

Abstract
Two electrochemical DNA hybridization biosensors (genosensors) for the detection of a 30-mer sequence unique to severe acute respiratory syndrome (SARS) virus are described in this work. Both genosensors rely on the hybridization of the oligonucleotide target with its complementary probe, which is immobilized on positively charged polylysine modified screen-printed carbon electrodes (SPCEs), through electrostatic interactions. In one design, a biotinylated target is used and the detection of the hybridization reaction is monitored using alkaline phosphatase labeled streptavidin (S-AP). This enzyme catalyzes the hydrolysis of the substrate 3-indoxyl phosphate (3-IP) to indigo, which is then solubilized to indigo carmine and detected by means of cyclic voltammetry (CV). In the other design, the target is labeled using an Au(I) complex, sodium aurothiomalate, and the duplex formation is detected by measuring, for first time, the current generated by the hydrogen evolution catalyzed by the gold label. Using 30 min of hybridization time, a detection limit of 8 pM is calculated for the enzymatic genosensor. Although this good sensitivity cannot be reached with the metal label (0.5 nM), the use of this label allows a considerable decrease of the analysis time. Both genosensors do not require the modification of the oligonucleotide probe and using stringent experimental conditions (60 min of hybridization time and 50% formamide in the hybridization buffer) can discriminate between a complementary oligonucleotide and an oligonucleotide with a three-base mismatch.
Keywords: Genosensor, Polylysine, Alkaline phosphatase, Sodium aurothiomalate, Screen-printed carbon electrode, SARS virus
1. Introduction
The detection of specific DNA sequences provides the basis for detecting a wide variety of infectious and inherited diseases. Conventional methods for the detection of specific DNA sequences are labor-intensive, time-consuming and require the use of carcinogenic or radioactive reagents. Electrochemical hybridization biosensors, coupling the selectivity of the DNA-based recognition layer with the sensitivity of the electrochemical detection step, may greatly simplify the protocols and reduce the time assay of the traditional methods. To prepare electrochemical DNA sensors, a crucial step is the immobilization of single-stranded DNA (ssDNA) probes onto the electrode surface. Many strategies have been developed for anchoring ssDNA on electrode surfaces, including physical adsorption (De la Escosura Muñiz et al., 2007, Fotja et al., 2006, Del Pozo et al., 2005, Erdem et al., 2004, Pividori et al., 2003, De los Santos Álvarez et al., 2002), covalent attachment (Zhu et al., 2005, Teh et al., 2005, Kara et al., 2005), film entrapment (Zhang et al., 2004, Zhang et al., 2003) or affinity binding (Lee et al., 2005, Mir and Katakis, 2005, Hernández Santos et al., 2005, Hernández Santos et al., 2004). An ideal method of immobilization should guarantee the orientation of the probes in the immobilized layers to facilitate the recognition event. For this purpose, in some designs, the DNA probes are modified (thiol (Ma et al., 2006, Abad Valle et al., 2005, Liu et al., 2005, Ozsoz et al., 2003), biotin (Mir and Katakis, 2005, Hernández Santos et al., 2005, Hernández Santos et al., 2004), etc.) and immobilized on the electrode surface by the 5′ or 3′ end. Another possibility to obtain the bases oriented toward the solution is to immobilize the ssDNA through the sugar-phosphate skeleton, using electrostatic adsorption (Lucarelli et al., 2002, Wang and Kawde, 2001) or cationic polymers, such as polypyrrole (Cai et al., 2003). In this work, the oligonucleotide probes are immobilized onto a screen-printed carbon electrode (SPCE) surface, previously modified with polylysine, through electrostatic interactions. Although the high affinity between DNA and (poly)lysine, has been exploited to immobilize DNA onto different solid surfaces in many biological applications (Bussiek et al., 2003, Segura et al., 2003) and in the development of enzymatic electrochemical sensors (Tominaga et al., 2005, Tsujimura et al., 2005), to the best of our knowledge polylysine has not been used yet to capture ssDNA through electrostatic interactions onto SPCEs for the development of DNA hybridization sensors. Polylysine has also been frequently used in the design of DNA microarrays, but in these devices the immobilization of the oligonucleotide is usually performed through covalent attachment with the free amino groups of the polylysine using bifunctional linkers (Wu et al., 2005, Strother et al., 2000). The base-pairing recognition event on the electrode surface can be detected using unlabeled DNA (intrinsic electroactivity of the DNA (De los Santos Álvarez et al., 2002, Erdem et al., 2004, Lucarelli et al., 2002, Wang and Kawde, 2001), electroactive indicators (Del Pozo et al., 2005, Teh et al., 2005, Zhu et al., 2005), …) or employing enzymatic (Abad Valle et al., 2005, Fotja et al., 2006, Hernández Santos et al., 2004, Kara et al., 2005, Mir and Katakis, 2005, Pividori et al., 2003, Zhang et al., 2004, Zhang et al., 2003) or metal (De la Escosura Muñiz et al., 2007, Lee et al., 2005, Liu et al., 2005, Ozsoz et al., 2003) labels. Although the use of labeled oligonucleotides makes the assay more complex and expensive, a very sensitive electrochemical detection is reached using these labels. In this work, two electrochemical DNA hybridization sensors are developed using both the enzyme alkaline phosphate (AP) and the Au(I) complex, sodium aurothiomalate, as label. This Au(I) complex has been previously used in our laboratory with success as electroactive label for immunosensor (De la Escosura Muñiz et al., 2004a, De la Escosura Muñiz et al., 2006a, De la Escosura Muñiz et al., 2006b) and genosensor (De la Escosura Muñiz et al., 2007) devices using glassy carbon electrodes as transducers. The electrochemical detection in these biosensors is based on the catalytic effect of ionic gold on silver electrodeposition (De la Escosura Muñiz et al., 2004b). In this work, the catalytic properties of the Au(I) complex toward the hydrogen evolution is exploited for first time to detect an hybridization event, and the catalytic current generated is chronoamperometrically measured. In the enzymatic assay the detection is performed by measuring the electroactive enzymatic product indigo carmine (IC), generated by AP in presence of the substrate 3-indoxyl phosphate (3-IP). This substrate has been proposed by our group as a suitable electrochemical substrate for AP and has been successfully applied in the development of biosensors (Fernández Sánchez and Costa García, 1998).
2. Materials and methods
2.1. Apparatus and electrodes
Voltammetric and amperometric studies were performed with an Autolab PGSTAT 12 (Eco Chemie, The Netherlands) potentiostatic/galvanostatic interfaced to an AMD K-6 400 computer system and controlled by Autolab GPES software version 4.7 for Windows 98. Screen-printed carbon electrodes (SPCEs) were purchased from Alderon Biosciences Inc. (Durham, NC, USA), together with an edge connector. The electrodes incorporate a conventional three-electrode configuration, which comprises a disk-shaped working (4 mm diameter), counter and silver pseudo-reference electrodes printed on polycarbonate substrate (4.5 cm × 1.5 cm). Both working and counter electrodes were made of heat-curated carbon composite inks. An insulating layer was printed over the electrode system, leaving uncovered a working electrode area of 7 mm × 5 mm and the electric contacts. A ring-shaped layer further printed around the working area constituted the reservoir of the electrochemical cell, with an actual volume of 50 μl.
2.2. Reagents and solutions
Tris(hydroxymethyl)aminomethane (Tris), 3-indoxyl phosphate disodium salt (3-IP), casein, formamide, polylysine (poly-l-lysine, molecular weight 15–30 kDa) and streptavidin conjugated to alkaline phosphatase (S-AP) were purchased from Sigma (Spain). 3-IP solutions (6 mM) were prepared in 0.1 M Tris pH 9.8, containing 20 mM MgCl2 and stored in opaque tubes at 4 °C. Working solutions of casein and S-AP were made in 0.1 M Tris pH 7.2, containing 2 mM MgCl2 in the last case. EDTA was obtained from Fluka (Spain). Sodium aurothiomalate was obtained from Aldrich (Spain). It was reconstituted in 0.15 M NaCl solution and protected from light. Dilutions of this stock solution were prepared daily in an unbuffered aqueous solution of 0.15 M NaCl, with pH adjusted to 7.5 with 0.1 M NaOH. Biotinylated polylysine (molecular weight 15–30 and 30–70 kDa) were kindly provided by Dr. Juan R. de los Toyos (Área de Inmunlogía, Facultad de Medicina, Universidad de Oviedo, Spain) in a 0.2 M NaHCO3 pH 8.5 solution. Analytical grade (Merck, Spain) NaCl, HCl, H2SO4, NaOH, NaHCO3 and sodium citrate were used. Ultra-pure water obtained with a Milli-Q plus 185 from Milipore Ibérica S.A. (Spain) was used for all solutions. Dialysis procedures were carried out with Slide-A-Lizer dialysis cassettes, 3500 MWCO from Pierce (USA). Synthetic 30-mer oligonucleotides were obtained from Eurogentec (Spain). The target sequence employed corresponds to a portion of the severe acute respiratory syndrome (SARS) virus, precisely the bases comprised between 26218 and 29247, both included. For selectivity studies, three-base mismatch strands were also purchased. Oligonucleotide probe: 5′-CTT-TTT-CTT-TTT-GTC-CTT-TTT-AGG-CTC-TGT-3′ (T m = 56 °C); Oligonucleotide target (unlabeled and 3′ biotinylated): 5′-ACA-GAG-CCT-AAA-AAG-GAC-AAA-AAG-AAA-AAG-3′ (T m = 56 °C); Three-base mismatch target 3′ biotinylated: 5′-ACA-GCG-CCT-AAA-AAC-GAC-AAA-AAG-AGA-AAG-3′ (T m = 59 °C). Oligonucleotides solutions were prepared in TE buffer, pH 8 (10 mM Tris–HCl buffer solution, 1 mM in EDTA) and maintained at −20 °C. Working solutions of the oligonucleotide probe were made in 0.1 M Tris, pH 7.2 buffer, while the oligonucleotide targets were diluted in a 2× SCC buffer (300 mM sodium chloride 30 mM sodium citrate), pH 7.2. These solutions were conserved at 4 °C. In selectivity studies, an amount of formamide was added to the hybridization solutions.
2.3. Methods
Fig. 1 shows a scheme of the analytical procedure.
Fig. 1.
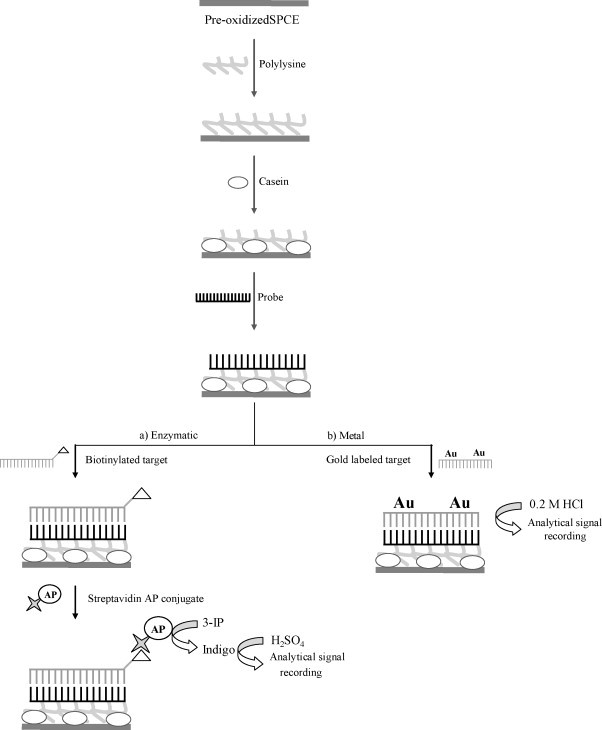
Schematic representation of the analytical procedure followed for the construction of the DNA hybridization sensor using (a) an enzymatic label (AP) and (b) a metal label (sodium aurothiomalate).
2.3.1. Labeling of the oligonucleotide target with sodium aurothiomalate
The conjugation of aurothiomalate to the oligonucleotide was carried out according to the procedure described in a previous work (De la Escosura Muñiz et al., 2007). Briefly, an aliquot of 50 μL of a 20 μM oligonucleotide target solution was mixed with an aliquot of 450 μL of a 390 ng/μL aurothiomalate solution. The reaction was carried out at 37 °C for 24 h. After that, the conjugate was purified by dialysis against 200 mL of 0.15 M NaCl, pH 7.5, unbuffered solution, for 48 h at room temperature.
2.3.2. Immobilization of oligonucleotide probe onto the electrode surface
An electrode pretreatment was carried out before each experiment with the aim of improving the sensitivity and repeatability of the results. For this purpose 50 μL of 0.1 M H2SO4 were dropped on the SPCEs and an anodic current of 25 μA was applied for 2 min. After a washing step with 0.1 M Tris buffer pH 7.2, the electrodes were modified with polylysine, leaving an aliquot of 10 μL of a 0.1 mg/mL polylysine solution overnight at 4 °C. Then, the electrodes were washed with 0.1 M Tris buffer pH 7.2 and the oligonucleotide probes were immobilized onto the electrode surface by applying +0.5 V for 2 min in a drop of 40 μL of a 100 nM oligonucleotide probe solution. After a new washing step with 0.1 M Tris buffer pH 7.2 free surfaces sites of the SPCEs surface were blocked using 40 μL of a 1% (w/v) solution of casein for 60 min. Finally, the electrodes were rinsed with 2× SCC buffer, pH 7.2.
2.3.3. Hybridization step
Hybridization was performed at room temperature (RT) by placing 30 μL of: (a) biotinylated oligonucleotide target (enzymatic assay) or (b) Au (I) labeled oligonucleotide target (metal assay), in 2× SCC buffer, pH 7.2, on the modified electrodes for 30 min.
2.3.4. Detection step for the enzymatic assay
After a washing step with 0.1 M Tris, pH 7.2, a drop of 40 μL of a 0.4 nM S-AP solution was placed on the electrode surface and left to react during 90 min. Then, the electrodes were washed with 0.1 M Tris, pH 9.8, containing 20 mM MgCl2, and finally the enzymatic reaction was carried out by dropping an aliquot of 30 μL of 6 mM 3-IP solution. After 15 min, the reaction was stopped by adding 4 μL of fuming sulfuric acid and 10 μL of ultrapure water. In this step, the corresponding indigo product is converted to its parent hydro soluble compound indigo carmine. This compound shows a reversible electrode process at −0.15 V (versus Ag pseudo-reference electrode) in 0.1 M H2SO4 (Díaz González et al., 2002). The anodic peak current registered using cyclic voltammetry (CV) constitutes the analytical signal for all the studies presented in this work, with the enzymatic label. Voltammograms were recorded from −0.25 to +0.20 V at a scan rate of 50 mV/s, after the SPCEs were held at −0.25 V for 25 s.
2.3.5. Detection step for the metal assay
A 50 μL portion of a 0.2 M HCl solution was dropped on the electrode surface, and the electrode was held at a potential of +1.35 V for 1 min. Then, the chronoamperometric detection was performed at −1.40 V, recording the electric current generated for 200 s.
2.3.6. Three-base-mismatch detection
The methodology used to detect a three-base mismatch is similar to the one explained above, increasing the hybridization time to 60 min and adding formamide (50%) to the hybridization buffer.
3. Results and discussion
3.1. Modification of SPCEs with polylysine
Biotinylated polylysine was used to verify the adsorption of the polymer on the SPCE surface. Different experimental conditions (times and temperatures) were tested using S-AP at a concentration of 0.4 nM and a streptavidin–biotin reaction time of 90 min. Best results were achieved when the adsorption of the polylysine was performed overnight at 4 °C onto the SPCEs previously oxidized by applying an anodic constant current of +25 μA in 0.1 M H2SO4 for 2 min. The same experimental conditions were previously found as optimal for the SPCEs modification with streptavidin (Díaz González et al., 2004). The electrode pretreatment is essential in both cases to obtain the higher analytical signals and the best precision of the results. Nonspecific adsorptions of S-AP on the electrode surface were minimized blocking the free surfaces sites of the SPCEs surface, after the polylysine adsorption, with a 1% casein solution for 15 min. The effect of the polylysine concentration on the voltammetric response was investigated using biotinylated polylysine of 15–30 kDa and 30–70 kDa. The peak current increases with increasing polymer concentration up to 0.1 mg/mL (data not shown). Beyond this point, the electrode response changes slightly indicating that a saturation state was reached on the SPCE surface. The same behavior was found for both molecular weights, but higher currents were registered with the biotinylated polylysine 15–30 kDa. Consequently, 0.1 mg/mL of polylysine 15–30 kDa was routinely used to modify the SPCEs for all the studies.
3.2. Immobilization of oligonucleotides on polylysine modified SPCEs
The oligonucleotides were captured on the electrode surface through electrostatic interactions with the adsorbed polylysine. To study this immobilization, different concentrations between 50 and 500 nM of a biotinylated oligonucleotide (5′-ACA-GAG-CCT-AAA-AAG-GAC-AAA-AAG-AAA-AAG-3′-biotin) were adsorbed onto the modified SPCEs. After a blocking step of 15 min with a 1% casein solution, oligonucleotides were detected using a 0.4 nM S-AP solution (90 min). As can be seen in Fig. 2 , for all the concentrations assayed, direct adsorption of the oligonucleotides onto unmodified SPCEs was not observed. For the polylysine modified SPCEs maximum analytical signals were reached with only 5 min of adsorption, indicating the rapid interaction between the oligonucleotides and the polylysine. Working with 5 min of adsorption, the peak current increase upon raising the oligonucleotide concentration up 100 nM, and then it starts to level off, which corresponds to the saturation state. To ensure that the analytical signals obtained are only due to the biotin–streptavidn interaction and there are not nonspecific adsorptions of the S-AP, a non biotinylated oligonucleotide was adsorbed on the polylysine modified SPCEs. Using the experimental conditions described above, no analytical signal was obtained, indicating the efficacy of the blocking step (data not shown). In some application the adsorption of the oligonucleotides onto carbon electrode is conducted under a positive potential (Lucarelli et al., 2002, Wang and Kawde, 2001). This positive potential favors the adsorption of the oligonucleotides through the sugar-phosphate backbone, with the bases being oriented toward the solution. The application of a positive potential can also be used with electrodes previously modified with polycations, to enhance the stability of the immobilized oligonucleotides (Cai et al., 2003). To study this possibility, different positive potentials were applied during the oligonucleotides adsorption onto the polylysine modified SPCEs. Best results were obtained by applying +0.5 V for 2 min in a 100 nM biotinylated oligonucleotide solution. The reproducibility of the results was notably improved but also higher analytical signals were registered.
Fig. 2.
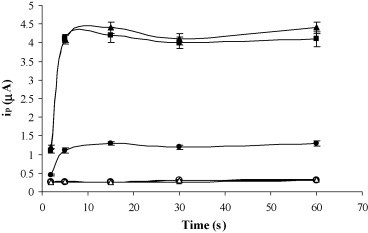
Effect of the oligonucleotide adsorption time on peak current using polylysine modified SPCEs ((●) 50 nM, (■) 100 nM and (▴) 500 nM biotinylated oligonucleotide) and unmodified SPCEs ((○)50 nM, (□) 100 nM and (▵) 500 nM biotinylated oligonucleotide). Polylysine adsorption 12 h 4 °C; [S-AP] = 0.4 nM; S-AP incubation time, 90 min. Current responses are given as average ± S.D. (n = 3).
3.3. Enzymatic genosensor
The response of the genosensor designed was evaluated using different concentrations of both the complementary and the noncomplementary strand. As can be seen in Fig. 3a, both strands were perfectly discriminated by the genosensor, but the currents registered for the noncomplementary strand were higher than those expected, since nonspecific adsorptions of S-AP on the modified electrodes were not observed. In order to know the origin of these high currents, nonspecific adsorptions of the oligonucleotide onto the polylysine modified SPCEs were studied. The analytical signals obtained when the probe was not incubated, were very similar than those registered for the noncomplementary strand (Fig. 3a). This fact proves that there are nonspecific adsorptions of the oligonucleotide onto the polylysine-modified electrodes. To minimize these nonspecific adsorptions, the blocking step of the genosensor device was revised using different blocking agents. Best results were found using the same casein solution (1% in 0.1 M Tris pH 7.2) but increasing the blocking step incubation time to 60 min (Fig. 3b). The experimental parameters that affect the response of the genosensor such as the oligonucleotide probe concentration, the hybridization time and the biotin–streptavidin interaction time were then optimized. Different concentrations of the oligonucleotide probe were immobilized using 30 min of hybridization time and a concentration of the complementary target of 2.0 nM. The concentration of S-AP used for this study was 0.4 nM and the biotin–streptavidin interaction time was 90 min. The peak current increases with increasing the probe concentration up to 100 nM (data not shown). Beyond this point, the genosensor response reaches a current plateau indicating that a saturation state was reached on the electrode surface. The rest of the experimental parameters were also studied, obtaining the best results for 30 min of hybridization time at room temperature, a 0.4 nM S-AP concentration and a biotin–streptavidin interaction time of 90 min. Under the experimental conditions optimized, the genosensor response varied linearly (r = 0.999) with the oligonucleotide target concentration between 20 and 200 pM, according to the following equation.
The calculated limit of detection was 8 pM, estimated as the concentration that gives an electrodic signal three times the standard deviation of the signal obtained when a noncomplementary strand is present in solution. This detection limit is similar than that obtained for the same oligonucleotide target using gold films as electrochemical transducer and thiolated oligonucleotide probe (6 pM, Abad Valle et al., 2005), but notably improves the detection limit obtained for a 20-mer oligonucleotide (Streptococcus pneumoniae), using the same SPCEs but immobilizing biotinylated probes onto the streptavidin modified electrodes (40 pM, Hernández Santos et al., 2004).
Fig. 3.
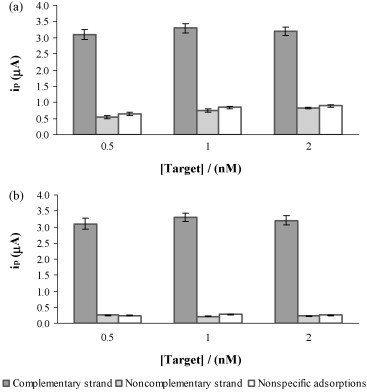
Effect of the blocking step time: (a) 30 min, (b) 60 min on the DNA hybridization sensor response. [Probe] = 100 nM, 2 min + 0.5 V; [S-AP] = 0.4 nM; hybridization time, 30 min; S-AP incubation time, 90 min; RT. Current response are given as average ± S.D. (n = 3).
A precision study was carried out using 50 pM of the oligonucleotide target. The current response was averaged (n = 4) and the intra-assay relative standard deviation (R.S.D.) was calculated. This was repeated in three consecutive days to determine the inter-assay R.S.D. The intra-assay R.S.D. was 5.5% with a mean anodic peak current of 0.70 μA while the inter-assay R.S.D. was 7.8% with a mean anodic 0.74 μA.
In order to test the selectivity of the assay, hybridization was carried out with a three-base mismatch complementary strand. Using the experimental hybridization parameters optimized (30 min 2× SCC, RT) there was no discrimination between the signals registered for the complementary strand and the strand with three-base mismatch. To obtain a 100% discrimination between both strands was necessary to increase the hybridization time to 60 min and carry out the hybridization in a 2× SCC buffer incorporating 50% formamide (Fig. 4 ). Under these more stringent conditions the genosensor is able to distinguish target concentrations ranging from 50 to 500 pM, with a regression equation of:
As it was expected, the sensitivity decrease and the calculated limit of detection for the oligonucleotide target were 24 pM.
Fig. 4.
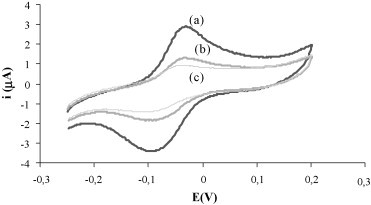
Cyclic voltammograms recorded using 2 nM of (a) complementary target (b) three-base-mismatch target and (c) noncomplementary strand. [Probe] = 100 nM, 2 min + 0.5 V; [S-AP] = 0.4 nM; hybridization buffer 2× SCC 50% formamide; hybridization time, 60 min; S-AP incubation time, 90 min; RT.
3.4. Genosensor based on aurothiomalate electroactive label
Following the procedure schematized in Fig. 1b, and using all the experimental parameters optimized with the enzymatic label, the response of the genosensor to Au(I) labeled oligonucleotide was evaluated. The procedure followed to obtain the analytical signal, based on the catalytic properties of the metal label toward hydrogen evolution, has been previously optimized in our group using a platinum(II) complex as label in the development of a genosensor using streptavidin modified SPCEs (Hernández Santos et al., 2005). In the presence of platinum on the electrode surface and fixing an adequate potential in acidic medium, the protons are catalytically reduced to hydrogen. The current generated by this catalytic reduction was measured and related with the labeled target concentration. While the catalytic properties of platinum complex have been used previously by some authors (Hernández Santos et al., 2005, Babkina et al., 2004), this work use for first time, to the best of our knowledge, the catalytic effect of the Au(I) complex, sodium aurothiomalate, on hydrogen evolution. Using a 5 nM concentration of the Au labeled target, the oligonucleotide probe concentration and the hybridization time were optimized. For all the studies, the detection was performed recording the electric current generated for 200 s at −1.40 V, in a drop of 50 μL of 0.2 M HCl solution, after the electrodes were held at +1.35 V for 1 min. As for the enzymatic genosensor, best results were reached working with a 100 nM oligonucleotide probe concentration and 30 min of hybridization time. Chronoamperograms recorded under the optimized experimental conditions for a 5 nM solution for both the complementary target and a noncomplementary strand are showed in Fig. 5 . As can be seen in this figure, the DNA hybridization sensor designed perfectly discriminates between a complementary/noncomplementary strand. A semilogarithmic relationship between the current registered for 200 s and the concentration of the oligonucleotide target was obtained from 1 to 20 nM, in accordance with the following equation.
The limit of detection, calculated as described above, was 0.5 nM, which is practically the same as the previously reported for a 20-mer oligonucleotide (Streptococcus pneumoniae), using a platinum(II) complex as label and immobilizing biotinylated probes onto the streptavidin modified SPCEs (0.6 nM, Hernández Santos et al., 2005). Repeatability was tested obtaining a R.S.D. value of 3.9% for five measurements with a mean current of −2.25 mA.
Fig. 5.
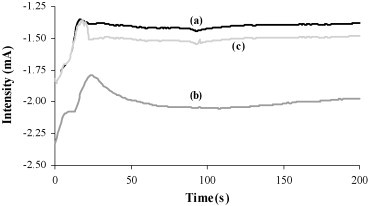
Chronoamperograms recorded using 5 nM of a gold labeled (a) noncomplementary strand, (b) complementary target and (c) three-base mismatch target. Electrodeposition potential, −1.40 V; [Probe] = 100 nM, 2 min + 0.5 V; hybridization time, 30 min; RT.
In Fig. 5 is also included the chronoamperogram for a three-base mismatch complementary strand. Once again no discrimination between the signals registered for the complementary strand and the strand with three-base mismatch was obtained, using 30 min of hybridization time at room temperature and 2× SCC as hybridization buffer. To detect three-base mismatch in the target strand was necessary to use the stringent hybridization conditions previously optimized for the enzymatic genosensor, 60 min of hybridization time in a 2× SCC buffer incorporating 50% formamide. Under these more stringent conditions, a new calibration curve was obtained. A semilogarithmic relationship between the current measured at 200 s and the concentration of the oligonucleotide target between 5 and 50 nM was found, according to the following equation.
The limit of detection was 2.5 nM.
4. Conclusions
Oligonucleotides can be easily immobilized onto polylysine modified SPCEs, through electrostatic interactions, by applying a constant potential of +0.5 V in a drop of 40 μL of the oligonucleotide probe solution for only 2 min. In contrast to other oriented immobilization methods, like the streptavidin–biotin interaction, the use of polylysine allows the rapid immobilization of unmodified oligonucleotides onto the electrode surface. The polylysine modified SPCEs have been successfully applied to the development of genosensors using an enzymatic and a metal label. A very sensitive detection of a 30-mer sequence unique to SARS virus was obtained using AP as label. As it was expected, the sensitivity obtained with the enzymatic label is better than that achieved with the metal label. Nevertheless the analysis time with the gold label is considerably shorter, about the half than that resulting for the enzymatic genosensor, because the analytical signal is achieved directly from the gold complex whereas in the enzymatic design two additional steps are necessary to obtain the analytical signal: the reaction between the biotinylated target and the S-AP (90 min) and the enzymatic reaction (15 min). Besides the metal assay is a cheaper alternative, since the oligonucleotide target is easily labeled with the Au(I) complex, sodium aurothiomalate, and is not necessary to use expensive reactives.
Acknowledgement
Financial support provided by project BIO2003-06008-C03-01 from the Spanish Ministry of Science and Technology is gratefully acknowledged.
References
- Abad Valle P., Fernández Abedul M.T., Costa García A. Biosens. Bioelectron. 2005;20:2251–2260. doi: 10.1016/j.bios.2004.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babkina S.S., Ulakhovich N.A., Zyavkina Y.I. Anal. Chim. Acta. 2004;502:23–30. [Google Scholar]
- Bussiek M., Mücke N., Langowski J. Nucleic Acids Res. 2003;31:e137. doi: 10.1093/nar/gng137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H., Zhu N., Jiamg Y., He P., Fang Y. Biosens. Bioelectron. 2003;18:1311–1319. doi: 10.1016/s0956-5663(03)00084-8. [DOI] [PubMed] [Google Scholar]
- De la Escosura Muñiz A., González García M.B., Costa García A. Biosens. Bioelectron. 2007;22:1048–1054. doi: 10.1016/j.bios.2006.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Escosura Muñiz A., González García M.B., Costa García A. Sens. Actuators B. 2006;114:473–481. [Google Scholar]
- De la Escosura Muñiz A., González García M.B., Costa García A. Anal. Bioanal. Chem. 2006;384:742–750. doi: 10.1007/s00216-005-0213-4. [DOI] [PubMed] [Google Scholar]
- De la Escosura Muñiz A., González García M.B., Costa García A. Anal. Chim. Acta. 2004;524:355–363. [Google Scholar]
- De la Escosura Muñiz A., González García M.B., Costa García A. Electroanalysis. 2004;16:1561–1568. [Google Scholar]
- De los Santos Álvarez P., Lobo Castañón M.J., Miranda Ordieres A.J., Tuñón Blanco P. Anal. Chem. 2002;74:3342–3347. doi: 10.1021/ac015749m. [DOI] [PubMed] [Google Scholar]
- Del Pozo M.V., Alonso C., Pariente F., Lorenzo E. Anal. Chem. 2005;77:2550–2557. doi: 10.1021/ac0489263. [DOI] [PubMed] [Google Scholar]
- Díaz González M., Fernández Sánchez C., Costa García A. Electroanalysis. 2002;14:665–670. [Google Scholar]
- Díaz González M., Hernández-Sánchez D., González García M.B., Costa García A. Talanta. 2004;65:565–573. doi: 10.1016/j.talanta.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Erdem A., Pividori M.I., del Valle M., Alegret S. J. Electroanal. Chem. 2004;567:29–37. [Google Scholar]
- Fernández Sánchez C., Costa García A. Electroanalysis. 1998;10:249–255. [Google Scholar]
- Fotja M., Brázdilová P., Cahová K., Pečinka P. Electroanalysis. 2006;18:141–151. [Google Scholar]
- Hernández Santos D., González García M.B., Costa García A. Anal. Chem. 2005;77:2868–2874. doi: 10.1021/ac048091w. [DOI] [PubMed] [Google Scholar]
- Hernández Santos D., Díaz González M., González García M.B., Costa García A. Anal. Chem. 2004;76:6887–6893. doi: 10.1021/ac048892z. [DOI] [PubMed] [Google Scholar]
- Kara P., Erdem A., Girousi S., Ozsoz M. J. Pharmaceut. Biomed. Anal. 2005;38:191–195. doi: 10.1016/j.jpba.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Lee T.M.H., Cai H., Hsing I.M. Analyst. 2005;130:364–369. doi: 10.1039/b413143f. [DOI] [PubMed] [Google Scholar]
- Liu J., Tian S., Tiefenauer L., Nielsen P.E., Knoll W. Anal. Chem. 2005;77:2756–2761. doi: 10.1021/ac048088c. [DOI] [PubMed] [Google Scholar]
- Lucarelli F., Marrazza G., Palchetti I., Cesaretti S., Mascini M. Anal. Chim. Acta. 2002;469:93–99. doi: 10.1016/j.aca.2008.03.027. [DOI] [PubMed] [Google Scholar]
- Ma K.S., Zhou H., Zoval J., Madou M. Sens. Actuators B. 2006;114:58–64. [Google Scholar]
- Mir M., Katakis I. Anal. Bioanal. Chem. 2005;381:1033–1035. doi: 10.1007/s00216-004-2950-1. [DOI] [PubMed] [Google Scholar]
- Ozsoz M., Erdem A., Kerman K., Ozkan D., Tugrui B., Topcuoglu N., Ekren H., Taylan M. Anal. Chem. 2003;75:2181–2187. doi: 10.1021/ac026212r. [DOI] [PubMed] [Google Scholar]
- Pividori M.I., Merkoçi A., Alegret S. Biosens. Bioelectron. 2003;19:473–484. doi: 10.1016/s0956-5663(03)00222-7. [DOI] [PubMed] [Google Scholar]
- Segura T., Volk M.J., Shea L.D. J. Contr. Rel. 2003;93:69–84. doi: 10.1016/j.jconrel.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Strother T., Cai W., Zhao X., Hamers R.J., Smith L.M. J. Am. Chem. Soc. 2000;122:1205–1209. [Google Scholar]
- Teh H.F., Gong H., Dong X.D., Zeng X., Tan A.L.K., Yang X., Tan S.N. Anal. Chim. Acta. 2005;551:23–29. [Google Scholar]
- Tominaga M., Soejima K., Matsumoto M., Taniguchi I. J. Electroanal. Chem. 2005;579:51–58. [Google Scholar]
- Tsujimura S., Kano K., Ikeda T. J. Electroanal. Chem. 2005;576:113–120. [Google Scholar]
- Wang J., Kawde A.N. Anal. Chim. Acta. 2001;431:219–224. [Google Scholar]
- Wu Q., Ma W., Shi R., Zhang B., Mao X., Zheng W. Eng. Life Sci. 2005;5:466–470. doi: 10.1002/elsc.200520097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Pothukuchy A., Shin W., Kim Y., Heller A. Anal. Chem. 2004;76:4093–4097. doi: 10.1021/ac0495034. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Kim H.H., Heller A. Anal. Chem. 2003;75:3267–3269. doi: 10.1021/ac034445s. [DOI] [PubMed] [Google Scholar]
- Zhu N., Chang Z., He P., Pang Y. Anal. Chim. Acta. 2005;545:21–26. [Google Scholar]