

Abstract
Viral genomes show unequalled diversity, ranging from single-stranded DNA to double-stranded RNA. Moreover, viruses can quickly adapt to the host's immune response and drug treatment. Although they tend to make optimal use of the host cell's reservoir of proteins, viruses need to carry some enzymatic functions with them, as they may not be available or accessible in the infected cell. Recently, progress has been made in our structural understanding of viral enzymes involved in all stages of the viral life cycle, which includes entry, hijack, replication and exit stages.
Introduction
The prime characteristic of viruses is their absolute dependence on a host for reproduction and, as such, they are obligate parasites and a plague to all living organisms. Back in 1898, the Dutch scientist Beyerinck and the German researchers Loeffler and Frosch established that, based on filtration experiments, the causative agents of certain (plant and animal) diseases were clearly smaller than all known bacteria. Only two years later, yellow fever was the first human viral disease to be recognized (Major Reed and assistants, US Army, Havana, 1900) and, to date, more than 1938 virus species belonging to 287 genera of 73 families have been identified [1]. Probably, viruses are as old as life on Earth, but a broad public awareness is only emerging now, in view of the current AIDS pandemic and the recent outbreaks of severe acute respiratory syndrome (SARS) and bird flu.
Compared to cells, viruses are indeed small, with (apparently) minimized genomes that mostly encode proteins not provided by the host cell. Not only do viruses try to make optimal use of the host's accessible reservoir of enzymes, but also they can quickly and efficiently adapt to any changes in the host, its immune response and drug treatment. Finally, because of their genomic flexibility, many viruses, in particular RNA viruses, are able to change their host range and cross species barriers. In contrast to living organisms, viral genomes show a breath-taking diversity, from single-stranded (ss) DNA to double-stranded (ds) RNA, with a size of up to 400 kb (e.g. Phycodnaviridae). An intriguing exception is the Acanthamoeba polyphaga mimivirus (APMV), which features a genome size of 1.2 mb. The genetic information is often programmed in a highly efficient way, displaying features such as overlapping genes and making full use of the possibility to encode both homo- and hetero-oligomers as the biologically active species. In principle, a small, naked, infectious DNA or RNA strand of a few thousand bases that can somehow enter a living cell, be replicated and translated by cellular enzymes, and finally leave the cell to infect new ones would suffice to qualify as a virus, that is, an obligate parasite in need of the host's energy-supplying and protein-synthesizing capabilities. Nevertheless, viruses generally bear an exterior coating (capsid or envelope) and have a variety of enzymes and auxiliary proteins, many of which are not available or accessible (due to compartmentalization) in the infected cell.
With the recent increase in public awareness, research into viral proteins has benefited from increased funding (e.g. the VIZIER project, www.vizier-europe.org, and the SEPSDA project on the SARS coronavirus, www.sepsda.info). As a result, a flood of structural information is becoming available that will help propel the discovery of antiviral lead compounds by structure-based drug design or virtual screening. In this review, current structural studies of viral enzymes are summarized and discussed.
We will discuss these studies within the framework of a viral life cycle composed of entry, hijack (taking control and dodging defense), replication (transcription and translation) and, finally, exit stages. Viral proteins are usually divided into structural and non-structural types; although most enzymes are classified as non-structural, some structural proteins may display enzymatic activity as well. Viral enzymes involved in the entry or exit steps will be discussed in one section.
Entry and exit: crossing cell barriers
Although some plant viruses make it into their target cells through open wounds, most viruses bind to a receptor molecule at the cell surface and then have to cross the cell membrane. Receptor binding is not an enzymatic process and therefore is not discussed here, but often the same viral glycoprotein that binds to cellular receptors through one domain (or fragment) catalyzes membrane fusion through another domain. The latter activity exhibited by these structural proteins can be considered ‘enzymatic’ (see [2] for a more detailed review). A few structures of such fusion cores described in the past two years will be mentioned here; these include relevant fragments of the SARS coronavirus (CoV) spike (S) protein (see [3] for a review), human parainfluenza virus 5 F protein [4], Semliki Forest virus glycoprotein E1 [5], Dengue virus envelope (E) protein ([6]; see also [7]) and glycoprotein B of herpes simplex virus 1 [8]. Upon pH shift and/or proteolytic cleavage, these proteins undergo conformational change, thereby exposing a hydrophobic fusion peptide that can insert into the host cell membrane.
For efficient egress of newly formed virions from the host cell, receptor molecules located at the surface of infected cells have to be modified or degraded. Influenza neuraminidase, a sialidase, cleaves sialic acid moieties from receptor molecules, thereby preventing the rebinding of viruses to the surface of the same, already infected, host cell. Very recently, the X-ray structure of avian influenza virus neuraminidase (a type-I neuraminidase) has been determined, revealing an extra cavity adjacent to the active site that closes upon ligand binding ([9•]; PDB codes 2HTY, 2HU0 and 2HU4; the latter two structures contain the drug oseltamivir). This cavity is not observed in the N2 and N9 (type-II) variants of the enzyme that were used for the structure-based design of oseltamivir (Tamiflu) and zanamivir (Relenza). Thus, this finding opens new avenues for the design of neuraminidase inhibitors.
Hijacking: engaging host defense and acquiring cell control
Once the virus is inside the host cell, it needs to take control of the cell and evade host defense strategies while initiating, in a timely manner, replication and translation processes. Viral offensive measures affect a wide variety of host defensive strategies, for example, modification of signaling pathways and cellular machines. Interferons play a key role in cell defense and viruses counteract accordingly. Recognition of viral nucleic acids through pattern-recognition receptors (e.g. Toll-like receptor 3 and cytosolic RNA helicase RIG-I; reviewed in [10]) is the first step in the induction of the innate immune system. But the viruses strike back: the NS3-NS4A serine protease of hepatitis C virus (HCV) cleaves Cardif, an adaptor molecule involved in the recognition of viral dsRNA by retinoic acid inducible gene-I (RIG-I; [11••]), thereby blocking interferon-β production. The simian virus 5 V protein hijacks DNA damage binding protein 1 (of the DDB1–Cul4A ubiquitin ligase complex) by inserting an entire helix into a pocket formed by two domains of the three-β-propeller DDB1 structure ([12••]; PDB code 2B5L), inducing degradation of STATs (signal transducer and activator of transcription) and thereby obstructing interferon signaling. Furthermore, the interferon-inducible and dsRNA-stimulated protein kinase PKR, a key player in the innate response to viral infection, is targeted by many DNA and RNA viruses (reviewed in [13]); for example, the human cytomegalovirus (HCMV) TRS1 gene product [14] seems to sequester PKR in the nucleus, away from both its activator, cytoplasmic dsRNA, and its substrate, eukaryotic initiation factor 2α (eIF2α). As a consequence, phosphorylation of eIF2α and the subsequent shutdown of protein synthesis are prevented.
Besides crippling or evading host defense, viruses need to slow down host mRNA translation to promote viral mRNAs. This is often achieved by modification of eukaryotic initiation factors (eIFs) or the poly(A)-binding protein (PABP). Like picornavirus and calicivirus proteases (see [15]), HIV-1 and -2 proteases efficiently cleave PABP [16]. The 2A cysteine proteinase of coxsackievirus 4B ([17]; see below) cleaves eukaryotic initiation factor eIF4G, thereby blocking host mRNA translation. Once host mRNA translation and host defense are perturbed, viral transcription and translation can proceed promptly. Viruses ensure that cap-independent eukaryotic initiation of viral (subgenomic) mRNA translation can proceed normally by employing specific viral proteins and/or internal ribosome entry sites (IRES) to recruit ribosomes. Instead of a 7-methylguanosine (m7G) cap structure at the 5′ end, (sub)genomic RNAs are, in several cases, covalently linked to viral protein VPg (viral protein genome-linked, see next section), which appears to substitute for the 5′-terminal m7G cap moiety in ribosome recruitment. Using GST-based pull-down assays, VPg was shown to interact with several eIFs and ribosomal protein S6 in norovirus-infected cells [18]. The 3′-untranslated region of turnip yellow mosaic virus (TYMV) harbors a tRNA-like structure (TLS) that acts as a molecular Trojan horse and is responsible for the internal initiation of polyprotein synthesis [19].
Replication: nucleic acid and protein synthesis
DNA and RNA viruses alike encode at least one protein involved in nucleic acid synthesis: whereas all RNA viruses have an RNA-dependent polymerase, some DNA virus genomes merely encode a helicase (e.g. parvoviruses and polyomaviruses) or primase (e.g. herpes simplex virus), and yet others encode a complete machinery for DNA replication (e.g. poxvirus). For translation, by contrast, they all depend on host cell ribosomes and translation factors. It would require a major effort to encode (large parts of) a translational machine: in bacteria, more than 150 different macromolecules (RNA and protein) are directly involved in protein biosynthesis, with nearly 60 of them organized in the ribosome. Located in the cytosol, the host cell translational apparatus is freely accessible to all types of viruses and there is no need for them to carry translation factors with them. This is in contrast to nucleic acid synthesis, for which many of the relevant host enzymes are contained in the nucleus. Mimivirus is a remarkable exception to this rule; this virus carries a substantial portion of the translational apparatus with it [20].
Viral enzymes are notoriously difficult to handle and crystallize, as many of them are part of larger assemblies; in a pure and concentrated state, the single, isolated proteins often tend to aggregate. Nevertheless, more and more three-dimensional structures are being determined and viral enzymes such as polymerases, helicases and especially proteases prove amenable to crystallization.
There are two main classes of viral RNA-dependent RNA polymerases (RdRps, reviewed in [21]), primer dependent and primer independent (de novo). The picornavirus and calicivirus RdRps represent one class; they display a more accessible active site cavity, enabling them to accommodate the small VPg protein that acts as a primer protein in RNA synthesis. The foot-and-mouth disease virus (FMDV) VPg protein lines the RNA-binding cleft of the corresponding RdRp ([22••]; PDB code 2D7S), positioning its Tyr3 OH group as a molecular mimic of the free 3′ OH group of a nucleic acid primer at the active site for nucleotidylylation, thereby initiating replication. In the presence of oligoadenylate and UTP, the product of the reaction, VPg–UMP, can be observed in the crystal structure ([22••]; PDB code 2F8E), in a position remarkably similar to the position of the template–primer RNA duplex ([23]; PDB code 1WNE, see also [24•, 25]) (Figure 1 ). After nucleotidylylation of VPg, larger structural rearrangements of the RdRp will follow, marking the transition from initiation to the elongation phase of RNA synthesis. By contrast (other class), reovirus and flavivirus RdRps contain additional structural elements that fill most of the active site cavity (see [21]), thereby enabling de novo RNA synthesis using a single NTP as a primer (see also [26••]).
Figure 1.
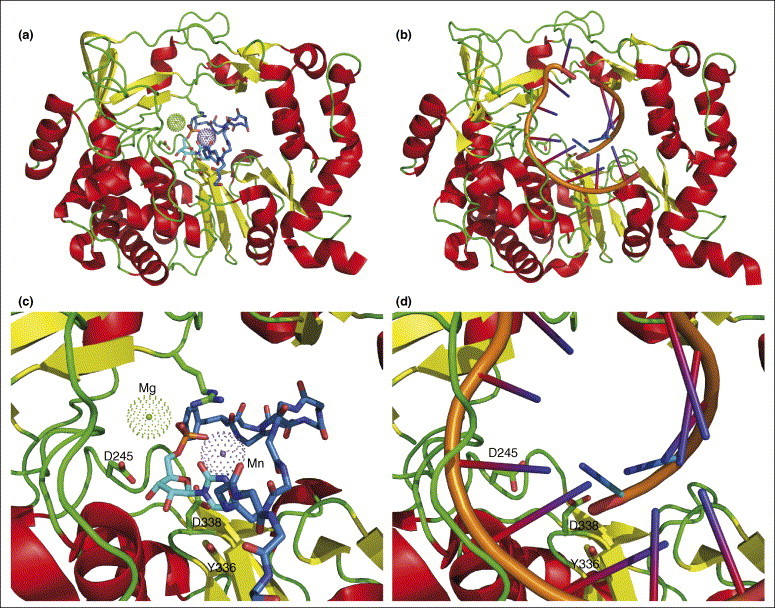
Comparison of polymerase-bound protein and RNA primers. VPg–UMP, overview in (a) and details in (c), is observed in the crystal structure ([22••]; PDB code 2F8E) in a position remarkably similar to that of the template–primer RNA duplex, overview in (b) and details in (d) ([23]; PDB code 1WNE). Except for Tyr3, only the mainchain atoms of VPg are shown for clarity: carbon atoms of VPg are colored blue and those of UMP cyan. Magnesium and manganese ions are shown as dotted balls in (a and c) and the template–primer RNA duplex is in stick form in (b and d). As a point of reference, three amino acids (of conserved elements in the active site) are shown as stick models: D245, Y336 and D338.
The RdRps are a major target for the development of antiviral compounds. In the ‘thumb’ domain of NS5B, the RdRp of HCV, the binding sites for thiophene-based non-nucleoside inhibitors (NNIs) ([27]; PDB codes 2D3U, 2D3Z and 2D41) and allosteric GTP (see [28•]) are in close proximity. Apparently, this part of the ‘thumb’ domain, located approximately 35 Å away from the polymerase active site (the ‘palm’ domain), fulfills an important regulatory function that can be modulated by GTP or NNI binding. The dimeric structure of HCV NS5A domain I (a large phosphoprotein and active component of the HCV replicase) reveals a groove with an exposed Trp84 residue ([29]; PDB code 1ZH1): the groove is large enough to accommodate an RNA helix. An N-terminal amphipathic α-helix places NS5A on the membrane, which is essential for the assembly of a functional viral replication complex [30, 31]. The polyadenylate polymerase heterodimer of vaccinia virus is made up of a catalytic component, polyadenylate polymerase VP55, and a cap-specific mRNA (nucleoside-2′-O-)-methyltransferase, processivity factor VP39 ([32•]; PDB code 2GA9): polyadenylation of mRNAs in poxviruses is crucial for virion maturation. Recently, the structures of the DNA polymerases of HCMV and herpes simplex virus have been reported [33, 34].
Since the global outbreak of SARS in 2003, much effort has been invested to elucidate the structures of components of the SARS-CoV replicase complex. However, the structure of the RdRp (also called non-structural protein 12, Nsp12) itself has remained elusive so far, due to difficulties with producing the full-length protein in sufficient quantities and crystallizing it. The crystal structure of the hexadecameric (8:8) complex between Nsp7 and Nsp8 has been determined recently ([35••]; PDB code 2AHM). The complex features a central channel of ∼30 Å diameter (Figure 2 ), sufficiently wide to accommodate dsRNA. Together with the positive electrostatic potential of the inner walls of the channel, this suggests that Nsp7–Nsp8 may function as a processivity factor for the RdRp, reminiscent of the β2-clamp of bacterial DNA polymerases. This is a nice example of successful functional assignment of an Nsp of unknown function resulting from the structural proteomics projects dealing with SARS-CoV; previously, the function of Nsp9 as an ssRNA-binding protein had been correctly deduced from its crystal structure ([36, 37]; PDB codes 1QZ8 and 1UW7). A new homodimeric form has recently been found for Nsp9 from human coronavirus 229E (R Ponnusamy et al., unpublished). Nsp10 is another coronaviral protein without known function, but it was recently demonstrated that it has two zinc fingers and binds dsRNA [38, 39, 40]. Upon dsRNA binding, the Nsp10 dodecamers observed in one crystal form ([39]; PDB codes 2G9T and 2GA6) apparently dissociate into monomers [38], as observed in the other crystal form ([40]; PDB code 2FYG).
Figure 2.
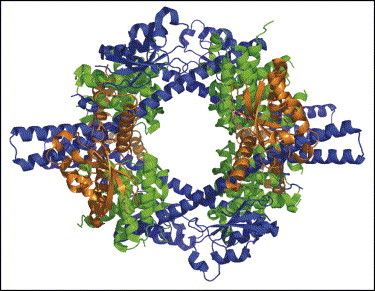
Ribbon diagram showing the quaternary structure of the Nsp7–Nsp8 hexadecamer complex ([35••]; PDB code 2AHM). The eight copies of Nsp7 are colored green and the eight copies of two markedly different conformations of Nsp8 are colored brown and blue. The positively charged, large channel (∼30 Å diameter) may well accommodate a double-stranded nucleic acid.
Several viruses encode a macrodomain (viral X-domain), as found in proteins associated with histones or involved in chromatin metabolism. Initially, a subdomain of Nsp3 of SARS-CoV was proposed to possess ADP-ribose 1′-phosphate phosphohydrolase (ADRP) activity ([41]; PDB code 2ACF). Very recently, the ADRP domain of Nsp3 was shown to bind efficiently to poly(ADP-ribose) in vitro, both in its free state and when bound to poly(ADP-ribose) polymerase 1 ([42•]; PDB code 2FAV) (Figure 3a). Its phosphohydrolase activity was redetermined as being poor and the true biological function of this protein in the viral life cycle remains to be elucidated. The membrane-bound replicase complex of the coronaviruses harbors an endonuclease, NendoU (Nsp15), a manganese-dependent enzyme specific for uridylate. The crystal structure of Nsp15 revealed that it possesses a novel fold and an active site that appears to characterize a separate endonuclease family ([43•, 44•]; PDB codes 2H85 and 2GTH) (Figure 3b). The biologically active unit of this enzyme is a dimer of trimers with six independent active sites.
Figure 3.
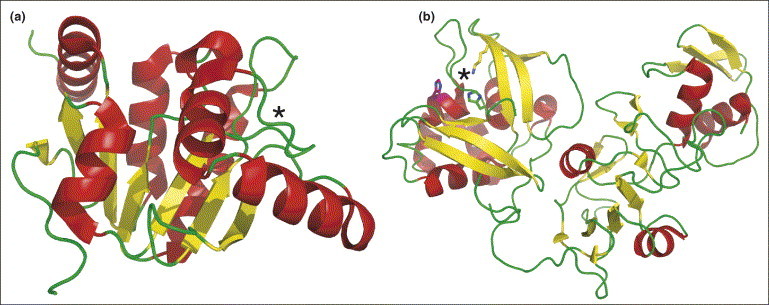
Structures of the monomeric forms of Nsp3 and Nsp15. (a) The nucleotide-binding site of Nsp3, a macrodomain (viral X-domain), is marked with an asterisk. Nsp3 has poor ADRP activity, but efficiently binds poly(ADP-ribose) in vitro ([42•]; PDB code 2FAV). (b) The active site of Nsp15, an endonuclease (NendoU) associated with the replicase complex of coronaviruses, is marked by an asterisk ([43•, 44•]; PDB codes 2H85 and 2GTH). The biological unit of this enzyme is a dimer of trimers with six independent active sites.
The structure of the coronavirus helicase remains to be determined, but recently structures became available for the helicases of other RNA viruses. A comparison of the NS3 HCV helicase [45] and both the yellow fever virus (YFV) ([46•]; PDB code 1YMF) (Figure 4 ) and Dengue virus ([47]; PDB code 2BMF) helicase structures revealed that only the C-terminal domains (third domain) are quite unrelated between the flaviviruses and HCV. The lack of nucleotide specificity nicely correlates with the observation that the adenine and ribose moieties of a bound ADP, in the YFV helicase–nucleotide complex [46•], protrude into solution. An arginine is expected to be critical for conformational switching upon NTP hydrolysis, although the mode of action is not well understood. One implication of modeling work seems to be that a 3′ single-stranded tail of about nine nucleotides would be required for the initiation of the unwinding reaction [47]. An interesting set of structural ‘snapshots’ has now become fairly complete for the helicase E1 protein subunit of papillomavirus [48••, 49]. An E1 monomer possesses two DNA-binding elements, a loop and a helix, that bind opposing DNA strands. Two E1 homodimers are initially loaded onto the DNA [49], each of which is further expanded to produce a hexamer that surrounds a single DNA strand [48••]: one strand of DNA passes through the hexamer channel, sliding along the DNA-binding loops that sequentially track the oligonucleotide backbone.
Figure 4.
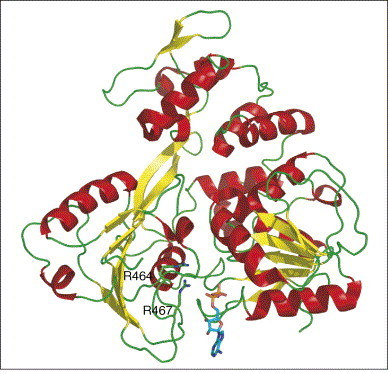
Structure of YFV helicase ([46•]; PDB code 1YMF, see also [47]; PDB code 2BMF). The adenine and ribose moieties of a bound ADP (shown as a stick model) protrude into solution [46•]; this observation nicely explains the lack of nucleotide specificity. An arginine (either R464 or R467) is expected to be critical for conformational switching upon NTP hydrolysis, although the mode of action is not well understood.
A few viral proteins have been shown to contain disulfide bonds, even though they fold in the reducing environment of the eukaryotic host cytoplasm. Enzymes involved in disulfide bond isomerization, and perhaps others that catalyze peptidyl-prolyl cis/trans isomerization, may therefore play a role in viral infectivity. This has been underscored by structural and functional investigations of vaccinia virus G4 disulfide oxidoreductase, an enzyme that displays a thioredoxin fold with the usual Cys-X-X-Cys motif [50].
Viral proteases are essential for processing virally encoded polyproteins during replication, in co- and post-translational steps. Because of their importance for viral replication, assembly and infectivity, they are potential targets of antiviral drugs. It seems that the proteases of at least some RNA viruses could qualify as targets of inhibitors with the desirable relatively broad specificity, due to similarities in their overall architecture and in their substrate-binding sites. The crystal structure of a birnavirus protease (VP4 of blotched snakehead virus) visualized, for the first time, a serine–lysine catalytic dyad in the viral world ([51•]; PDB code 2GEF). The amino group of the lysine residue is almost completely buried and therefore probably deprotonated. Bacterial proteases using a similar active site include LexA and the Lon protease, although the latter enzyme oligomerizes and requires ATP hydrolysis for proteolytic activity. Named after the 3C domain of the picornavirus polyprotein, a large number of proteases from RNA viruses have a chymotrypsin-like fold, but most of them have a cysteine rather than a serine residue as the nucleophile. Two crystal structures of closely related norovirus 3C-like proteases revealed that the active site features a catalytic Cys–His–Glu triad ([52, 53]; PDB codes 1WQS and 2FYQ). The cysteine can be replaced by serine, suggesting that the enzyme works through a general base mechanism involving a non-charged Cys–His pair rather than a thiolate–imidazolium ion pair (as has been shown to exist in papain-like proteinases).
The picornavirus genome comprises a single-stranded, messenger-oriented RNA molecule of 7.5 kb that is used as the template for both replication and translation. Processing of the single polyprotein encoded by this genome is initiated by the 2A proteinase (2Apro), which cleaves the bond between the structural protein VP1 and its own N terminus, and is subsequently completed by the 3C proteinase (3Cpro) or its 3CD precursor. In addition to polyprotein processing, 2Apro of coxsackievirus B3 is involved in cleaving dystrophin, leading to acquired dilated cardiomyopathia, and in interrupting host protein synthesis by cleaving eukaryotic initiation factors eIF4GI and eIF4GII, as well as the PABP. An NMR structure has been published of 2Apro of coxsackievirus B4, revealing the expected chymotrypsin-like architecture, with a proper Cys–His–Asp catalytic triad ([17]; PDB code 1Z8R). Such a triad has also been seen in the 3C proteinases of FMDV ([54]; PDB code 2BHG) and tobacco etch virus ([55, 56]; PDB codes 1Q31 and 1LVM). This is significant, because earlier structures of picornavirus 3C proteinases revealed irregularities with regard to the acidic partner of the catalytic triad; either the aspartate residue was found oriented away from the active site histidine, such as in hepatitis A virus 3Cpro ([57]; PDB code 1QA7), or it was replaced by glutamate, such as in poliovirus 3Cpro [58], in human rhinovirus 3Cpro ([59]; PDB code 1CQQ) or in the recently determined structure of 3C proteinase of coxsackievirus 3B (K Anand et al., unpublished).
All of the viral proteases mentioned thus far more or less prefer glutamine over glutamate residues at the P1 position of the substrate, although this distinction is not so clear in case of the enzyme from FMDV. However, the chymotrypsin-like serine protease from Sesbania mosaic virus (SeMV), an ssRNA plant sobemovirus, is specific for glutamate at P1. The activity of this enzyme is modulated by the binding of VPg, whose Trp43 residue is believed to interact with an exposed aromatic patch near the C terminus of the SeMV proteinase ([60•, 61•]; PDB code 1ZYO). Thus, at least three different functions have been described for VPg-like peptides in viral replication: it can substitute for the 5′-terminal m7G cap moiety in binding to initiation factors and in ribosome recruitment; it can act as a primer in viral RNA synthesis; and it can increase the activity of certain viral proteases. Another chymotrypsin-like viral protease whose catalytic activity is augmented by a peptide cofactor is the NS3-NS4A protease of HCV ([62]; PDB code 1A1R). This enzyme continues to be the target of numerous efforts in drug design (e.g. [63]).
In addition to the chymotrypsin-like β-barrel domains I and II, the coronavirus main proteinases (Mpro, also called 3C-like proteinases, 3CLpro) have a C-terminal helical domain that is involved in dimerization of the enzyme [64, 65, 66]. The catalytic center consists of a Cys–His dyad; the position of the third member of a proper triad is taken by a highly conserved water molecule that is involved in multiple hydrogen-bonding interactions. Another unique feature not present in the 3C proteases is that the N terminus of one Mpro monomer is involved in shaping the substrate-binding site of the other; therefore, dimerization is essential for catalytic activity [64, 65, 66]. Among this protease family, SARS-CoV Mpro is now by far the best investigated because of its paramount importance as a target for the treatment of SARS [67, 68]. A pH-dependent switch has been proposed to be responsible for the activation of this enzyme; at low pH, at least one monomer of the homodimeric enzyme tends to be in a catalytically incompetent conformation, with the oxyanion hole and the S1 pocket collapsed [66, 69•], whereas both monomers appear to be active at higher pH [66, 70]. The transition between the two conformations is believed to depend on the protonation/deprotonation of a histidine residue (His163) that interacts with the P1 glutamine residue of the substrate; this has been reproduced using molecular dynamics simulations [69•]. Several structures of complexes with peptidomimetic inhibitors have been determined for the enzyme [66, 71•, 72, 73] and compounds that acylate the active site cysteine residue were also described as potent inhibitors [74, 75]. Peptidyl aldehydes have been designed and synthesized as potentially reversible inhibitors [76]. In silico screening efforts have resulted in the identification of a few non-covalent binders that act as competitive inhibitors [77].
The Mpro performs 11 of the 14 cleavage reactions necessary to process coronaviral polyproteins and to generate the components of the replicase machinery. The remaining three cleavage sites in the N-proximal portion of the polyproteins are substrates of viral papain-like proteinases (PLpro). Most coronaviruses have two of these enzymes (PL1pro and PL2pro), but the SARS virus genome only encodes the latter. Most interestingly, it has been found that this protease not only cleaves the polyproteins after LXGG sequences, but also has a deubiquitinating function [78, 79, 80]. The crystal structure of SARS-CoV PLpro ([81••]; PDB code 2FE8) revealed that the enzyme comprises four distinct domains (Figure 5 ), three of which form an extended right-handed architecture in which they have been nicknamed ‘thumb, palm and finger’, although they are different from the corresponding features of DNA polymerases. The N-terminal 62 residues form a distinct ubiquitin-like domain. The active site comprises a catalytic Cys–His–Asp triad, the cysteine of which is contributed by the thumb domain and the two other residues by the palm domain. The tip of the finger domain carries a structural zinc ion that is ligated by four cysteine sidechains. The closest structural relatives of SARS-CoV PLpro are the cellular deubiquitinating enzymes HAUSP (herpes-associated ubiquitin-specific protease) and USP14 (ubiquitin-specific protease 14). In addition to ubiquitin, SARS-CoV PLpro also removes ISG15 from target proteins. ISG15 is a ubiquitin-like molecule induced by interferon α/β as part of the innate immune response against viral infections [82]. Apparently, SARS-CoV protects itself from interferon-triggered innate immune response by interfering with ISG15 or ubiquitin conjugation pathways. But what is the role of the N-terminal ubiquitin-like domain carried by PLpro itself? Ratia et al. [81••] have entertained the interesting idea that it may serve as a decoy to detract cellular ubiquitinating enzymes from modifying proteins of SARS-CoV.
Figure 5.
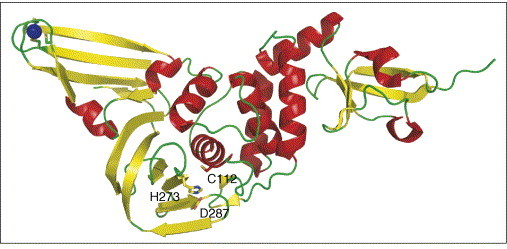
Structure of the SARS-CoV PLpro deubiquitinase ([81••]; PDB code 2FE8). The 62-residue N-terminal region forms a distinct ubiquitin-like domain (right-hand side). The other three domains form an extended right-handed architecture with thumb, palm and finger regions. The active site comprises a catalytic Cys–His–Asp triad (labeled), the cysteine of which is contributed by the thumb domain and the two other residues by the palm domain. The tip of the finger domain (left-hand side) has a structural zinc ion that is ligated by four cysteine sidechains.
Finally, several viruses encode glycosyltransferases that are involved in the post-translational modification of the structural proteins. Paramecium bursaria Chlorella virus (PCBV) is probably the first virus known to encode enzymes involved in sugar metabolism: the N-terminal domain of PBCV GDP-d-mannose 4,6 dehydratase adopts a modified Rossmann fold, displaying a seven-stranded parallel β-sheet with a 3-2-1-4-5-6-7 topology, and the smaller C-terminal domain is probably involved in nucleotide-sugar substrate binding [83]. The Sulfolobus turreted icosahedral virus (STIV) glycosyltransferase [84] possesses the glycosyltransferase superfamily A fold (GT-A), displaying both the canonical DXD motif and a putative catalytic base.
Conclusions
Structural virology has made significant progress over the past few years. The outbreak of SARS in 2003 has triggered increased research activity in the field, including a few structural proteomics projects. Viral enzymes are promising targets for antiviral drug discovery and such drugs are in great need in view of the dramatic increase in the number of viral outbreaks in recent years. It has to be realized that, in the case of an outbreak, it will take at least six months and probably twelve until a vaccine will be ready for use; hence, we will have to resort to drugs that have been discovered and preclinically developed before the outbreak. Of course, such drugs should exhibit broad antiviral activity and, even though real broad-band antivirals may remain an illusion (as opposed to the existing broad-band antibacterials), preliminary results with inhibitors of RNA virus proteases suggest that there may be at least groups of viruses susceptible to one and the same drug. Structural results on viral target enzymes are of course essential to design inhibitors or discover them by in silico screening, and it is indeed satisfying that three-dimensional structures of viral enzymes are determined with increasing pace.
A nice side effect of the structural biology of viral proteins, especially complexes thereof, is that a lot can also be learnt about the cellular machines or pathways that are targeted by viruses (e.g. see [11••]). Also, it is increasingly realized that molecular mimicry seems to be used by viral components, for example, the small VPg proteins that are able to reprogram or redirect large molecular machines. The large diversity and dynamics of viral genomes seem to be reflected in the three-dimensional structures of viral proteins and their complexes.
Update
Very recently, Imbert et al. [85] have shown that Nsp8 (and its complex with Nsp7) may in fact constitute a second RdRp in SARS-CoV.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgements
The authors acknowledge support by the European Commission through VIZIER (Comparative Structural Genomics of Viral Enzymes Involved in Replication; contract LSHG-CT-2004-511960) and SEPSDA (Sino-European Project on SARS Diagnostics and Antivirals; contract SP22-CT-2004-003831). RH thanks the Innovation Fund of the Government of Schleswig-Holstein and the Fonds der Chemischen Industrie for continuous support. Figures were prepared with the PyMol molecular graphics system (DeLano Scientific LLC, South San Francisco, CA, USA).
References
- 1.Fauquet C.M., Mayo M.A., Maniloff J., Desselberger U., Ball L.A., editors. Virus Taxonomy - VIIIth Report of the International Committee on Taxonomy of Viruses. Academic Press; London: 2005. [Google Scholar]
- 2.Schibli D.J., Weissenhorn W. Class I and class II viral fusion protein structures reveal similar principles in membrane fusion. Mol Membr Biol. 2004;21:361–371. doi: 10.1080/09687860400017784. [DOI] [PubMed] [Google Scholar]
- 3.Bartlam M., Yang H., Rao Z. Structural insights into SARS coronavirus proteins. Curr Opin Struct Biol. 2005;15:664–672. doi: 10.1016/j.sbi.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin H.S., Wen X., Paterson R.G., Lamb R.A., Jardetzky T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roussel A., Lescar J., Vaney M.C., Wengler G., Wengler G., Rey F.A. Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure. 2006;14:75–86. doi: 10.1016/j.str.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 6.Modis Y., Ogata S., Clements D., Harrison S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 7.Mukhopadhyay S., Kuhn R.J., Rossmann M.G. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 8.Heldwein E.E., Lou H., Bender F.C., Cohen G.H., Eisenberg R.J., Harrison S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 9•.Russell R.J., Haire L.F., Stevens D.J., Collins P.J., Lin Y.P., Blackburn G.M., Hay A.J., Gamblin S.J., Skehel J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]; The first X-ray structure of an avian influenza type-I neuraminidase reveals an extra cavity adjacent to the active site observed in structures of type-II variants. The latter were used for structure-based design of oseltamivir and zanamivir. Clearly, this cavity can be exploited for structure-based design of new neuraminidase type-I specific inhibitors.
- 10.Meylan E., Tschopp J. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol Cell. 2006;22:561–569. doi: 10.1016/j.molcel.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 11••.Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]; The authors describe a new CARD-containing adaptor protein, Cardif, which interacts with RIG-I, a helicase that senses double-stranded (viral) RNA. Cardif can recruit several kinases, thereby stimulating NF-κB and IRF3 promoter activation. Interestingly, in order to perturb the host's immune response (e.g. interferon-β production), Cardif is targeted and inactivated by the NS3-NS4A serine protease of HCV.
- 12••.Li T., Chen X., Garbutt K.C., Zhou P., Zheng N. Structure of DDB1 in complex with a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitin ligase. Cell. 2006;124:105–117. doi: 10.1016/j.cell.2005.10.033. [DOI] [PubMed] [Google Scholar]; The quaternary structure of the complex between simian virus 5 V protein and DNA damage binding protein 1 (of the DDB1–Cul4A ubiquitin ligase complex) reveals how a single viral protein can reprogram a cellular pathway, inducing degradation of STATs and thereby obstructing interferon signaling. This is achieved by the insertion a single helix of the 5 V protein into a pocket formed by two domains of the three-β-propeller DDB1 structure.
- 13.Langland J.O., Cameron J.M., Heck M.C., Jancovich J.K., Jacobs B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–110. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Hakki M, Marshall EE, De Niro KL, Geballe AP: Binding and nuclear relocalization of PKR by human cytomegalovirus TRS1.J Virol 2006, in press. [DOI] [PMC free article] [PubMed]
- 15.Lloyd R.E. Translational control by viral proteinases. Virus Res. 2006;119:76–88. doi: 10.1016/j.virusres.2005.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alvarez E., Castello A., Menendez-Arias L., Carrasco L. HIV protease cleaves poly(A)-binding protein. Biochem J. 2006;396:219–226. doi: 10.1042/BJ20060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baxter N.J., Roetzer A., Liebig H.D., Sedelnikova S.E., Hounslow A.M., Skern T., Waltho J.P. Structure and dynamics of coxsackievirus B4 2A proteinase, an enzyme involved in the etiology of heart disease. J Virol. 2006;80:1451–1462. doi: 10.1128/JVI.80.3.1451-1462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daughenbaugh K.F., Wobus C.E., Hardy M.E. VPg of murine norovirus binds translation initiation factors in infected cells. Virol J. 2006;3:33. doi: 10.1186/1743-422X-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barends S., Bink H.H., van den Worm S.H., Pleij C.W., Kraal B. Entrapping ribosomes for viral translation: tRNA mimicry as a molecular Trojan horse. Cell. 2003;112:123–129. doi: 10.1016/s0092-8674(02)01256-4. [DOI] [PubMed] [Google Scholar]
- 20.Raoult D., Audic S., Robert C., Abergel C., Renesto P., Ogata H., La Scola B., Suzan M., Claverie J.M. The 1.2-megabase genome sequence of Mimivirus. Science. 2004;306:1344–1350. doi: 10.1126/science.1101485. [DOI] [PubMed] [Google Scholar]
- 21.Ferrer-Orta C., Arias A., Escarmis C., Verdaguer N. A comparison of viral RNA-dependent RNA polymerases. Curr Opin Struct Biol. 2006;16:27–34. doi: 10.1016/j.sbi.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 22••.Ferrer-Orta C., Arias A., Agudo R., Perez-Luque R., Escarmis C., Domingo E., Verdaguer N. The structure of a protein primer-polymerase complex in the initiation of genome replication. EMBO J. 2006;25:880–888. doi: 10.1038/sj.emboj.7600971. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first reported structure of a protein primer–polymerase complex involved in the initiation of viral genome replication. Protein primer VPg lines the RNA-binding cleft of RdRp, positioning its Tyr3 OH group as a molecular mimic of the free 3′ OH group of a nucleic acid primer at the active site for nucleotidylylation. Moreover, VPg–UMP, the product of the nucleotidylylation reaction, is observed within the crystal structure.
- 23.Ferrer-Orta C., Arias A., Perez-Luque R., Escarmís C., Domingo E., Verdaguer N. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template–primer RNA. J Biol Chem. 2004;279:47212–47221. doi: 10.1074/jbc.M405465200. [DOI] [PubMed] [Google Scholar]
- 24•.Appleby T.C., Luecke H., Shim J.H., Wu J.Z., Cheney I.W., Zhong W., Vogeley L., Hong Z., Yao N. Crystal structure of complete rhinovirus RNA polymerase suggests front loading of protein primer. J Virol. 2005;79:277–288. doi: 10.1128/JVI.79.1.277-288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; Based on the X-ray structure of human rhinovirus 16 3D RNA-dependent RNA polymerase, the authors argue in favor of a so-called front-loading model for VPg binding and uridylylation.
- 25.Love R.A., Maegley K.A., Yu X., Ferre R.A., Lingardo L.K., Diehl W., Parge H.E., Dragovich P.S., Fuhrman S.A. The crystal structure of the RNA-dependent RNA polymerase from human rhinovirus: a dual function target for common cold antiviral therapy. Structure. 2004;12:1533–1544. doi: 10.1016/j.str.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 26••.Choi K.H., Groarke J.M., Young D.C., Kuhn R.J., Smith J.L., Pevear D.C., Rossmann M.G. The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc Natl Acad Sci USA. 2004;101:4425–4430. doi: 10.1073/pnas.0400660101. [DOI] [PMC free article] [PubMed] [Google Scholar]; The structure of bovine viral diarrhea virus polymerase with a bound GTP molecule, required in addition to the initiation NTP, reveals that the GTP in fact substitutes for a primer RNA in de novo (primer-independent) initiation of replication.
- 27.Biswal B.K., Wang M., Cherney M.M., Chan L., Yannopoulos C.G., Bilimoria D., Bedard J., James M.N. Non-nucleoside inhibitors binding to hepatitis C virus NS5B polymerase reveal a novel mechanism of inhibition. J Mol Biol. 2006;361:33–45. doi: 10.1016/j.jmb.2006.05.074. [DOI] [PubMed] [Google Scholar]
- 28•.Biswal B.K., Cherney M.M., Wang M., Chan L., Yannopoulos C.G., Bilimoria D., Nicolas O., Bedard J., James M.N. Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J Biol Chem. 2005;280:18202–18210. doi: 10.1074/jbc.M413410200. [DOI] [PubMed] [Google Scholar]; Two conformations are reported for RdRp genotype 2a from HCV: an open, active, form and a closed, inactive, one. NNIs apparently bind exclusively to an allosteric patch of the open form, located 35 Å away from the active site.
- 29.Tellinghuisen T.L., Marcotrigiano J., Rice C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature. 2005;435:374–379. doi: 10.1038/nature03580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moradpour D., Brass V., Penin F. Function follows form: the structure of the N-terminal domain of HCV NS5A. Hepatology. 2005;42:732–735. doi: 10.1002/hep.20851. [DOI] [PubMed] [Google Scholar]
- 31.Sapay N., Montserret R., Chipot C., Brass V., Moradpour D., Deleage G., Penin F. NMR structure and molecular dynamics of the in-plane membrane anchor of nonstructural protein 5A from bovine viral diarrhea virus. Biochemistry. 2006;45:2221–2233. doi: 10.1021/bi0517685. [DOI] [PubMed] [Google Scholar]
- 32•.Moure C.M., Bowman B.R., Gershon P.D., Quiocho F.A. Crystal structures of the vaccinia virus polyadenylate polymerase heterodimer: insights into ATP selectivity and processivity. Mol Cell. 2006;22:339–349. doi: 10.1016/j.molcel.2006.03.015. [DOI] [PubMed] [Google Scholar]; The first structure of a viral polyadenylate polymerase is presented. The reported vaccinia virus heterodimeric structure is made up of a catalytic component (polymerase VP55) and a cap-specific mRNA (nucleoside-2′-O-)-methyltransferase (VP39). The VP55 protein reveals an unusual architecture that differs from many known polymerases, including the eukaryotic poly(A) polymerases.
- 33.Appleton B.A., Brooks J., Loregian A., Filman D.J., Coen D.M., Hogle J.M. Crystal structure of the cytomegalovirus DNA polymerase subunit UL44 in complex with the C terminus from the catalytic subunit. Differences in structure and function relative to unliganded UL44. J Biol Chem. 2006;281:5224–5232. doi: 10.1074/jbc.M506900200. [DOI] [PubMed] [Google Scholar]
- 34.Liu S., Knafels J.D., Chang J.S., Waszak G.A., Baldwin E.T., Deibel M.R., Jr., Thomsen D.R., Homa F.L., Wells P.A., Tory M.C. Crystal structure of the herpes simplex virus 1 DNA polymerase. J Biol Chem. 2006;281:18193–18200. doi: 10.1074/jbc.M602414200. [DOI] [PubMed] [Google Scholar]
- 35••.Zhai Y., Sun F., Li X., Pang H., Xu X., Bartlam M., Rao Z. Insights into SARS-CoV transcription and replication from the structure of the nsp7-nsp8 hexadecamer. Nat Struct Mol Biol. 2005;12:980–986. doi: 10.1038/nsmb999. [DOI] [PMC free article] [PubMed] [Google Scholar]; This fine paper provides insight into the complexity of the coronavirus replicase machinery. The crystal structure of the hexadecameric complex between Nsp7 and Nsp8 is described in great detail. Nsp8 adopts two very different conformations in the complex.
- 36.Egloff M.P., Ferron F., Campanacci V., Longhi S., Rancurel C., Dutartre H., Snijder E.J., Gorbalenya A.E., Cambillau C., Canard B. The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a single-stranded RNA-binding subunit unique in the RNA virus world. Proc Natl Acad Sci USA. 2004;101:3792–3796. doi: 10.1073/pnas.0307877101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sutton G., Fry E., Carter L., Sainsbury S., Walter T., Nettleship J., Berrow N., Owens R., Gilbert R., Davidson A. The nsp9 replicase protein of SARS-coronavirus, structure and functional insights. Structure. 2004;12:341–353. doi: 10.1016/j.str.2004.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matthes N., Mesters J.R., Coutard B., Canard B., Snijder E.J., Moll R.G., Hilgenfeld R. The non-structural protein Nsp10 of mouse hepatitis virus binds zinc ions and nucleic acids. FEBS Lett. 2006;580:4143–4149. doi: 10.1016/j.febslet.2006.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su D., Lou Z., Sun F., Zhai Y., Yang H., Zhang R., Joachimiak A., Zhang X.C., Bartlam M., Rao Z. Dodecamer structure of severe acute respiratory syndrome coronavirus nonstructural protein nsp10. J Virol. 2006;80:7902–7908. doi: 10.1128/JVI.00483-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joseph J.S., Saikatendu K.S., Subramanian V., Neuman B.W., Brooun A., Griffith M., Moy K., Yadav M.K., Velasquez J., Buchmeier M.J. Crystal structure of nonstructural protein 10 from the severe acute respiratory syndrome coronavirus reveals a novel fold with two zinc-binding motifs. J Virol. 2006;80:7894–7901. doi: 10.1128/JVI.00467-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saikatendu K.S., Joseph J.S., Subramanian V., Clayton T., Griffith M., Moy K., Velasquez J., Neuman B.W., Buchmeier M.J., Stevens R.C., Kuhn P. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1′′-phosphate dephosphorylation by a conserved domain of nsP3. Structure. 2005;13:1665–1675. doi: 10.1016/j.str.2005.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.Egloff M.P., Malet H., Putics A., Heinonen M., Dutartre H., Frangeul A., Gruez A., Campanacci V., Cambillau C., Ziebuhr J. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J Virol. 2006;80:8493–8502. doi: 10.1128/JVI.00713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors show that Nsp3, a macrodomain or viral X-domain found in proteins associated with particular histones or involved in chromatin metabolism, actually displays poor ADRP activity (its proposed function). Instead, Nsp3 is shown to efficiently bind poly(ADP-ribose) in vitro. As a consequence, the true biological function of this protein remains to be elucidated.
- 43•.Ricagno S., Egloff M.P., Ulferts R., Coutard B., Nurizzo D., Campanacci V., Cambillau C., Ziebuhr J., Canard B. Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc Natl Acad Sci USA. 2006;103:11892–11897. doi: 10.1073/pnas.0601708103. [DOI] [PMC free article] [PubMed] [Google Scholar]; The replicase complex of the nidoviruses includes an RNA endoribonuclease. The SARS-CoV Nsp15 protein exhibits a unique fold and assembles into a hexamer with six peripheral active sites. The spatial arrangement of the catalytic residues in the active site defines a separate endoribonuclease family, endoU.
- 44•.Xu X., Zhai Y., Sun F., Lou Z., Su D., Xu Y., Zhang R., Joachimiak A., Zhang X.C., Bartlam M., Rao Z. New antiviral target revealed by the hexameric structure of mouse hepatitis virus nonstructural protein nsp15. J Virol. 2006;80:7909–7917. doi: 10.1128/JVI.00525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; The mouse hepatitis virus Nsp15 protein was one the first members of the endoribonuclease endoU family to have its structure unveiled. The fully biological active unit is a dimer of trimers that is specific for uridylate. No structural homologues could be identified for the butterfly-shaped Nsp15 monomer as a whole or for any of its single domains.
- 45.Yao N., Hesson T., Cable M., Hong Z., Kwong A.D., Le H.V., Weber P.C. Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol. 1997;4:463–467. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- 46•.Wu J., Bera A.K., Kuhn R.J., Smith J.L. Structure of the Flavivirus helicase: implications for catalytic activity, protein interactions, and proteolytic processing. J Virol. 2005;79:10268–10277. doi: 10.1128/JVI.79.16.10268-10277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; This structure reveals that the adenine and ribose moieties of a bound ADP, in the YFV helicase–nucleotide complex, protrude into solution; this nicely correlates with the reported lack of nucleotide specificity.
- 47.Xu T., Sampath A., Chao A., Wen D., Nanao M., Chene P., Vasudevan S.G., Lescar J. Structure of the Dengue virus helicase/nucleoside triphosphatase catalytic domain at a resolution of 2.4 Å. J Virol. 2005;79:10278–10288. doi: 10.1128/JVI.79.16.10278-10288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48••.Enemark E.J., Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]; The E1 protein subunit of papillomavirus possesses two DNA-binding elements, a loop and a helix. Initially, two E1 homodimers are loaded onto separate strands of the dsDNA. The authors report the hexameric structure, disclosing that only one strand of DNA passes through the central channel. The DNA-binding loops of the subunits are proposed to sequentially track the oligonucleotide backbone. This hexameric structure represents the last step in assembling the helicase complex around ssDNA.
- 49.Enemark E.J., Stenlund A., Joshua-Tor L. Crystal structures of two intermediates in the assembly of the papillomavirus replication initiation complex. EMBO J. 2002;21:1487–1496. doi: 10.1093/emboj/21.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su H.P., Lin D.Y., Garboczi D.N. The structure of g4, the poxvirus disulfide oxidoreductase essential for virus maturation and infectivity. J Virol. 2006;80:7706–7713. doi: 10.1128/JVI.00521-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51•.Feldman A.R., Lee J., Delmas B., Paetzel M. Crystal structure of a novel viral protease with a serine/lysine catalytic dyad mechanism. J Mol Biol. 2006;358:1378–1389. doi: 10.1016/j.jmb.2006.02.045. [DOI] [PubMed] [Google Scholar]; The first example of the unusual Ser–Lys catalytic dyad in a viral protease. A useful comparison with other proteases featuring this dyad, such as Lon, LexA and E. coli signal peptidase, is presented.
- 52.Nakamura K., Someya Y., Kumasaka T., Ueno G., Yamamoto M., Sato T., Takeda N., Miyamura T., Tanaka N. A norovirus protease structure provides insights into active and substrate binding site integrity. J Virol. 2005;79:13685–13693. doi: 10.1128/JVI.79.21.13685-13693.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeitler C.E., Estes M.K., Venkataram Prasad B.V. X-ray crystallographic structure of the Norwalk virus protease at 1.5 Å resolution. J Virol. 2006;80:5050–5058. doi: 10.1128/JVI.80.10.5050-5058.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Birtley J.R., Knox S.R., Jaulent A.M., Brick P., Leatherbarrow R.J., Curry S. Crystal structure of foot-and-mouth disease virus 3C protease. J Biol Chem. 2005;280:11520–11527. doi: 10.1074/jbc.M413254200. [DOI] [PubMed] [Google Scholar]
- 55.Nunn C.M., Jeeves M., Cliff M.J., Urquhart G.T., George R.R., Chao L.H., Tscuchia Y., Djordjevic S. Crystal structure of tobacco etch virus protease shows the protein C terminus bound within the active site. J Mol Biol. 2005;350:145–155. doi: 10.1016/j.jmb.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 56.Phan J., Zdanov A., Evdokimov A.G., Tropea J.E., Peters H.K., Kapust R.B., Li M., Wlodawer A., Waugh D.S. Structural basis for the substrate specificity of tobacco etch virus protease. J Biol Chem. 2002;277:50564–50572. doi: 10.1074/jbc.M207224200. [DOI] [PubMed] [Google Scholar]
- 57.Bergmann E.M., Cherney M.M., McKendrick J., Frormann S., Luo C., Malcolm B.A., Vederas J.C., James M.N. Crystal structure of an inhibitor complex of the 3C proteinase from hepatitis A virus (HAV) and implications for the polyprotein processing in HAV. Virology. 1999;265:153–163. doi: 10.1006/viro.1999.9968. [DOI] [PubMed] [Google Scholar]
- 58.Mosimann S.C., Cherney M.M., Sia S., Plotch S., James M.N. Refined X-ray crystallographic structure of the poliovirus 3C gene product. J Mol Biol. 1997;273:1032–1047. doi: 10.1006/jmbi.1997.1306. [DOI] [PubMed] [Google Scholar]
- 59.Matthews D.A., Smith W.W., Ferre R.A., Condon B., Budahazi G., Sission W., Villafranca J.E., Janson C.A., McElroy H.E., Gribskov C.L., Worland S. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell. 1994;77:761–771. doi: 10.1016/0092-8674(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 60•.Satheshkumar P.S., Gayathri P., Prasad K., Savithri H.S. “Natively unfolded” VPg is essential for Sesbania mosaic virus serine protease activity. J Biol Chem. 2005;280:30291–30300. doi: 10.1074/jbc.M504122200. [DOI] [PubMed] [Google Scholar]; This paper describes the interesting observation that VPg is necessary for the activity of SeMV serine protease. The repertoire of described VPg activities continues to grow.
- 61•.Gayathri P., Satheshkumar P.S., Prasad K., Nair S., Savithri H.S., Murthy M.R. Crystal structure of the serine protease domain of Sesbania mosaic virus polyprotein and mutational analysis of residues forming the S1-binding pocket. Virology. 2006;346:440–451. doi: 10.1016/j.virol.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; This publication presents the crystal structure of the plant viral protease described in [60•]. The VPg is not bound to the enzyme in this structure, but a binding site has been assigned with a reasonable degree of certainty.
- 62.Kim J.L., Morgenstern K.A., Lin C., Fox T., Dwyer M.D., Landro J.A., Chambers S.P., Markland W., Lepre C.H. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell. 1996;87:343–355. doi: 10.1016/s0092-8674(00)81351-3. [DOI] [PubMed] [Google Scholar]
- 63.Ontoria J.M., Di Marco S., Conte I., Di Francesco M.E., Gardelli C., Koch U., Matassa V.G., Poma M., Steinkühler C., Volpari C., Harper S. The design and enzyme-bound crystal structure of indoline based peptidomimetic inhibitors of hepatitis C virus NS3 protease. J Med Chem. 2004;47:6443–6446. doi: 10.1021/jm049435d. [DOI] [PubMed] [Google Scholar]
- 64.Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 66.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Groneberg D.A., Hilgenfeld R., Zabel P. Molecular mechanisms of severe acute respiratory syndrome (SARS) Respir Res. 2005;6:8–23. doi: 10.1186/1465-9921-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anand K., Yang H., Bartlam M., Rao Z., Hilgenfeld R. Coronavirus main proteinase: target for antiviral drug therapy. In: Schmidt A., Wolf M.H., Weber O., editors. Coronaviruses with Special Emphasis on First Insights Concerning SARS. Birkhäuser; Basel: 2005. pp. 173–199. [Google Scholar]
- 69•.Tan J., Verschueren K.H.G., Anand K., Shen J., Yang M., Xu Y., Rao Z., Bigalke J., Heisen B., Mesters J.R. pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J Mol Biol. 2005;354:25–40. doi: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes two new crystal forms of SARS-CoV main proteinase that provide valuable information on the dynamics of the enzyme. Also, the pH-dependent activation switch, observed previously by determining structures at different pH values, is simulated by molecular dynamics and shown to be triggered by protonation and deprotonation of two histidine residues in the substrate-binding site.
- 70.Xu T., Ooi A., Lee H.C., Wilmouth R., Liu D.X., Lescar J. Structure of the SARS coronavirus main proteinase as an active C2 crystallographic dimmer. Acta Crystallogr F. 2005;61:964–966. doi: 10.1107/S1744309105033257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71•.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]; Starting from the previous proposal that vinylogous alkyl ester derivatives of substrate-like peptides may be potent inhibitors of coronavirus proteinases, the optimization of such compounds and their crystal structures with the main proteinases of several coronaviruses are described. This useful study demonstrates how antiviral drugs with the desired, relatively broad, activity can be designed.
- 72.Lee T.W., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J Mol Biol. 2005;353:1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang S., Chen S.J., Hsu M.F., Wu J.D., Tseng C.T., Liu Y.F., Chen H.C., Kuo C.W., Wu C.S., Chang L.W. Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J Med Chem. 2006;49:4971–4980. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- 74.Wu C.Y., King K.Y., Kuo C.J., Fang J.M., Wu Y.T., Ho M.Y., Liao C.L., Shie J.J., Liang P.H., Wong C.H. Stable benzotriazole esters as mechanism-based inactivators of the severe acute respiratory syndrome 3CL protease. Chem Biol. 2006;13:261–268. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hilgenfeld R., Pumpor K. Sometimes intermediates do the job! Chem Biol. 2006;13:235–236. doi: 10.1016/j.chembiol.2006.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Al-Gharabli S.I., Shah S.T., Weik S., Schmidt M.F., Mesters J.R., Kuhn D., Klebe G., Hilgenfeld R., Rademann J. An efficient method for the synthesis of peptide aldehyde libraries employed in the discovery of reversible SARS coronavirus main protease (SARS-CoV Mpro) inhibitors. ChemBioChem. 2006;7:1048–1055. doi: 10.1002/cbic.200500533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen L., Gui C., Luo X., Yang Q., Günther S., Scandella E., Drosten C., Bai D., He X., Ludewig B. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J Virol. 2005;79:7095–7103. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sulea T., Lindner H.A., Purisima E.O., Menard R. Deubiquitination, a new function of the severe acute respiratory syndrome coronavirus papain-like protease? J Virol. 2005;79:4550–4551. doi: 10.1128/JVI.79.7.4550-4551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lindner H.A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Menard R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81••.Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc Natl Acad USA. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]; The crystal structure of the papain-like protease, the second protease of the SARS coronavirus, is presented here. Interestingly, the enzyme has been shown to have deubiquitinating activity, in addition to its protease function. In agreement with this, the three-dimensional structure is reminiscent of cellular deubiquitinases such as HAUSP.
- 82.Zhao C., Denison C., Huibregtse J.M., Gygi S., Krug R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci USA. 2005;102:10200–10205. doi: 10.1073/pnas.0504754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rosano C., Zuccotti S., Sturla L., Fruscione F., Tonetti M., Bolognesi M. Quaternary assembly and crystal structure of GDP-D-mannose 4,6 dehydratase from Paramecium bursaria Chlorella virus. Biochem Biophys Res Commun. 2006;339:191–195. doi: 10.1016/j.bbrc.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 84.Larson E.T., Reiter D., Young M., Lawrence C.M. Structure of A197 from Sulfolobus turreted icosahedral virus: a crenarchaeal viral glycosyltransferase exhibiting the GT-A fold. J Virol. 2006;80:7637–7644. doi: 10.1128/JVI.00567-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Imbert I., Guillemot J.C., Bourhis J.M., Bussetta C., Coutard B., Egloff M.P., Ferron F., Gorbalenya A.E., Canard B. A second, non-canonical RNA-dependent RNA polymerase in SARS coronavirus. EMBO J. 2006;25:4933–4942. doi: 10.1038/sj.emboj.7601368. [DOI] [PMC free article] [PubMed] [Google Scholar]