

Graphical abstract

Keywords: Carbocyclic C-nucleosides, Enantioselective synthesis, Antiviral activity
Abstract
The enantiomerically pure carbocyclic purine and pyrimidine C-nucleosides 1–4 were synthesized via the key intermediate, 2,3-(isopropylidenedioxy)-4-(trityloxymethyl)-4-cyclopenten-1-ol (5), which was prepared from d-ribose in eight steps. Synthesized compounds were evaluated as potential antiviral agents against HIV, SARSCoV, Punta Toro, West Nile, and Cowpox viruses. However, only 9-deazaneplanocin A (1) exhibited moderate anti-HIV activity.
1. Introduction
Synthesis of C-nucleoside and carbocyclic nucleoside analogs has been inspired by their interesting biological activities as well as chemical and enzymatic stability. C-Nucleosides are a unique class of compounds in which the heterocycle is connected to the sugar moiety by a C–C bond instead of a C–N bond of the natural nucleosides. As a result, they are resistant to the chemical and enzymatic hydrolytic cleavage of the glycosidic bond. C-Nucleosides have received considerable attention due not only to their chemical stability, but also for their interesting biological activities of naturally occurring compounds such as showdomycin, formycins, oxazinomycin, pyrazomycin, etc.1 Also several biologically active synthetic C-nucleosides such as 9-deazaadenosine,2 pseudoisocytidine,3 and thiazofurin4 have been reported to possess both antitumor and antiviral activities. Carbocyclic nucleosides are another class of metabolically stable nucleosides, in which a methylene group replaces the oxygen in the furan ring of the natural nucleosides.5 The natural carbocyclic nucleosides such as neplanocin6 and aristeromycin7 were isolated from microorganisms and were found to possess various biological activities including antiviral and antitumor activities. The synthetic carbocyclic nucleosides, abacavir,8 entecavir,9 and lobucavir,10 have shown promising antiviral activities. Recently, abacavir and entecavir were approved by the FDA as an anti-HIV agent and anti-hepatitis B virus agent, respectively.
On the basis of these chemical and biological properties of C-nucleosides and carbocyclic nucleosides, it was of interest to synthesize hybrid nucleosides, carbocyclic C-nucleosides. Although some carbocyclic nucleosides and C-nucleosides are naturally occurring, so far no natural carbocyclic C-nucleosides have been identified. The history of the synthesis of carbocyclic C-nucleosides dates back to 1960s.11 Despite the long history of both carbocyclic and C-nucleosides, only a few carbocyclic C-nucleosides have been synthesized,12 probably due to the synthetic difficulties of these nucleosides. As part of our ongoing program toward synthesis of novel nucleosides, we were exploring the synthesis of cyclopentenyl carbocyclic nucleosides with unnatural bases.13 Therefore, it was of great interest to synthesize optically active carbocyclic C-nucleosides, possessing a cyclopentenyl moiety as analogs of neplanocin. Herein, we report the enantiomeric synthesis of purine and pyrimidine cyclopentenyl C-nucleosides 1–4 (Fig. 1 ) from the key intermediate, 2,3-(isopropylidenedioxy)-4-(trityloxymethyl)-4-cyclopenten-1-ol (5), and their antiviral activities.
Figure 1.

Structure of target compounds 1–4.
2. Results and discussion
2.1. Synthesis
For the synthesis of target purine and pyrimidine carbocyclic C-nucleosides, we utilized the key intermediate 5 as a chiral starting material, which was prepared from d-ribose in eight steps using recently reported procedure from our group.13a In order to synthesize 9-deazaneplanocin A (1) (Scheme 1 ), the key intermediate 5 was converted to mesylate 6, followed by SN2 reaction with ethylcyanoacetate in the presence of sodium hydride to give compound 7 in 52% yield plus disubstituted product 8 in 10% yield. The optimal temperature was found to be 55 °C for this reaction, since at 70 °C the starting material decomposed. The assignment of β-configuration of 7 was performed by proton NOE experiments.13a Selective reduction of ester group of 7 with DIBAL-H, followed by condensation with aminoacetonitrile bisulfate in the presence of sodium acetate, gave the pyrrole precursor 9.12a Protection of the amino nitrogen of 9 as a carbamate with ethylchloroformate followed by cyclization catalyzed by 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU) and subsequent deprotection of carbamate with sodium carbonate led to the substituted pyrrole 10. Condensation of 10 with formamidine acetate in ethanol yielded the protected 9-deazaneplanocin 11 in 61%, which was deprotected with methanolic hydrochloric acid to afford the target compound, 9-deazaneplanocin A (1).
Scheme 1.
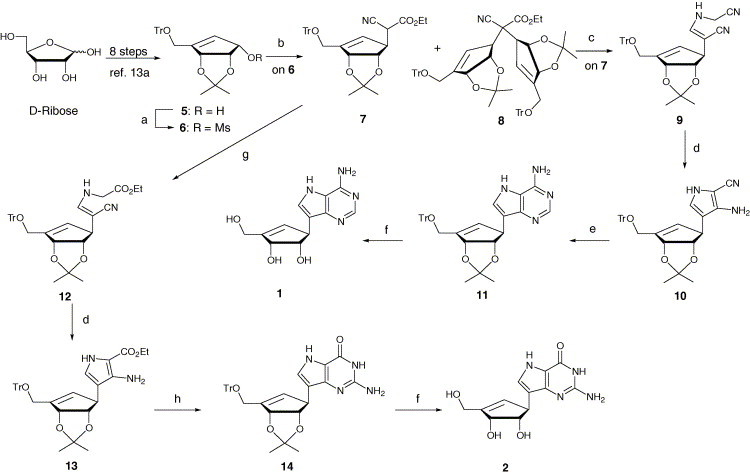
Reagents and conditions: (a) MsCl, Et3N, CH2Cl2, 0 °C, 1 h; (b) NCCH2CO2Et, NaH, THF, rt then to 55 °C, 40 h; (c) i—DIBAL-H, Et2O, −78 °C, 30 min; ii—H2NCH2CN·H2SO4, NaOAc·3H2O, MeOH, rt, 24 h; (d) i—ClCO2Et, DBU, CH2Cl2 0 °C then to reflux, 6 h; ii—K2CO3, MeOH, rt, 1 h; (e) HC( NH)NH2·AcOH, EtOH, reflux, 8 h; (f) i—12% HCl/MeOH, rt, 2 h; ii—NaHCO3/MeOH, rt, 2 h; (g) i—DIBAL-H, Et2O, −78 °C, 30 min; ii—H2NCH2CO2Et·HCl, NaOAc·3H2O, MeOH, rt, 24 h; (h) i—BzN C S, CH2Cl2, 0 °C, 1 h; ii—MeI, DBN, CH2Cl2, rt, 2 h; iii—NH3/MeOH, 95 °C, 16 h.
To synthesize the carbocyclic 9-deazaguanosine (2) (Scheme 1), substituted ethylcyanoacetate 7 was treated with DIBAL-H followed by condensation with ethylglycinate hydrochloride in the presence of sodium acetate to provide 12. N-Protection of 12 with ethyl chloroformate, followed by ring closure with DBU and subsequent deprotection of carbamate, provided 13. Treatment of 13 with N-benzoylisothiocyanate and methyl iodide, followed by treatment with methanolic ammonia in a steel bomb at 95 °C, gave the protected carbocyclic 9-deazaguanosine (14) in 64% yield. Deprotection of 14 with methanolic hydrochloric acid gave the target compound 2.
The carbocyclic isocytosine (3) was prepared in four steps from the intermediate 6 (Scheme 2 ). The SN2 reaction on 6 with diethylmalonate in the presence of sodium hydride gave 15 in 72% yield. Mono-decarbethoxylation of 15 in the presence of lithium chloride in DMSO gave 16 14 in 86% yield, which was then treated with lithium diisopropylamide and ethyl formate in THF, followed by condensation with guanidine carbonate in the presence of sodium ethoxide in ethanol yielded 17 in 64%. Deprotection of 17 with methanolic hydrochloric acid gave the HCl salt of target compound 3.
Scheme 2.
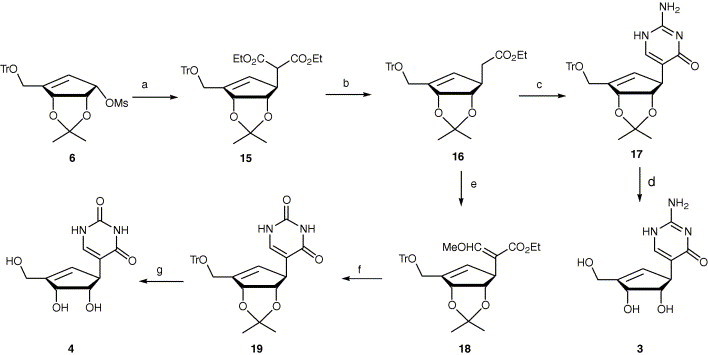
Reagents and conditions: (a) CH2(CO2Et)2, NaH, THF, 38 °C, 48 h; (b) LiCl, DMSO, 175 °C, 3 h; (c) i—LDA, THF, −78 °C, 2 h then ethyl formate, 12 h; ii—guanidine hydrochloride, NaOEt, EtOH, reflux, 12 h; (d) 10% HCl/MeOH, 2 h; (e) i—LDA, THF, −78 °C, 2 h then ethyl formate, 12 h; ii—MeI, DMF, rt, 4 h; (f) urea, KOtBu, THF, reflux, 48 h; (g) i—10% HCl/MeOH, 2 h; ii—NaHCO3/MeOH, rt, 2 h.
Preparation of the carbocyclic pseudouridine analog 4 was accomplished from 16 in three steps (Scheme 2). Compound 16 was treated with lithium diisopropylamide and ethylformate, followed by methyl iodide in DMF, to give the, compound 18 15 in 64% yield, which was condensed with urea in the presence of potassium tert-butoxide to afford 19. Deprotection of 19 with methanolic hydrochloric acid gave the target compound 4 in 72% yield. All the compound structures were assigned based on NMR, UV, mass spectroscopy, and elemental analysis.
2.2. Biological results and conclusions
The synthesized carbocyclic C-nucleosides 1–4 were tested for anti-HIV activity as well as cytotoxicity using AZT as the positive control, and the results are summarized in Table 1 . Anti-HIV activity was determined in human peripheral blood mononuclear (PBM) cells infected with HIV-1 (strain LAI). Among the synthesized nucleosides, only 9-deazaneplanocin A (1) showed moderate anti-HIV-1 activity (EC50 2.0 μM) and the other compounds did not display any anti-HIV activity. Compounds 1–4 were also evaluated against a wide variety of viruses, including vaccinia, cowpox, severe acute respiratory syndrome (SARS), Punta Toro virus, West Nile virus, yellow fever, herpes simplex virus 1 and 2, varicella zoster virus, Tacaribe, Venezulean equine encephalitis, Dengue, and hepatitis C. Compounds 2 (EC50 2.5 μM) and 4 (EC50 65 μM) showed moderate activity against Punta Toro virus (strain Adames, LLC-MK2 cell line) without any cytotoxicity. Compound 3 was slightly active against West Nile virus (New York isolate) (EC50 11 μM) and compound 4 (EC50 49 μM) showed marginal activity against SARSCoV (Table 2 ). None of other compounds showed any antiviral activity against all other tested viruses.
Table 1.
In vitro anti-HIV-1LAI activity and toxicity of target nucleosides 1–4
| Compound | Anti-HIV-1LAI activity |
Cytotoxicity (IC50, μM) |
|||
|---|---|---|---|---|---|
| EC50 (μM) | EC90 (μM) | PBM | CEM | Vero | |
| 1 | 2.0 ± 1.1a | 43.2 ± 28.8a | 15.6 | 10.6 | >100 |
| 2 | >100 | >100 | >100 | >100 | >100 |
| 3 | >100 | >100 | >100 | >100 | >100 |
| 4 | >100 | >100 | >100 | >100 | >100 |
| AZT | 0.0029 | 0.02 | >100 | 14.3 | 50.6 |
Standard deviation.
Table 2.
Antiviral activities of synthesized nucleosides 1–4 against SARS, Punta Toro, Cowpox, and West Nile viruses
| Compound | SARSCoV (μM) |
Punta Toro (μM) |
WNV (μM) |
Cowpox (μM) |
||||
|---|---|---|---|---|---|---|---|---|
| EC50 | IC50 | EC50 | IC50 | EC50 | IC50 | EC50 | IC50 | |
| 1 | ND | ND | ND | ND | >100 | >100 | >100 | >100 |
| 2 | >100 | >100 | 2.5 | >100 | >100 | >100 | >100 | >100 |
| 3 | >100 | >100 | >100 | >100 | 11 | >100 | >100 | >100 |
| 4 | 49 | >100 | 65 | >100 | >100 | >100 | >100 | >100 |
| Neplanocin A | ND | ND | ND | ND | >51 | ND | 8.4 | >300 |
ND, not determined.
In summary, enantioselective synthesis of purine and pyrimidine cyclopentenyl C-nucleosides 1–4 was achieved from the common key intermediate 5, which was prepared from commercially available d-ribose. Synthesized compounds were tested as potential antiviral agents. However, only 9-deazaneplanocin A (1) exhibited moderate anti-HIV activity.
3. Experimental
3.1. Materials and methods
Melting points were determined on a Mel-temp II apparatus and are uncorrected. Nuclear magnetic resonance spectra were recorded on a Varian Inova 500 spectrometer at 500 MHz for 1H NMR and 125 MHz for 13C NMR with tetramethylsilane as the internal standard. Chemical shifts (δ) are reported as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), or br s (broad singlet). UV spectra were recorded on a Beckman DU-650 spectrometer. Optical rotations were measured on a Jasco DIP-370 digital polarimeter. High-resolution mass spectra were recorded on a Micromass Autospec high-resolution mass spectrometer. TLC was performed on Uniplates (silica gel) purchased from Analtech Co. Column chromatography was performed using silica gel 60 (220–440 mesh) for flash chromatography or silica gel G (TLC grade, >440 mesh) for vacuum flash column chromatography or Radisep amine functionalized column (Combiflash, Isco, Inc.). Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA.
3.1.1. (1S,2R,3R)-[2,3-(Isopropylidenedioxy)-4-(trityloxymethyl)-4-cyclopenten-1-yl]-methane sulfonate (6)
To a solution of 5 (12 g, 28.0 mmol) in anhydrous CH2Cl2 (100 mL), Et3N (11.64 mL, 84.0 mmol) was added under N2 atmosphere at 0 °C. After 5 min, methanesulfonyl chloride (3.24 mL, 42.0 mmol) was slowly added to the reaction mixture at 0 °C and then stirred for 1 h at room temperature. To the reaction mixture, Et3N (20 mL) was added until the solution became basic and then solvent was removed under reduced pressure. The residue was purified by silica gel chromatography (hexane–EtOAc = 8:2–7:3) to give compound 6 (13.2 g, 93%) as a white solid. Mp 92 °C; (c 1.0, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.45–7.42 (m, 6H, Tr-H), 7.32–7.22 (m, 9H, Tr-H), 6.02 (d, 1H, J = 2 Hz, H-6), 5.43 (m, 1H, H-1), 4.86 (m, 2H, H-2, 3), 3.96 (d, 1H, J = 15.2 Hz, H-5a), 3.69 (d, 1H, J = 15.2 Hz, H-5b), 3.15 (s, 3H, –CH3), 1.37 (s, 3H, –CH3), 1.35 (s, 3H, CH3); 13C NMR (CDCl3, 125 MHz) δ 147.8, 143.7, 128.5, 127.9, 127.2, 124.0, 113.5, 86.4, 80.8, 77.2, 61.0, 39.0, 27.5, 27.1; HRMS-ESI (m/z): (M+H)+ calcd for C29H30O6S, 507.1842; found, 507.1843.
3.1.2. (2R/S,1′S,2′S,3′R)-Ethyl-2-[2′,3′-(isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl]-2-cyanoacetate (7) and (1′S,2′S,3′R)-ethyl-2-bis[2′,3′-(isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl]-2-cyanoacetate (8)
To a solution of NaH (1.87 g, 78.2 mmol) in THF (200 mL) was added ethylcyanoacetate (8.33 mL, 78.2 mmol) in THF (100 mL) under nitrogen atmosphere at 0 °C. After 1 h, 6 (13.2 g, 26.0 mmol) in THF (0.8 L) was added to the reaction mixture. The mixture was stirred at 50 °C for 40 h and then cooled to room temperature, H2O (100 mL) was added and extracted with ethyl acetate (2× 300 mL). The combined organic layers were dried over MgSO4, concentrated, and then purified on silica gel column chromatography (hexane–EtOAc = 9:1–8:2) to give compound 7 (7.09 g, 52%) and compound 8 (2.43 g, 10%) as white solids. Compound 7: mp 50 °C; (c 0.5, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.45–7.43 (m, 6H, Tr-H), 7.31–7.22 (m, 9H, Tr-H), 5.87 (d, 1H, J = 11 Hz, H-6′), 5.08 (d, 1H, J = 5.5 Hz, H-3′), 4.68 (d, 0.55H, J = 6.0 Hz, H-2′), 4.62 (d, 0.45H, J = 6.0 Hz, H-2′), 4.31 (m, 2H, –OCH2–), 3.84 (d, 1H, J = 15 Hz, H-5′a), 3.70 (m, 2H, H-5′b, –CHCN), 3.48 (br d, 1H, J = 2.5 Hz, H-1′), 1.35–1.31 (m, 9H, –CH3, –C(CH3)2); 13C NMR (CDCl3, 125 MHz) δ 165.0, 164.8, 147.27, 147.25, 143.9, 128.6, 128.5, 127.9, 123.4, 123.2, 115.2, 115.1, 111.4, 111.3, 87.1, 84.4, 84.3, 81.9, 81.4, 63.19, 63.14, 61.4, 51.8, 51.5, 41.2, 40.8, 27.4, 26.0, 25.9, 14.0; HRMS-ESI (m/z): (M+Na)+ calcd for C33H33NO5, 546.2256; found, 546.2248.
Compound 8: mp 87 °C; (c 1, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.45–7.43 (m, 12H, Tr-H), 7.32–7.21 (m, 18H, Tr-H), 6.22 (s, 1H, H-6′) 5.78 (s, 1H, H-6″), 5.10 (d, 1H, J = 5.5 Hz, H-3′), 5.01 (d, 1H, J = 6.0 Hz, H-3″), 4.91 (d, 1H, J = 6.0 Hz, H-2′), 4.63 (d, 1H, J = 6.0 Hz, H-2″), 4.33 (m, 2H, –OCH2–), 3.88 (d, 1H, J = 15.5 Hz, H-5′a), 3.82 (d, 1H, J = 14.5 Hz, H-5″a), 3.74 (d, 1H, J = 15 Hz, H-5′b), 3.72 (d, 1H, J = 15 Hz, H-5″b) 3.64 (br s, 1H, H-1′), 3.51 (s, 1H, H-1″) 1.38, 1.35 (2× s, 2× 3H, –C(CH3)2), 130, 1.22 (2× s, 2× 3H, –C(CH3)2). 1.31 (t, 3H, J = 6.5 Hz, –CH3); 13C NMR (CDCl3, 125 MHz) δ 167.3, 148.0, 147.4, 143.9, 143.8, 128.6, 128.5, 127.9, 127.1, 122.7, 121.7, 117.1, 111.4, 111.1, 87.1, 84.1, 83.9, 81.4, 81.1, 63.3, 61.5, 57.2, 56.4, 55.5, 27.1, 27.3, 26.1, 25.7, 14.2; HRMS-ESI (m/z): (M+H)+ calcd for C61H59NO8, 933.4319; found, 933.4317.
3.1.3. (1′S,2′S,3′R)-3-Amino-2-cyano-4-(2′,3′-(isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl)-1H-pyrrole (10)
To a solution of 7 (3.50 g, 6.68 mmol) in anhydrous ether (60 mL), DIBAL-H (1 M in hexane, 13.36 mL, 13.36 mmol) was added at −78 °C over 10 min. The mixture was stirred at −78 °C for 30 min and quenched with MeOH (90 mL) at −78 °C. The mixture was stirred for 30 min and concentrated under reduced pressure. The resultant solid cake was suspended in EtOAc–MeOH (30:2) and then stirred for 2 h and filtered. After removal of the filtrate under reduced pressure, the residue was dissolved in MeOH (70 mL). Aminoacetonitrile bisulfate (2.57 g, 16.71 mmol), NaOAc·3H2O (2.18 g, 16.04 mmol) were added and the mixture was stirred at rt for 24 h. The solvent was evaporated and the residue was co-evaporated with MeOH (3× 60 mL). The obtained residue was suspended in EtOAc (100 mL) and stirred for 30 min. After filtration and concentration, the residue was purified using silica gel column chromatography (hexane–EtOAc = 8:2–7:3) to give compound 9 (1.54 g, 45%) as a white solid. To a solution of 9 (1.54 g, 2.97 mmol) in anhydrous CH2Cl2 (75 mL), DBU (8.89 mL, 59.5 mmol) and ethyl chloroformate (2.26 g, 20.8 mmol) were added at 0 °C and the mixture was heated to reflux for 12 h. After removal of the solvent, the residue was dissolved in MeOH (30 mL) and water (1 mL), and then treated with K2CO3 (55 mg, 0.42 mmol). The new mixture was stirred for 1 h at rt and solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (hexane–EtOAc = 8:2) to give 10 (730 mg, 21% from 7) as a white solid. Mp 92 °C; (c 0.5, CHCl3); UV (MeOH) λ max, nm: 249; 1H NMR (CDCl3, 500 MHz) δ 7.87 (s, 1H, NH), 7.46 (m, 6H, Tr-H), 7.27 (m, 9H, Tr-H), 6.42 (s, 1H, H-5), 5.97 (s, 1H, H-6′), 5.09 (d, 1H, J = 5.5 Hz, 1H, H-3′), 4.49 (d, 1H, J = 5.5 Hz, H-2′), 3.90 (d, 1H, J = 14 Hz, H-5′a), 3.79 (s, 1H, H-1′), 3.73 (d, 1H, J = 14 Hz, H-5′b), 3.71 (br s, 2H, NH2), 1.40 (s, 3H, CH3), 1.31 (s, 3H, CH3); 13C NMR (CDCl3, 125 MHz) δ 150.0, 143.9, 143.2, 141.6, 128.6, 127.8, 127.18, 127.12, 120.9, 114.6, 113.7, 111.2, 87.1, 85.0, 84.7, 61.4, 47.0, 27.5, 26.0; HRMS-ESI (m/z): (M+Na)+ calcd for C33H31N3O3, 540.2262; found, 540.2286.
3.1.4. (1′S,2′S,3′R)-4-Amino-7-[2′,3′-(isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl]-5H-pyrrolo[3,2-d]pyrimidine (11)
A solution of 10 (350 mg, 0.67 mmol) and formamidine acetate (352 mg, 3.38 mmol) in EtOH (20 mL) was heated under reflux for 8 h. The solvent was removed by reduced pressure and the residue was purified by silica gel column chromatography (CH2Cl2–MeOH = 98:2) to give 11 (224 mg, 61%) as a white solid. Mp 132 °C; (c 0.40, MeOH); UV (MeOH) λ max, nm: 274; 1H NMR (DMSO-d 6, 500 MHz) δ 10.82 (s, 1H, NH), 8.11 (s, 1H, H-2), 7.46–7.25 (m, 16H, Tr-H, H-6), 6.75 (s, 2H, –NH2), 5.98 (s, 1H, H-6′), 5.22 (d, 1H, J = 5.5 Hz, H-3′), 4.65 (d, 1H, J = 6 Hz, H-2′), 4.16 (s, 1H, H-1′), 3.70 (d, 1H, J = 13.5 Hz, H-5′a), 3.60 (d, 1H, J = 14 Hz, H-5′b), 1.31 (s, 3H, –CH3), 1.24 (s, 3H, –CH3); 13C NMR (DMSO-d 6, 125 MHz) δ 150.6, 150.3, 145.0, 144.1, 141.2, 128.7, 128.5, 128.4, 127.6, 125.6, 115.5, 114.1, 110.4, 86.8, 85.0, 84.7, 61.4, 46.1, 27.9, 26.3; HRMS-ESI (m/z): (M+H)+ calcd for C34H32N4O3, 545.2553; found, 545.2558; Anal. Calcd for C34H32N4O3: C, 74.98; H, 5.92; N, 10.29; found: C, 74.96; H, 5.88; N, 10.01.
3.1.5. (1′S,2′S,3′R)-4-Amino-7-(2′,3′-dihydroxy-4′-hydroxymethyl-4′-cyclopenten-1′-yl)-5H-pyrrolo[3,2-d]pyrimidine (9-deazaneplanocin) (1)
A solution of 11 (200 mg, 0.36 mmol) in 12% methanolic HCl (10 mL) was stirred at rt for 2 h. The solvent was evaporated under reduced pressure and the residue was co-evaporated with MeOH (3× 30 mL). The product was dissolved in MeOH (15 mL), neutralized with solid NaHCO3, stirred for 2 h, filtered, and concentrated to dryness under reduced pressure. The residue was purified by column chromatography (amine functionalized silica gel) (CH2Cl2–MeOH = 5:1) to give 1 (60 mg, 63%) as a white solid. Mp 202 °C; (c 0.30, MeOH); UV (H2O) λ max, nm: 276 (18,530, pH 2), 275 (11,760, pH 7), 274 (11,720, pH 11); 1H NMR (DMSO-d 6, 500 MHz) δ 10.76 (s, 1H, NH), 8.11 (s, 1H, H-2), 7.33 (d, 1H, J = 2.0 Hz, H-6), 6.73 (s, 2H, NH2), 5.83 (d, 1H, J = 2 Hz, H-6′), 5.41 (br s, 1H, OH), 4.75 (t, 1H, J = 5.5 Hz, OH), 4.53 (d, 1H, J = 6.5 Hz, OH), 4.37 (t, 1H, J = 5.5 Hz, H-3′), 4.08 (m, 3H, H-2′,5′a,b), 3.92 (d, 1H, J = 2.5 Hz, H-1′); 13C NMR (DMSO-d 6, 125 MHz) δ 150.6, 149.9, 145.3, 144.9, 129.1, 125.6, 116.1, 114.1, 78.3, 73.6, 58.9, 46.0; HRMS-ESI (m/z): (M+Na)+ calcd for C12H14N4O3, 263.1148; found, 263.1170; Anal. Calcd for C12H14N4O3·0.8H2O: C, 52.03; H, 5.69; N, 20.02; found: C, 52.03; H, 5.69; N, 19.98.
3.1.6. (1′S,2′S,3′R)-[3-Amino-4-(2′,3′-(isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl)-1H-pyrrole]-2-carboxylic acid ethyl ester (13)
To a solution of 7 (5.2 g, 9.93 mmol) in anhydrous ether (80 mL), DIBAL-H (1 M in hexane, 19.8 mL) was added at −78 °C over 10 min. The mixture was stirred at −78 °C for 30 min and quenched with MeOH (90 mL) at −78 °C. The mixture was stirred for 30 min and concentrated under reduced pressure. The resultant solid cake was suspended in EtOAc–MeOH (30:2) and then stirred for 1 h and filtered. After removal of the filtrate under reduced pressure, the residue was dissolved in MeOH (70 mL). Ethylglycinate hydrochloride (3.32 g, 23.8 mmol) and NaOAc·3H2O (3.37 g, 24.8 mmol) were added and the mixture was stirred at rt for 24 h. The solvent was evaporated and the residue was co-evaporated with MeOH (3× 60 mL). The obtained residue was suspended in EtOAc (100 mL) and stirred for 30 min. After filtration and concentration under vacuum, the residue was purified using silica gel column chromatography (hexane–EtOAc = 8:2–7:3) to give compound 12 (2.12 g, 38%) as a white solid. To a solution of 12 (2.12 g, 3.75 mmol) in dry CH2Cl2 (75 mL) were added DBU (11.23 mL, 75.14 mmol) and ethyl chloroformate (2.50 mL, 26.30 mmol) at 0 °C and then the mixture was heated to reflux for 6 h. After removal of the solvent, the residue was dissolved in MeOH (30 mL) and water (1 mL), and then treated with K2CO3 (50 mg). This mixture was stirred for 1 h at rt and solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (hexane–EtOAc = 4:1) to give 13 (0.986 g, 18% from 7) as a white solid. Mp 86 °C; (c 0.50, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.49–7.46 (m, 6H, Tr-H), 7.32–7.22 (m, 9H, Tr-H), 6.45 (br s, 1H, H-5), 6.0 (s, 1H, H-6′), 5.10 (d, 1H, J = 5.5 Hz, H-3′), 4.53 (d, 1H, J = 6.5 Hz, H-2′), 4.31 (q, 2H, –CH2), 3.90 (d, 1H, J = 14 Hz, H-5′a), 3.81 (s, 1H, H-1′), 3.73 (d, 1H, J = 14 Hz, H-5′b), 1.41 (s, 3H, –CH3), 1.35 (t, 3H, J = 7 Hz, –CH3), 1.31 (s, 3H, –CH3); 13C NMR (CDCl3, 125 MHz) δ 162.1, 144.0, 142.7, 128.6, 127.8, 127.5, 127.0, 120.2, 113.2, 111.0, 87.05, 85.2, 84.8, 61.4, 47.0, 27.6, 26.0, 14.7; HRMS-ESI (m/z): (M+Na)+ calcd for C35H36N2O5, 587.2521; found, 587.2438.
3.1.7. (1′S,2′S,3′R)-9-(2′,3′-(Isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl)-9-deazaguanosine (14)
N-Benzoylisothiocyanate (0.214 mL, 1.59 mmol) was added to a solution of 13 (0.6 g, 1.06 mmol) in CH2Cl2 (20 mL) at 0 °C. The reaction mixture was stirred for 1 h at rt and concentrated under vacuum. The residue was purified by silica gel column chromatography (hexane–EtOAc = 4:1) to give a residue (1.55 g). After being dried under high vacuum, the residue (1.55 g, 2.12 mmol) was dissolved in CH2Cl2 (15 mL) and treated consecutively with DBN (1.39 mL, 11.68 mmol) and MeI (0.30 mL, 4.67 mmol). The resulting reaction mixture was stirred for 2 h at rt, diluted with CH2Cl2 (30 mL), washed with water (2× 20 mL), dried over MgSO4, and concentrated. The residue was purified by silica gel column chromatography (hexane–EtOAc = 4:1) to give a residue. The residue was dissolved in MeOH (10 mL), saturated with NH3 (g) at −78 °C in a steel bomb under anhydrous conditions. The mixture was then heated for 16 h at 95 °C under tight sealing. After being cooled to rt, the mixture was concentrated under vacuum and the residue was purified by silica gel chromatography (hexane–EtOAc = 1:4) to give 14 (380 mg, 64%) as a white solid. Mp 168°C; (c 0.50, MeOH); 1H NMR (CD3OD, 500 MHz) δ 7.48–7.44 (m, 6H, Tr-H), 7.31–7.20 (m, 9H, Tr-H), 6.89 (s, 1H, H-8), 5.93 (s, 1H, H-6′), 5.20 (d, 1H, J = 7 Hz, H-3′), 4.60 (d, 1H, J = 6.5 Hz, H-2′), 4.11 (s, 1H, H-1′), 3.80 (d, 1H, J = 14 Hz, H-5′a), 3.74 (d, 1H, J = 14 Hz, H-5′b), 1.34 (s, 3H, –CH3), 1.28 (s, 3H, –CH3); HRMS-ESI (m/z): (M+H)+ calcd for C34H32N4O4, 561.2503; found, 561.2501; Anal. Calcd for C34H32N4O4: C, 72.84; H, 5.75; N, 9.99; found: C, 72.77; H, 5.69; N, 9.98.
3.1.8. (1′S,2′S,3′R)-9-(2′,3′-Dihydroxy-4′-hydroxymethyl-4′-cyclopenten-1′-yl)-deazaguanosine (2)
A solution of 14 (220 mg, 0.39 mmol) in 12% methanolic HCl (10 mL) was stirred at rt for 2 h. The solvent was evaporated under reduced pressure and the residue was co-evaporated with MeOH (3× 30 mL). The product was dissolved in MeOH (15 mL), neutralized with solid NaHCO3, stirred for 2 h, filtered, and concentrated to dryness under reduced pressure. The residue was purified by combiflash column chromatography (amine functionalized silica gel) (CH2Cl2–MeOH = 5:1) to give 2 (76 mg, 70%) as a white solid. Mp 198 °C; (c 0.25, MeOH); UV (H2O) λ max, nm: 273 (14,946, pH 2), 272 (12,040, pH 7), 270 (11,003, pH 11); 1H NMR (DMSO-d 6, 500 MHz) δ 11.16 (s, 1H, NH), 6.90 (s, 1H, H-8), 6.24 (s, 2H, NH2), 5.76 (d, 1H, J = 1.5 Hz, H-6′), 4.78 (br s, 1H, –NH), 4.34 (d, 1H, J = 5.5 Hz, H-3′), 4.05 (t, 2H, J = 16 Hz, H-5′a,b), 3.94 (t, 1H, J = 6.0 Hz, H-2′), 3.75 (d, 1H, J = 2.5 Hz, H-1′), 3.42 (br s, 3H, 3× OH); 13C NMR (DMSO-d 6, 125 MHz) δ 155.6, 151.8, 145.4, 144.8, 128.2, 123.8, 115.3, 113.2, 78.5, 73.2, 59.0, 46.0; HRMS-ESI (m/z): (M+Na)+ calcd for C12H14N4O4, 301.0912; found, 301.0921; Anal. Calcd for C12H14N4O4·0.2H2O: C, 51.13; H, 5.15; N, 19.88; found: C, 51.12; H, 5.15; N, 19.84.
3.1.9. (1′S,2′S,3′R)-2-[2′,3′-(isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl]-2-diethyl-malonate (15)
To a solution of NaH (1.98 g, 82.9 mmol) in THF (200 mL), diethyl malonate (20.9 mL, 138.17 mmol) in THF (100 mL) was added under nitrogen atmosphere at rt. After 1 h, compound 6 (14 g, 27.6 mmol) in THF (0.8 L) was added to the reaction mixture. The mixture was stirred at 38 °C for 48 h and then cooled to room temperature, H2O (100 mL) was added and extracted with ethyl acetate (2× 300 mL). The combined organic layers were dried over MgSO4, concentrated, and then purified on silica gel column chromatography (hexane–EtOAc = 9:1) to give compound 15 (11.35 g, 72%) as a colorless liquid. (c 1, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.45–7.42 (m, 6H, Tr-H), 7.30–7.20 (m, 9H, Tr-H), 5.86 (d, J = 1 Hz, 1H, H-6′), 5.01 (d, 1H, J = 7.5 Hz, H-3′), 4.70 (d, 1H, J = 7 Hz, H-2′), 4.20 (m, 4H, –OCH2–), 3.80 (d, 1H, J = 18.5 Hz, H-5′a), 3.62 (d, 1H, J = 18.5 Hz, H-5′b), 3.46 (m, 2H, H-1′, –CH–), 1.35 (s, 3H, –CH3), 1.31 (s, 3H, –CH3), 1.29–1.24 (m, 6H, 2× CH3); 13C NMR (CDCl3, 125 MHz) δ 168.2, 168.1, 144.4, 143.9, 128.5, 127.9, 127.8, 127.0, 125.8, 110.6, 86.8, 84.5, 82.0, 61.6, 61.5, 61.3, 54.2, 50.9, 27.5, 26.0, 14.1, 14.0; HRMS-ESI (m/z): (M+Na)+ calcd for C35H38O7, 593.2515; found, 593.2519.
3.1.10. (1′R,2′S,3′R)-Ethyl-2-[2′,3′-(isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl]-acetate (16)
To a solution of 15 (7.5 g, 13.4 mmol) in DMSO (50 mL), LiCl (1.67 g, 39.4 mmol) and H2O (2 drops) were added. The mixture was heated up to 175 °C for 3 h. After cooling the mixture, H2O (50 mL) was added and extracted with ethyl acetate (2× 200 mL), washed with brine, dried over MgSO4, and concentrated under reduced pressure. The product was purified using silica gel column chromatography (hexane–EtOAc = 9:1) to give 16 (5.69 g, 86%) as a white solid. Mp 119 °C; (c 1, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.46 (m, 6H, Tr-H), 7.30–7.20 (m, 9H, Tr-H), 5.86 (s, 1H, H-6′), 5.01 (d, 1H, J = 5.5 Hz, H-3′), 4.48 (d, 1H, J = 5.5 Hz, H-2′), 4.15 (q, 2H, –OCH2–), 3.83 (d, 1H, J = 14.5 Hz, H-5′a), 3.65 (d, 1H, J = 14.5 Hz, H-5′b), 3.19 (t, 1H, J = 6 Hz, H-1′), 2.40 (dd, 2H, J = 2, 7.5 Hz, –CH2–), 1.36 (s, 3H, –CH3), 1.31 (s, 3H, –CH3), 1.26 (t, 3H, J = 7 Hz, –CH3); 13C NMR (CDCl3, 125 MHz) δ 171.9, 144.0, 142.7, 128.6, 128.2, 127.8, 127.0, 110.6, 86.9, 84.5, 83.7, 61.4, 60.6, 47.3, 38.0, 27.6, 26.1, 14.2; HRMS-ESI (m/z): (M+H)+ calcd for C32H34O5, 499.2484; found, 499.2479.
3.1.11. (1′S,2′S,3′R)-5-(2′,3′-(Isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl)-isocytosine (17)
To a solution of lithium diisopropylamide (2 M in hexane, 3.95 mL) in THF (20 mL), a solution of 16 (2.63 g, 5.27 mmol) in THF (40 mL) was added at −78 °C under nitrogen atmosphere. The mixture was stirred at the same temperature for 1 h and to this was added ethyl formate (1.69 mL, 21.09 mmol). After stirring at −78 °C for an additional hour, the mixture was stirred at rt for 12 h. To the mixture satd NH4Cl solution (50 mL) was added and extracted with EtOAc (2× 200 mL), washed with water (50 mL) and brine solution (50 mL), dried over MgSO4, and evaporated to dryness to obtain a crude residue. To a solution of guanidine carbonate (2.70 g, 14.99 mmol) in EtOH (50 mL), EtONa/EtOH (21 wt% solution, 6.47 mL, 19.99 mmol) was added and stirred for 10 min. To this mixture, the above-obtained residue in ethanol (10 mL) was added and refluxed for 12 h. The mixture was filtered, concentrated, and purified using silica gel column chromatography (EtOAc–MeOH = 95:5) to give 17 (1.78 g, 64 %) as a white solid. Mp 156 °C; (c 0.8, MeOH); 1H NMR (DMSO-d 6, 500 MHz) δ 10.93 (s, 1H, NH), 7.44–7.27 (m, 16H, Tr-H, H-6), 6.46 (s, 2H, NH2), 5.78 (s, 1H, H-6′), 5.08 (d, 1H, J = 5.5 Hz, H-3′), 4.40 (d, 1H, J = 5.5 Hz, H-2′), 3.76 (s, 1H, H-1′), 3.67 (d, 1H, J = 14.0 Hz, H-5′a), 3.57 (d, 1H, J = 14.0 Hz, H-5′b), 1.27 (s, 3H, –CH3), 1.22 (s, 3H, –CH3); 13C NMR (DMSO-d 6, 125 MHz) δ 162.9, 155.7, 144.2, 142.8, 128.5, 128.4, 127.5, 126.9, 114.8, 110.0, 86.8, 84.7, 84.4, 61.6, 48.4, 28.0, 26.4; HRMS-ESI (m/z): (M+Na)+ calcd for C32H31N3O4, 544.2212; found, 544.2262; Anal. Calcd for C32H31N3O4: C, 73.68; H, 5.99; N, 8.06; found: C, 73.57; H, 5.95; N, 8.01.
3.1.12. (1′S,2′S,3′R)-5-(2′,3′-Dihydroxy-4′-hydroxymethyl-4′-cyclopenten-1′-yl)-isocytosine (3)
A mixture of 17 (250 mg) and 10% methanolic hydrogen chloride (10 mL) solution was stirred at rt for 2 h. Concentration in vacuum gave a white solid which was co-evaporated with EtOH (3× 5 mL). The residue was triturated three times with 5 mL of ether, the compound 3 was obtained as a white powder (112 mg, 85%). Mp 186 °C (color change); (c 0.1, MeOH); UV (H2O) λ max, nm: 264 (6507, pH 2), 290 (4217, pH 7), 279 (5737, pH 11); 1H NMR (DMSO-d 6, 500 MHz) δ 12.42 (s, 1H, NH), 8.28 (br s, 2H, NH2), 7.34 (s, 1H, H-6), 5.49 (d, 1H, J = 1.5 Hz, H-6′), 4.56 (br s, 3H, 3× OH), 4.31 (d, 1H, J = 5.0 Hz, H-3′), 4.05 (m, 2H, H-5′a,b), 3.90 (t, 1H, J = 5.0 Hz, H-2′), 3.56 (d, 1H, J = 2.5 Hz, H-1′); 13C NMR (DMSO-d 6, 125 MHz) δ 160.8, 152.6, 147.7, 137.1, 125.4, 117.8, 76.1, 73.7, 58.9, 47.8; HRMS-ESI (m/z): (M+H)+ calcd for C10H13N3O4, 240.0985; found, 240.0963; Anal. calcd for C10H13N3O4·HCl: C, 43.57; H, 5.12; N, 15.24; found: C, 43.46; H, 5.09; N, 15.03.
3.1.13. (1′S,2′S,3′R)-Ethyl-2-(2′,3′-(isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl)-3-methoxy acrylate (18)
To a solution of lithium diisopropylamide (2 M in hexane, 3.95 mL) in THF (20 mL) was added a solution of 16 (2.63 g, 5.27 mmol) in THF (40 mL) at −78 °C under nitrogen atmosphere. The mixture was stirred at the same temperature for 1 h and then ethyl formate (1.69 mL, 21.09 mmol) was added. After stirring at −78 °C for an additional hour, the mixture was warmed up to 0 °C and allowed to stir for 12 h at rt. The resulting mixture was evaporated to dryness and the residue was dissolved in dry DMF (15 mL) and treated with MeI (0.98 mL, 15.83 mmol) under nitrogen. After stirring for 4 h at rt, DMF was removed under reduced pressure and water (30 mL) was added, extracted with ethyl acetate (2× 100 mL). The combined organic layers were washed with brine (50 mL), dried over MgSO4, concentrated under reduced pressure, and purified by silica gel column chromatography (hexane–EtOAc = 9:1) to give 18 (1.82 g, 64%) as a white solid. Mp 58 °C; (c 1.0, CHCl3); 1H NMR (CDCl3, 500 MHz) δ 7.45 (m, 6H, Tr-H), 7.36 (s, 1H, CH–OMe), 7.30–7.20 (m, 9H, Tr-H), 5.71 (s, 1H, H-6′), 5.16 (d, 1H, J = 5.6 Hz, H-3′), 4.68 (d, 1H, J = 6 Hz, H-2′), 4.12 (q, 2H, –OCH2), 4.04 (br s, 1H, H-1′), 3.83 (s, 3H, –OCH3), 3.82 (d, 1H, J = 14.0 Hz, H-5′a), 3.64 (d, 1H, J = 14.8 Hz, H-5′b), 1.38 (s, 3H, –CH3), 1.31 (s, 3H, –CH3), 1.23 (t, 3H, J = 6.8 Hz, –CH3); 13C NMR (CDCl3, 125 MHz) δ 167.3, 159.6, 144.2, 141.9, 128.6, 127.8, 126.9, 126.5, 110.3, 110.2, 86.9, 86.2, 84.3, 61.8, 61.6, 59.9, 47.1, 27.7, 26.2, 14.3; HRMS-ESI (m/z): (M+H)+ calcd for C34H36O6, 541.2591; found, 541.2590.
3.1.14. (1′S,2′S,3′R)-5-(2′,3′-(Isopropylidenedioxy)-4′-(trityloxymethyl)-4′-cyclopenten-1′-yl)uracil (19)
A mixture of urea (133 mg, 2.22 mmol) and KOtBu (246 mg, 2.22 mmol) in THF (5 mL) was stirred at rt for 30 min and then 18 (0.6 g, 1.11 mmol) in THF (8 mL) was added under N2 atmosphere. The mixture was heated under reflux for 48 h and evaporated under reduced pressure. The product was purified using silica gel column chromatography (hexane–EtOAc = 8:2) to give 19 (208 mg, 36%) as a white solid. Mp 135 °C; +14.69 (c 0.55, MeOH); 1H NMR (DMSO-d 6, 500 MHz) δ 11.15 (s, 1H, NH), 10.78 (s, 1H, NH), 7.42–7.26 (m, 15H, Tr-H), 6.97 (s, 1H, H-6), 5.75 (s, 1H, H-6′), 5.05 (d, 1H, J = 6 Hz, H-3′), 4.38 (d, 1H, J = 6 Hz, H-2′), 3.72 (s, 1H, H-1′), 3.66 (d, 1H, J = 13.5 Hz, H-5′a), 3.56 (d, 1H, J = 13.5 Hz, H-5′b), 1.25 (s, 3H, –CH3), 1.20 (s, 3H, –CH3); 13C NMR (DMSO-d 6, 125 MHz) δ 164.3, 151.7, 144.2, 143.8, 138.3, 128.5, 128.4, 127.6, 126.0, 112.4, 110.2, 86.8, 84.5, 83.9, 61.5, 48.0, 27.8, 26.4; HRMS-ESI (m/z): (M+Na)+ calcd for C32H30N2O5, 545.2052; found, 545.2058.
3.1.15. (1′S,2′S,3′R)-5-(2′,3′-Dihydroxy-4′-hydroxymethyl-4′-cyclopenten-1′-yl)uracil (4)
A mixture of 19 (200 mg) and 10% methanolic HCl (10 mL) solution was stirred at rt for 2 h. Concentration under reduced pressure gave a white solid, which was co-evaporated with EtOH (3× 3 mL). The residue was dissolved in methanol (5 mL), neutralized with solid NaHCO3, and stirred for 2 h. The mixture was filtered and purified using amine functionalized silica gel column chromatography (CHCl3–MeOH = 8.5:1.5). The compound 4 (65 mg, 72%) was obtained as a white powder. Mp 192 °C; (c 0.15, MeOH); UV (H2O) λ max, nm: 263 (8400, pH 2), 262 (8800, pH 7), 285 (6500, pH 11); 1H NMR (DMSO-d 6, 500 MHz) δ 11.16 (s, 1H, NH), 10.75 (s, 1H, NH), 7.01 (s, 1H, H-6), 5.89 (d, 1H, J = 2 Hz, H-6′), 4.52 (br s, 3H, 3× OH) 4.26 (d, 1H, J = 5.0 Hz, H-3′), 4.10 (t, 2H, J = 13.5 Hz, H-5′a,b), 3.96 (t, 1H, J = 5.0 Hz, H-2′), 3.82 (d, 1H, J = 2.5 Hz, H-1′); 13C NMR (DMSO-d 6, 125 MHz) δ 164.8, 151.6, 147.7, 139.1, 127.4, 125.8, 76.1, 73.7, 58.9, 47.8; HRMS-ESI (m/z): (M+H)+ calcd for C10H12N2O5, 240.0746; found, 240.0743; Anal. calcd for C10H12N2O5: C, 50.00; H, 5.04; N, 11.66; found: C, 49.89; H, 5.03; N, 11.67.
3.2. Antiviral assay
HIV drug susceptibility assay16 and cytotoxicity assay17 (PBM, CEM and Vero cells) were performed as previously described.
Acknowledgments
This research was supported in part by U.S. Public Health Service Research Grants UO19-AI-056540, 5R37-AI-25899, 5R37-AI-41980, 5P-AI-50409 from the National Institutes of Health and the Department of Veterans Affairs. We thank Dr. Earl R. Kern of University of Alabama School of Medicine, Dr. Dale L. Bernard and Dr. Robert W. Sidwell of Institute for Antiviral Research, Utah State University, for performing certain antiviral assays.
References and notes
- 1.(a) Buchanan J.G. Fortscher. Chem. Org. Naturst. 1983;44:243. doi: 10.1007/978-3-7091-8714-2_4. [DOI] [PubMed] [Google Scholar]; (b) Wu Q., Simons C. Synthesis. 2004;10:1533. [Google Scholar]; (c) Chen X., Sauer D.R., Schneller S.W. Curr. Med. Chem. 1994;1:105. [Google Scholar]
- 2.(a) Chu M.Y., Zuckerman L.B., Sato S., Carbtree G.W., Bogden A.E., Lim M.-I., Klein R.S. Biochem. Pharmacol. 1984;33:1229. doi: 10.1016/0006-2952(84)90174-6. [DOI] [PubMed] [Google Scholar]; (b) Zimmerman T.P., Deeprose R.D., Wolberg G., Stopford C.R., Duncan G.S., Miller W.H., Miller R.L., Lim M.-I., Ren W.-Y., Klein R.S. Biochem. Pharmacol. 1983;32:1211. doi: 10.1016/0006-2952(83)90274-5. [DOI] [PubMed] [Google Scholar]
- 3.Burchenal J.H., Ciovacco K., Kalaher K., O’Toole T., Kiefner R., Dowing M.D., Chu C.K., Watanabe K.A., Wempen I., Fox J.J. Cancer Res. 1976;36:1520. [PubMed] [Google Scholar]
- 4.(a) Fuertes M., Garcia-Lopez T., Garcia-Munoz E., Stud M. J. Org. Chem. 1976;41:4074. [Google Scholar]; (b) Srivastava P.C., Pickering M.V., Allen L.B., Streeter D.G., Campelbell M.T., Witkowski J.T., Sidwell R.W., Robins R.K. J. Med. Chem. 1977;20:256. doi: 10.1021/jm00212a014. [DOI] [PubMed] [Google Scholar]
- 5.(a) Rodriguez J.B., Comin M.J. Mini-Rev. Med. Chem. 2003;3:95. doi: 10.2174/1389557033405331. [DOI] [PubMed] [Google Scholar]; (b) Schneller S.W. Curr. Top. Med. Chem. 2002;2:1087. doi: 10.2174/1568026023393228. [DOI] [PubMed] [Google Scholar]; (c) Crimmins M.T. Tetrahedron. 1998;54:9229. [Google Scholar]
- 6.(a) Yaginuma S., Muto N., Tsujino M., Sudate Y., Hayashi M., Otani M. J. Antibiot. 1981;34:359. doi: 10.7164/antibiotics.34.359. [DOI] [PubMed] [Google Scholar]; (b) Hayashi M., Yaginuma S., Muto N., Tsujino M. Nucleic Acids Symp. Ser. 1980;(No. 8):S65. [PubMed] [Google Scholar]
- 7.Kusaka T., Yamamoto H., Shibata M., Muroi M., Kishi T., Mizuno K. J. Antibiot. 1968;21:255. doi: 10.7164/antibiotics.21.255. [DOI] [PubMed] [Google Scholar]
- 8.Daluge S.M., Good S.S., Faletto M.B., Miller W.H., St. Clair M.H., Boone L.R., Tisdale M., Parry N.R., Reardon J.E., Dornsife R.E., Averett D.R., Krenitsky T.A. Antimicrob. Agents Chemother. 1977;41:1082. doi: 10.1128/aac.41.5.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Marquez V.V., Lim M.-I. Med. Res. Rev. 1986:6. doi: 10.1002/med.2610060102. [DOI] [PubMed] [Google Scholar]; (b) Bisacchi G.S., Chao S.T., Bachard C., Daris J.P., Innaimo S.F., Jacobs G.A., Kocy O., Lapointe P., Martel A., Merchant Z., Slusarchyk W.A., Sundeen J.E., Yung M.G., Colonno R.J., Zahler R. Bioorg. Med. Chem. Lett. 1997;7:127. [Google Scholar]; (c) Colonno, R. J.; Innaimo, S. F.; Seifer, M.; Genovesi, E.; Clark, J.; Yamanaka, R.; Hamatake, B.; Terry, B.; Strandring, D.; Bisacchi, G.; Sundeen, J.; Zahler, R. Abstracts of 10th Conference on Antiviral Research, Atlanta, 1997; Vol. 34, A51, p 32.
- 10.(a) Hayashi S., Norbeck D.W., Rosenbrook W., Fine R.L., Matsukara M., Plattner J.J., Broder S., Mitsuya H. Antimicrob. Agents Chemother. 1990;34:287. doi: 10.1128/aac.34.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maruyama T., Hanai Y., Sato Y., Snoeck R., Andrei G., Hosoya M., Balzarini J., De Clercq E. Chem. Pharm. Bull. 1993;41:516. doi: 10.1248/cpb.41.516. [DOI] [PubMed] [Google Scholar]; (c) Maruyama T., Sato Y., Horri T., Shiota H., Nitta K., Sirasaka T., Mitsuya H., Honjo M. Chem. Pharm. Bull. 1990;38:2719. doi: 10.1248/cpb.38.2719. [DOI] [PubMed] [Google Scholar]; (d) Yokota T., Konno K., Chonan E., Mochizuki S., Kojima K., Shigeta S., De Clercq E. Antimicrob. Agents Chemother. 1990;34:1326. doi: 10.1128/aac.34.7.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fissecks J.D., Market Creegan B. J. Org. Chem. 1967;32:3595. [Google Scholar]
- 12.(a) Tuncbilek M., Schneller S.W. Bioorg. Med. Chem. 2003;11:3331. doi: 10.1016/s0968-0896(03)00268-2. [DOI] [PubMed] [Google Scholar]; (b) Chun B.K., Song G.Y., Chu C.K. J. Org. Chem. 2001;66:4852. doi: 10.1021/jo010224f. [DOI] [PubMed] [Google Scholar]; (c) Katagiri N., Haneda T., Kaneko C. Chem. Pharm. Bull. 1986;34:4875. [Google Scholar]; (d) Katagiri N., Honeda T., Hayasaka E., Watanabe N., Kaneko C. J. Org. Chem. 1988;53:226. [Google Scholar]; (e) Katagiri N., Tomura M., Haneda T., Kaneko C. Heterocycles. 1990;30:211. [Google Scholar]; (f) Katagiri N., Tomura M., Haneda T., Kaneko C. J. Chem. Soc. Chem. Commun. 1987:1422. [Google Scholar]; (g) Cookson R.C., Dufield P.J., Scopes D.I.C. J. Chem, Soc. Perkin Trans. 1. 1986:393. [Google Scholar]; (h) Takahashi T., Kotsubo H., Koizumi T. Tetrahedron: Asymmetry. 1991;2:1035. [Google Scholar]; (i) Dishington A.P., Humber D.C., Stoodely R.J. J. Chem. Soc. Perkin Trans. 1. 1993:57. [Google Scholar]; (j) Hildbrand S., Leuman C., Scheffold R. Helv. Chim. Acta. 1996;79:702. [Google Scholar]
- 13.(a) Cho J.H., Bernard D.L., Sidwell R.W., Kern E.R., Chu C.K. J. Med. Chem. 2006;49:1140. doi: 10.1021/jm0509750. [DOI] [PubMed] [Google Scholar]; (b) Balakumar A., Kim H.J., Prichard M.N., Kern E.R., Chu C.K. Bioorg. Med. Chem. Lett. 2006;16:285. doi: 10.1016/j.bmcl.2005.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tokuda M., Fujita H., Nitta M., Suginome H. Heterocycles. 1996;42:385. [Google Scholar]
- 15.Sato T., Noyori R. Bull. Chem. Soc. Jpn. 1983;56:2700. [Google Scholar]
- 16.Schinazi R.F., Sommadossi J.P., Saalmann V., Cannon D.L., Xie M.W., Hart G.C., Smith G.A., Hahn E.F. Antimicrob. Agents Chemother. 1990;34:1061. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuyver L.J., Lostia S., Adams M., Mathew J., Pai B.S., Grier J., Tharnish P., Choi Y., Chong Y., Choo H., Chu C.K., Otto M.J., Schinazi R.F. Antimicrob. Agents Chemother. 2002;46:3854. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]