 12.4, CH 2-Z), 5.04 (d, 1H, J = 12.4, CH 2-Z), 4.34 (m, 1H, 1′-H), 3.54 (m, 1H, 4-H), 3.31 (d, 1H, J = 13.9, 2-CH 2), 3.01 (d, 1H, J = 13.9, 2-CH 2), 2.95 (m, 1H, 4-H), 2.84 (m, 2H, CH 2-iBu), 2.32 (m, 1H, 3-H), 2.01 (m, 1H, 3-H), 1.72 (m, 1H, CH-iBu), 1.26 (d, 3H, J = 7.1, 1′-CH 3), 0.85 (d, 6H, J = 6.6, CH 3-iBu). MS: 452.3 [M+1]+, 474.3 [M+Na]+. Anal. Calcd for C26H33N3O4: C, 69.16; H, 7.37; N, 9.31. Found: C, 68.91; H, 7.29; N, 9.15.
4.2.2. Series B and C
General procedure for the solid-phase coupling reactions: A solution of the corresponding Fmoc-Xaa-OH (0.3 mmol) and HOBt (0.3 mmol) in anhydrous DMF (1 mL), was treated with DIC (0.3 mmol) and the resulting mixture was added over the Fmoc-deprotected resin (0.1 mmol). After 18 h of slow stirring at rt, the excess of reagents was eliminated by successive washes with DMF/DCM/DMF/DCM (4 × 0.5 min) to afford intermediates 5a–g. The efficiency of the coupling reactions was monitored by the Kaiser′s test. A similar procedure was followed for the subsequent incorporation of azetidines 2 and 8.
Synthesis of N-substituted derivatives of series C: After removal of Fmoc group from resin 9, propylene oxide (15 equiv, except for the reaction with isocyanates) was added. The reaction was cooled at 0 °C and 10 equiv of the corresponding isocyanate, chloroformate, acyl- or sulfonyl-chloride were added. After 18 h of slow stirring at room temperature, the excess of reagents was eliminated by successive washes with DMF/DCM/DMF/DCM (4 × 0.5 min) to afford compounds 10b–i. The efficiency of the coupling reactions was monitored by the chloranyl test.
Cleavage reactions: The resin-bound compounds were cleaved by treatment with the TFA/H2O/TIPS (95:4:1) cocktail for 3 h at rt. The filtrate was kept and the resin was washed with DCM (3 × 3 mL). The filtrates, containing final products, were combined and evaporated under vacuum. The resulting residue was dissolved in water and lyophilised before chromatography, using the solvent system indicated in each case.
4.2.2.1. (2S,1′R)-2-Benzyl-1-benzyloxycarbonyl-2-[(1′-carbamoyl)ethyl]-carbamoylazetidine, (S,R)-7a
Foam. 5–17% Acetone in CH2Cl2. Yield: 70%. [α]D = +20.0 (c 1.3, CHCl3). HPLC: R t = 8.72 min (A/B = 35:65). 1H NMR (DMSO-d 6), rotamers ratio: 2.2:1. Major rotamer δ: 8.06 (d, 1H, J = 7.2, 1′-NH), 7.41–7.13 (m, 11H, Ph, CONH 2), 7.05 (br s, 1H, CONH 2), 5.23 (d, 1H, J = 12.5, CH 2-Z), 5.03 (d, 1H, J = 12.5, CH 2-Z), 4.28 (m, 1H, 1′-H), 3.49 (m, 1H, 4-H), 3.37 (d, 1H, J = 13.8, 2-CH 2), 2.99 (d, 1H, J = 13.8, 02-CH 2), 2.79 (m, 1H, 4-H), 2.30 (m, 1H, 3-H), 2.11 (m, 1H, 3-H), 1.16 (d, 3H, J = 6.7, 1′-CH3). MS: 396.1 [M+1]+. Anal. Calcd for C22H25N3O4: C, 66.82; H, 6.37; N, 10.63. Found: C, 66.73; H, 6.43; N, 10.55.
4.2.2.2. (2R,1′S)-2-Benzyl-1-benzyloxycarbonyl-2-(1′-carbamoyl-2′-methyl)propyl]carbamoylazetidine, (R,S)-7c
Syrup. 5–17% Acetone in CH2Cl2. Yield: 24%. [α]D = −9.3 (c 1.2, CHCl3). HPLC: R t = 19.84 min (A/B = 35:65). 1H NMR (DMSO-d 6), rotamers ratio: 2.5:1. Major rotamer δ: 7.88 (d, 1H, J = 8.7, 1′-NH), 7.48 (br s, 1H, CONH 2), 7.38–7.10 (m, 10H, Ph), 7.07 (br s, 1H, CONH 2), 5.25 (d, 1H, J = 12.3, CH 2-Z), 5.06 (d, 1H, J = 12.3, CH 2-Z), 4.21 (m, 1H, 1′-H), 3.45 (m, 1H, 4-H), 3.36 (d, 1H, J = 13.9, 2-CH 2), 2.98 (d, 1H, J = 13.9, 2-CH 2), 2.78 (m, 1H, 4-H), 2.37 (m, 1H, 3-H), 2.16 (m, 2H, 2′-H, 3-H), 0.87 (d, 3H, J = 6.6, 2′-CH 3), 0.76 (d, 3H, J = 6.8, 3′-H). MS: 424.2 [M+1]+, 446.3 [M+Na]+. Anal. Calcd for C24H29N3O4: C, 68.06; H, 6.90; N, 9.92. Found: C, 68.17; H, 7.01; N, 9.85.
4.2.2.3. (2R,1′S)-2-Benzyl-1-benzyloxycarbonyl-2-[(1′-carbamoyl-3′-methyl)butyl]carbamoylazetidine, (R,S)-7d
Syrup. 5–25% acetone in CH2Cl2. Yield: 27%. [α]D = −6.3 (c 0.9, CHCl3). HPLC: R t = 13.54 min (A/B = 40:60). 1H NMR (DMSO-d 6), rotamers ratio: 2.2:1. Major rotamer δ: 7.94 (d, 1H, J = 8.5, 1′-NH), 7.48–7.15 (m, 11H, Ph, CONH 2), 6.99 (br s, 1H, CONH 2), 5.24 (d, 1H, J = 12.4, CH 2-Z), 5.07 (d, 1H, J = 12.4, CH 2-Z), 4.35 (m, 1H, 1′-H), 3.48 (m, 1H, 4-H), 3.28 (d, 1H, J = 13.9, 2-CH 2), 3.02 (d, 1H, J = 13.9, 2-CH 2), 2.78 (m, 1H, 4-H), 2.35 (m, 1H, 3-H), 2.14 (m, 2H, 3-H, 2′-H), 1.58 (m, 2H, 2′-H and 3′-H), 0.87 (d, 3H, J = 6.6, 3′-CH 3), 0.83 (d, 3H, J = 6.7, 4′-H). MS: 438.3 [M+1]+, 460.2 [M+Na]+. Anal. Calcd for C25H31N3O4: C, 68.63; H, 7.14; N, 9.60. Found: C, 68.71; H, 7.23; N, 9.52.
4.2.2.4. (2S,1′S)-2-Benzyl-2-[(1′-carbamoyl)ethyl]carbamoyl-1-(1-naphthyloxy)carbonylazetidine, (S,S)-10e
Syrup. Acetone/CH2Cl2, 1:6. Yield: 52%. [α]D = +18.1 (c 0.8, CHCl3). HPLC: R t = 13.31 min (A/B = 40:60). 1H NMR (DMSO-d 6), rotamers ratio: 2.2:1. Major rotamer δ: 8.16 (d, 1H, J = 7.7, 1′-NH), 7.72–7.35 (m, 13H, Ar, CONH 2), 7.31 (br s, 1H, CONH 2), 4.32 (m, 1H, 1′-H), 3.64 (m, 1H, 4-H), 3.27 (d, 1H, J = 13.8, 2-CH 2), 3.18 (d, 1H, J = 13.8, 2-CH 2), 2.98 (m, 1H, 4-H), 2.33 (m, 1H, 3-H), 2.28 (m, 1H, 3-H), 1.28 (d, 3H, J = 7.6, 1′-CH 3). MS: 432.2 [M+1]+. Anal. Calcd for C25H25N3O4: C, 69.59; H, 5.84; N, 9.74. Found: C, 69.66; H, 5.88; N, 9.68.
4.2.2.5. (2S,1′S)-2-Benzyl-2-[(1′-carbamoyl)ethyl]carbamoyl-1-(2-naphtyl)-sulfonylazetidine, (S,S)-10f
Solid. Acetone/CH2Cl2, 1:6. Yield: 42%. Mp: 154–156 °C. [α]D = −41.2 (c 0.3, CHCl3). HPLC: R t = 17.07 min (A/B = 40:60). 1H NMR (DMSO-d 6): 8.51 (br s, 1H, 1′-NH), 8.21–7.64 (m, 7H, Naph), 7.41 (br s, 1H, CONH 2), 7.28–7.18 (m, 5H, Ph), 7.21 (br s, 1H, CONH 2), 4.33 (m, 1H, 1′-H), 3.73 (m, 1H, 4-H), 3.62 (m, 1H, 4-H), 3.48 (d, 1H, J = 13.8, 2-CH 2), 3.24 (d, 1H, J = 13.8, 2-CH 2), 2.48 (m, 1H, 3-H), 2.27 (m, 1H, 3-H), 1.36 (d, 3H, J = 7.3, 1′-CH 3). MS: 452.2 [M+1]+. Anal. Calcd for: C24H25N3O4S: C, 63.84; H, 5.58; N, 9.31. Found: C, 63.88; H, 5.61; N, 9.28.
4.2.2.6. (2S,1′S)-2-Benzyl-2-[(1′-carbamoyl)ethyl]carbamoyl-1-phenetyl-carbonylazetidine, (S,S)-10g
Foam. 15–33% acetone in CH2Cl2. Yield: 28%. [α]D = +14.2 (c 0.2, CHCl3). HPLC: R t = 10.03 min (A/B = 40:60). 1H NMR (DMSO-d 6), rotamers ratio: 4.5:1. Major rotamer δ: 8.67 (d, 1H, J = 7.6, 1′-NH), 7.2 (br s, 1H, CONH 2), 7.28–7.16 (m, 10H, Ph), 7.05 (br s, 1H, CONH 2), 4.26 (m, 1H, 1′-H), 3.63 (m, 1H, 4-H), 3.33 (d, 1H, J = 13.6, 2-CH 2), 2.98 (d, 1H, J = 13.6, 2-CH 2), 2.98 (m, 1H, 4-H), 2.82 (m, 2H, CH2CH 2Ph), 2.34 (m, 2H, CH 2CH2Ph), 2.24 (m, 1H, 3-H), 2.07 (m, 1H, 3-H), 1.36 (d, 3H, J = 7.4, 1′-CH 3). MS: 394.2 [M+1]+. Anal. Calcd for C23H27N3O3: C, 70.21; H, 6.92; N, 10.68. Found: C, 70.09; H, 7.07; N, 10.57.
4.2.2.7. (2S,1′S,1″S)-2-Benzyl-2-[(1″-carbamoyl)ethyl]carbamoyl-1-[(1′-phenyl)ethyl]carbamoylazetidine, (S,S)-10i
Solid. Acetone/CH2Cl2, 1:4. Yield: 35%. Mp: 137–139 °C. [α]D = +4.0 (c 0.9, CHCl3). HPLC: R t = 6.67 min (A/B = 40.60). 1H NMR (DMSO-d 6), rotamers ratio: 4.7:1. Major rotamer δ: 8.12 (br s, 1H, 1″-NH), 7.38–7.14 (m, 12H, 1′-NH, Ph, CONH 2), 7.06 (br s, 1H, CONH 2), 4.97 (m, 1H, 1′-H), 4.34 (m, 1H, 1″-H), 3.49 (m, 1H, 4-H), 3.35 (d, 1H, J = 13.6, 2-CH 2), 3.27 (d, 1H, J = 13.6, 2-CH 2), 2.82 (m, 2H, 4-H and 3-H), 2.36 (m, 1H, 3-H), 1.36 (d, 3H, J = 7.4, 1′-CH 3), 1.24 (d, 3H, J = 7.1, 1″-CH 3). MS: 409.2 [M+1]+. Anal. Calcd for C23H28N4O3: C, 67.63; H, 6.91; N, 13.72. Found: C, 67.69; H, 7.02; N, 13.67.
4.3. Antiviral assays
The antiviral assays, other than the anti-HIV, -HCMV and -VZV assays, were based on inhibition of virus-induced cytopathicity in HEL [herpes simplex virus type 1 (HSV-1) (KOS), HSV-2 (G), vaccinia virus, vesicular stomatitis virus, cytomegalovirus (HCMV) and varicella-zoster virus (VZV), Vero (parainfluenza-3, reovirus-1, Sindbis and Coxsackie B4), HeLa (vesicular stomatitis virus, Coxsackie virus B4, and respiratory syncytial virus), CrFK (feline coronavirus (FIPV) and feline herpes virus) or MDCK [influenza A (H1N1; H3N2) and influenza B] cell cultures. Confluent cell cultures (or nearly confluent for MDCK cells) in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures). After a 1-h virus adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations (fivefold compound dilutions) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. The minimal cytotoxic concentration (MCC) of the compounds was defined as the compound concentration that caused a microscopically visible alteration of cell morphology. The methodology of the anti-HIV assays was as follows: human CEM (∼3 × 105 cells/cm3) cells were infected with 100 CCID50 of HIV(IIIB) or HIV-2 (ROD)/mL and seeded in 200 μL wells of a microtiter plate containing appropriate dilutions of the test compounds. After 4 days of incubation at 37 °C, HIV-induced CEM giant cell formation was examined microscopically.
4.3.1. Anti-HCMV assays
Confluent human embryonic lung (HEL) fibroblasts were grown in 96-well microtiter plates and infected with the human cytomegalovirus (HCMV) strain Davis at 100 PFU per well. After a 2-h incubation period, residual virus was removed and the infected cells were further incubated with medium containing different concentrations of the test compounds (in duplicate). After incubation for 7 days at 37 °C, virus-induced cytopathogenicity was monitored microscopically after ethanol fixation and staining with Giemsa. Antiviral activity was expressed as the EC50 or compound concentration required to reduce virus-induced cytopathogenicity by 50%. EC50’s were calculated from graphic plots of the percentage of cytopathogenicity as a function of concentration of the compounds.
4.3.2. Cytostatic toxicity assays
Cytostatic activity measurements were based on the inhibition of HEL cell growth. HEL cells were seeded at 5 × 103 cells/well into 96-well microtiter plates and allowed to proliferate for 24 h. Then, medium containing different concentrations of the test compounds was added. After 3 days of incubation at 37 °C, the cell number was determined with a Coulter Counter. The cytostatic concentration was calculated as the CC50 or compound concentration required to reduce growth by 50% relative to the number of cells in the untreated controls. CC50 values were estimated from graphic plots of the number of cells (percentage of control) as a function of the concentration of the test compounds. Cytotoxicity was expressed as minimum cytotoxic concentration (MCC) or compound concentration that causes a microscopically detectable alteration of cell morphology.
Acknowledgements
This work was supported by the Spanish Ministry of Science and Innovation (SAF 2006-01205 and SAF 2009-09323) and the K.U.L. (GOA no. 10/14). P.P.-F. and M.T.A. thank the CSIC for a pre-doctoral I3P fellowship and a postdoctoral I3P contract, respectively. Thanks to Mrs. Lies Van den Heurck, Mrs. Anita Camps, Mr. Steven Carmans, Mrs. Leentje Persoons, Mrs. Leen Ingels and Mrs. Frieda De Meyer for excellent technical assistance.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2010.12.052.
Supplementary data
References and notes
- 1.Trincado D.E., Scott G.M., White P.A., Hunt C., Rasmussen L., Rawlinson W.D. J. Med. Virol. 2000;61:481. doi: 10.1002/1096-9071(200008)61:4<481::aid-jmv11>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 2.Melnick J.L., Hu C., Burek J., Adam E., DeBakey M.E. J. Med. Virol. 1994;42:170. doi: 10.1002/jmv.1890420213. [DOI] [PubMed] [Google Scholar]
- 3.Zhou Y.F., Leon M.B., Waclawiw M.A., Popma J.J., Yu Z.X., Finkel T., Epstein S.E. N. Eng. J. Med. 1996;335:624. doi: 10.1056/NEJM199608293350903. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths P.D. J. Antimicrob. Chemother. 2002;49:243. doi: 10.1093/jac/49.2.243. [DOI] [PubMed] [Google Scholar]
- 5.Biron K.K. Antiviral Res. 2006;71:154. doi: 10.1016/j.antiviral.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Baldanti F. J. Antimicrob. Chemother. 2003;52:324. doi: 10.1093/jac/dkg354. [DOI] [PubMed] [Google Scholar]
- 7.Biron K.K., Harvey R.J., Chamberlain S.C., Good S.S., Smith A.A., III, Davis M.G., Talarico C.L., Miller W.H., Ferris R., Dornsife R.E., Stanat S.C., Drach J.C., Townsend L.B., Koszalka G.W. Antimicrob. Agents Chemother. 2002;46:2365. doi: 10.1128/AAC.46.8.2365-2372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones T.R., Lee S.W., Johann S.V., Razinkov V., Visalli R.J., Feld B., Bloom J.D., O’Connell J. J. Virol. 2004;78:1289. doi: 10.1128/JVI.78.3.1289-1300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) De Castro S., Peromingo M.-T., Naesens L., Andrei G., Snoeck R., Balzarini J., Velázquez S., Camarasa M.-J. J. Med. Chem. 2008;51:5823. doi: 10.1021/jm800050t. [DOI] [PubMed] [Google Scholar]; (b) De Castro S., García-Aparicio C., Andrei G., Snoeck R., Balzarini J., Camarasa M.-J., Velázquez S. J. Med. Chem. 2009;52:1582. doi: 10.1021/jm8014662. [DOI] [PubMed] [Google Scholar]
- 10.(a) Welch A.R., Woods A.S., McNally L.M., Cotter R.J., Gibson W.A. Proc. Natl. Acad. Sci. U.S.A. 1991;88:10792. doi: 10.1073/pnas.88.23.10792. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shieh H.-S., Kurumbail R.G., Stevens A.M., Stegeman R.A., Sturman E.J., Pak J.Y., Wittwer A.J., Palmier M.O., Wiegand R.C., Holwerda B.C., Stallings W.C. Nature. 1996;383:279. doi: 10.1038/383279a0. [DOI] [PubMed] [Google Scholar]; (c) Waxman L., Darke P.L. Antiviral Chem. Chemother. 2000;11:1. doi: 10.1177/095632020001100101. [DOI] [PubMed] [Google Scholar]
- 11.LaPlante S.R., Bonneau P., Aubry N., Cameron D.R., Déziel R., Grand-Maitre C., Plouffe C., Tong L., Kawai S. J. Am. Chem. Soc. 1999;121:2974. [Google Scholar]
- 12.(a) Pinto I.L., Jarvest R.L., Clarke B., Dabrowski C.E., Fenwick A., Gorczyca M.M., Jennings L.J., Lavery P., Sternberg E.J., Tew D.G., West A. Bioorg. Med. Chem. Lett. 1999;9:449. doi: 10.1016/s0960-894x(99)00005-0. [DOI] [PubMed] [Google Scholar]; (b) Borthwick A.D., Crame A.J., Erlt P.F., Exall A.M., Haley T.M., Hart G.J., Mason A.M., Pennell A.M.K., Sing O.M.P., Weingarten G.G., Woolven J.M. J. Med. Chem. 2002;45:1. doi: 10.1021/jm0102203. [DOI] [PubMed] [Google Scholar]
- 13.(a) Yoakim C., Ogilvie W., Cameron D.R., Chabot C., Guse I., Haché B., Naud J., O’Meara J.A., Plante R., Déziel R. J. Med. Chem. 1998;41:2882. doi: 10.1021/jm980131z. [DOI] [PubMed] [Google Scholar]; Bonneau P., Hasani F., Plouffe C., Malenfant E., Laplante S.R., Guse I., Ogilvie W.W., Plante R., Davidson W.C., Hopkins J.L., Morelock M.M., Cordingley M.G., Déziel R. J. Am. Chem. Soc. 1999;121:2965. [Google Scholar]
- 14.(a) Gerona-Navarro G., Pérez de Vega M.J., García-López M.T., Andrei G., Snoeck R., De Clercq E., Balzarini J., González-Muñiz R. Bioorg. Med. Chem. Lett. 2004;14:2253. doi: 10.1016/j.bmcl.2004.02.010. [DOI] [PubMed] [Google Scholar]; (b) Gerona-Navarro G., Pérez de Vega M.J., García-López M.T., Andrei G., Snoeck R., De Clercq E., Balzarini J., González-Muñiz R. J. Med. Chem. 2005;48:2612. doi: 10.1021/jm0492812. [DOI] [PubMed] [Google Scholar]
- 15.(a) Gerona-Navarro G., Bonache M.A., Alías M., Pérez de Vega M.J., García-López M.T., López P., Cativiela C., González-Muñiz R. Tetrahedron Lett. 2004;45:2193. [Google Scholar]; (b) Gerona-Navarro G., Bonache M.A., Herranz R., García-López M.T., González-Muñiz R. J. Org. Chem. 2001;66:3538. doi: 10.1021/jo015559b. [DOI] [PubMed] [Google Scholar]
- 16.(a) Baeza J.L., Gerona-Navarro G., Pérez de Vega M.J., García-López M.T., González-Muñiz R., Martín-Martínez M. Tetrahedron Lett. 2007;48:3689. [Google Scholar]; (b) Baeza J.L., Gerona-Navarro G., Pérez de Vega M.J., García-López M.T., González-Muñiz R., Martín-Martínez M. J. Org. Chem. 2008;73:1704. doi: 10.1021/jo701746w. [DOI] [PubMed] [Google Scholar]; (c) Baeza J.L., Gerona-Navarro G., Thomson K., Pérez de Vega M.J., Infantes L., García-López M.T., González-Muñiz R., Martín-Martínez M. J. Org. Chem. 2009;74:8203. doi: 10.1021/jo901712x. [DOI] [PubMed] [Google Scholar]
- 17.Belvisi L., Gennari C., Mielgo A., Potenza D., Scolastico C. Eur. J. Org. Chem. 1999:389. [Google Scholar]
- 18.Stevens E.S., Sugawara N., Bonora G.M., Toniolo C. J. Am. Chem. Soc. 1980;102:7048. [Google Scholar]
- 19.Snoeck R., Andrei G., Bodaghi B., Lagneaux L., Daelemans D., De Clercq E., Neytes J., Schols D., Naesens L., Michelson S., Bron D., Otto M.J., Bousseau A., Nemecek C., Roy C. Antiviral Res. 2002;55:413. doi: 10.1016/s0166-3542(02)00074-8. [DOI] [PubMed] [Google Scholar]
- 20.McGuigan C., Brancale A., Andrei G., Snoeck R., De Clercq E., Balzarini J. Bioorg. Med. Chem. Lett. 2003;13:4511. doi: 10.1016/j.bmcl.2003.08.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.