

Graphical abstract
Keywords: Nucleoside derivatives, Sulfinyl epimers, Ascidian, Herdmania momus
Abstract
Investigation of the secondary metabolites of the ascidian Herdmania momus led to the isolation and characterization of four new nucleoside derivatives (1–4). Structural studies showed that these derivatives represent a series of rare methylsulfinyladenosine derivatives of interconvertible transesterification isomers and/or sulfinyl epimers. The antiviral activities of these rare nucleosides were evaluated against a series of human pathogenic viruses.
Marine ascidians are a rich source of chemically diverse secondary metabolites, with remarkable biological activities.1, 2 such as amino acid derivatives and complex alkaloids.3, 4, 5, 6, 7, 8, 9 In our previous study on bioactive compounds from the solitary tunicate Herdmania momus, a series of amino acid derivatives (herdmanines A–M) were isolated.3, 4 As a part of continuing study on the metabolites of the same tunicate, four new complex nucleosides (1–4) containing the rare methylsulfinyladenosine were isolated. Details of the isolation, structure elucidation, and of the results of the antiviral screening of these metabolites are described herein.
The combined MeOH and CH2Cl2 extracts were sequentially partitioned between CH2Cl2 and H2O, and n-BuOH and H2O. The n-BuOH-soluble fraction was subjected to chromatographic separation and purification to yield a series of nucleoside derived metabolites (1–4).10 Compounds 1 and 2 were found to be interconvertible, and on standing at room temperature after HPLC separation, each compound afforded the same equilibrium ratio of compound 1–2 (56:44). Compounds 3 and 4 were also interconvertible, and on standing at room temperature produced an identical equilibrium mixture (56:44) as that of 1 and 2. Spectral analysis of these interconvertible compounds was performed on equilibrium mixtures 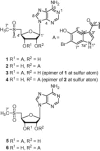 .
.
The equilibrium mixture of compounds 1 and 2 was a white powder. The (+)-FABMS spectrum of this mixture showed isotopic peaks of [M+2H]+ and [M+3H]+ at m/z 554/552 and 555/553, respectively, with an isotopic peak ratio of 1:1. This pseudomolecular ionic configuration is shared with various other nucleosides and amines.11 (+)-HRFABMS analysis of the mix of compounds 1 and 2 yielded the molecular formula C20H19 79BrN6O6S, with 14 degrees of unsaturation. The 1H and 13C NMR spectra of this equilibrium mixture revealed isomeric pairs of signals corresponding to two different molecular structures. Each set of signals can be delineated based on COSY, HMQC, and HMBC data. 1H and 13C NMR spectra indicated the presence of a nucleoside moiety. More specifically, characteristic chemical shifts of C-2 (δ C 145.8), C-4 (δ C 149.4), C-5 (δ C 125.5), C-6 (δ C 157.7), and C-8 (δ C 140.2) for one set of 13C NMR signals (compound 1) suggested that the purine base was adenine, and this was further confirmed by HMBC correlations between H-2 and C-4 and C-6, and between H-8 and C-4 and C-5 (Fig. 1 ). Successive COSY correlations between H-1′and H-5′confirmed that the four oxygenated methines and the oxygenated methylene of compound 1 were linked to form a furanose moiety. The relative stereochemistry of the pentofuranose was deduced from 1H–1 H NOESY correlations and 1H–1 H coupling constants.12, 13, 14, 15 Furthermore, a NOESY crosspeak between H-1′ and H-4′ indicated that both protons were on the same face of the ring. The sugar moiety was deduced to be β-d-ribose, as proven by the coupling constants (H-1′, H-2′, and H-3′) and the comparable specific rotation value of the compound 1/2 mixture , which matches those reported for modified ribose units.15 The unusual 1H and 13C NMR chemical shifts of CH3-7 and CH2-5 were indicative of a sulfoxide group in compound 1, and the HMBC correlation between the methyl protons (H-7) and C-5′ indicated a methylsulfinylfuranose moiety.15, 16 The chemical shifts for the anomeric proton and carbon signals (δ H 6.16; δ C 90.2) were consistent with the presence of a C–N glycosidic linkage. HMBC correlations between H-1 and C-4 and C-8 confirmed connection between the furanose unit and the adenine moiety at the expected N-9 position. Further 1H NMR analyses and correlations observed in the HMBC spectrum of the mixture indicated the presence of the 6-bromo-5-hydroxy-3-carboxylindolyl moiety, which was previously recognized as a part of the structure of herdmanine E from the same tunicate.4 The correlation between H-3′ and C-8″ suggested that these two moieties were joined by an ester linkage (Fig. 1). Based on the above, compound 1 was characterized as (2S,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-hydroxy-2-[(methylsulfinyl)methyl]tetrahydrofuran-3-yl-(6-bromo-5-hydroxy-1H-indole)-3-carboxylate and given the trivial name momusine A.
Figure 1.
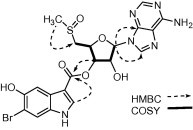
Selected HMBC and COSY correlations for compound 1.
The 1H and 13C NMR spectra of another component (compound 2) were differed from those of 1 only in splitting patterns and chemical shifts of some of the ribose ring proton signals. The signal for H-3′ (δ H 4.84) was shifted upfield, whilst that of H-2′ (δ H 5.94) was shifted downfield as compared to those of compound 1 (H-3′, δ H 5.63; H-2′, δ H 5.21), indicating an acyl substitution at C-2′, and thereby, deshielding induced by the carbonyl group. This was further confirmed by the HMBC correlation between H-2′ and C-8″ (δ C 164.5) in compound 2.
Compounds 1 and 2 were interconvertible at room temperature affording an equilibrium mixture of 56:44 ratio. Likewise, a previous study reported that 3′-O-acetyl-ADP-ribose is an intramolecular transesterification product of 2′-O-acetyl-ADP-ribose produced by the Sir2 family of histone/protein deacetylases and the mixture of 3′- and 2′-aceatate exist in equilibrium (55:45).17 Other examples of non-enzymatic acyl migration under mild conditions have also been reported.18, 19, 20, 21 Furthermore, several research groups have studied the stability and/or acyl migration of many O-acyl glucuronides, and in all cases, acyl migration occurred when the hydroxyl groups of the glucuronic acid were essentially on the same face of the ring.
The equilibrium mixture of compounds 3 and 4 was also a white powder. The molecular formulas of compounds 3 and 4 were determined to be C20H19BrN6O6S based on NMR and MS data. The 1H and 13C NMR signals of each set of components were delineated based on COSY, HMQC, and HMBC. One set (compound 3) of 1H NMR signals was almost identical to those of compound 1, with the exception of a slight chemical shift difference at H-5′ (Table 1 ). In addition, the carbon signals of C-5′ (δ C 55.0) and C-7′ (δ C 37.3) of compound 3 were shifted slightly upfield versus compound 1 (C-5′, δ C 57.5; C-7′, δ C 38.0) (Table 2 ). According to the above evidence, compounds 1 and 3 were deduced to be epimers as has been previously described for 5-deoxy-5-methylsulphinyladenosine16 and the sulfoxide derivatives,22 which only differ in their configuration with respect to the position of the sulfoxide group.
Table 1.
1H (500 MHz, CD3OD) NMR data for compounds 1–4a
| Position |
1 |
2 |
3 |
4 |
|---|---|---|---|---|
| δH (J in Hz) | ||||
| 2 | 8.11 (s) | 8.08 (s) | 8.11 (s) | 8.08 (s) |
| 8 | 8.29 (s) | 8.26 (s) | 8.25 (s) | 8.23 (s) |
| 1′ | 6.16 (d, 5.5) | 6.34 (d, 4.0) | 6.16 (d, 6.0) | 6.32 (d, 4.0) |
| 2′ | 5.21 (t, 5.5) | 5.94 (dd, 5.5, 4.0) | 5.20 (t, 5.5) | 5.92 (t, 5.5) |
| 3′ | 5.63 (dd, 5.5, 4.5) | 4.84 (m) | 5.67 (dd, 5.5, 4.0) | 4.82 (m) |
| 4′ | 4.75 (m) | 4.55 (ddd, 9.5, 6.5, 2.5) | 4.78 (m) | 4.63 (ddd, 10.5, 7.5, 4.0) |
| 5a′ | 3.58 (dd, 13.0, 10.5) | 3.46 (dd, 13.0, 10.5) | 3.53 (dd, 14.0, 7.0) | 3.46 (dd, 13.5, 9.0) |
| 5b′ | 3.32 (m) | 3.32 (m) | 3.48 (dd, 13.5, 5.0) | 3.34 (m) |
| 7′ | 2.72 (s) | 2.73 (s) | 2.70 (s) | 2.71 (s) |
| 2″ | 8.08 (s) | 8.04 (s) | 8.08 (s) | 8.04 (s) |
| 4″ | 7.65 (s) | 7.59 (s) | 7.66 (s) | 7.59 (s) |
| 7″ | 7.59 (s) | 7.57 (s) | 7.58 (s) | 7.56 (s) |
Chemical shifts were assigned using COSY and HMBC spectral data.
Table 2.
13C (100 MHz, CD3OD) NMR data for compounds 1–4a
| Position |
1 |
2 |
3 |
4 |
|---|---|---|---|---|
| δC | ||||
| 2 | 145.8 | 145.8 | 145.8 | 145.8 |
| 4 | 149.4 | 149.4 | 149.3 | 149.3 |
| 5b | 125.5 | 125.5 | 125.5 | 125.5 |
| 6 | 157.7 | 157.7 | 157.7 | 157.7 |
| 8 | 140.2 | 140.3 | 140.2 | 140.2 |
| 1′ | 90.2 | 88.6 | 90.2 | 88.6 |
| 2′ | 72.6 | 74.9 | 72.4 | 74.5 |
| 3′ | 74.6 | 72.9 | 74.7 | 72.6 |
| 4′ | 76.9 | 78.3 | 77.0 | 78.2 |
| 5′ | 57.5 | 57.4 | 55.0 | 54.5 |
| 7′ | 38.0 | 38.1 | 37.3 | 37.3 |
| 2″ | 133.8 | 133.8 | 133.8 | 133.8 |
| 3″ | 106.8 | 106.8 | 106.8 | 106.8 |
| 3a″ | 126.7 | 126.7 | 126.7 | 126.7 |
| 4″ | 106.0 | 106.0 | 106.0 | 106.0 |
| 5″ | 149.4 | 149.4 | 149.3 | 149.3 |
| 6″ | 105.0 | 105.0 | 105.4 | 105.0 |
| 7″ | 115.8 | 115.8 | 115.8 | 115.8 |
| 7a″ | 131.9 | 131.9 | 131.8 | 131.8 |
| 8″ | 164.5 | 164.5 | 164.5 | 164.5 |
Chemical shifts were assigned based on gHSQC and gHMBC spectral data.
No signal was observed in the 13C spectrum.
Comparisons of the 1H and 13C NMR data of 4 with those of 1-3 suggested that 4 was a 2′–3′ transesterification derivative of compound 3, and a sulfoxide epimer of compound 2. Consequently, compounds 1, 3 and 2, 4 were deduced to be two pairs of epimers (Fig. 2 A), and this was confirmed by oxidizing the sulfinyl group in mixtures with H2O2 and subsequent HPLC analysis.23 The mixtures of 1/2 (Fig. 2B) and of 3/4 (Fig. 2C) were converted to sulfonyladenosine derivatives, and each mixture yielded identical two-component (5 and 6) mixtures (Fig. 2D and E, respectively). This finding indicated that compounds 1, 3 and 2, 4 are epimeric at the sulfoxide group. 5′-Deoxy-5′-methylsulfinyladenosine epimers have been isolated from Ganoderma lucidum,16 and reported in onion extracts.24, 25 Distinct biological activities have also been described for epimers differentiated by sulfoxide-asymmetry.24, 25 Usually the sulfoxide configuration of natural products is (S).26 In the present study, we tried to determine the absolute stereochemistry of the sulfoxide moiety in compounds 1–4 by using MsrA (methionine sulfoxide reductase A, Protein Data Bank code: 1NWA), which selectively reduces S-configured sulfoxides.27 However, neither mixture 1/2 nor mixture 3/4 was reduced by MrsA,28 suggesting that these new nucleosides are not good substrates for MsrA and cannot bind properly to the active enzyme site.
Figure 2.
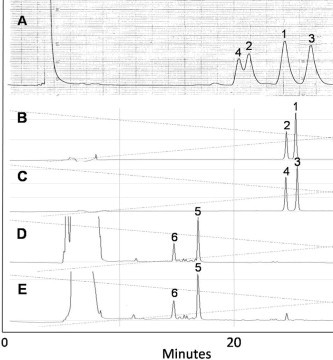
RP HPLC analysis of compounds 1–6. (A) Chromatogram of initial separation of compounds 1–4 (tR 86.5, 75.6, 93.4, and 71.2 min, respectively; YMC-packed C-8 column, 35% MeOH, flow rate 1.2 mL/min, UV, 220 nm); (B) chromatogram of 1/2 (tR 25.39 min/24.57 min); (C) chromatogram of 3/4 (tR 25.54 min/24.54 min); (D) chromatogram of the oxidation products of 1/2 (5/6, tR 16.86/14.76 min); (E) chromatogram of the oxidation products of 3/4 (5/6, tR 16.86/14.76 min). RP HPLC condition for B–E was as follows; YMC-packed C-18 column, linear gradient elution system from 5% MeOH (0.1% TFA) to 90% MeOH (0.1% TFA) in 60 min, flow rate 0.5 mL/min, UV, 220 nm.
Rare sponge-derived nucleosides, such as, spongothymidine and spongouridine served as antiviral leads and culminated in antiviral drugs like acyclovir and zanamivir. In some antiviral 5′-deoxyribonucleosides, substitution of a methylsulfonyl group at the 5′-position increased antiviral activity while securing low cytotoxicity.29 Moreover, 5-hydroxy-6-bromoindole-3-carboxylic acid derivatives display antiviral activities against a number of viruses including influenza A/B/C viruses, avian coronavirus, infectious bronchitis virus, Marek’s disease virus, and hepatitis B/C viruses.30, 31, 32, 33 Therefore, in an expectation of antiviral activity of compounds 1–4,34 we evaluated them for antiviral activity against human pathogenic viruses. However, none of the isomers showed antiviral activity against human rhinoviruses (HRV14, HRV17, or HEV71, EC50 >100 μg/mL, CC50 >100 μg/mL), coxsackieviruses (CoxB1 or CoxB3, EC50 >100 μg/mL, CC50 >100 μg/mL), or poliovirus (PV3, EC50 >100 μg/mL, CC50 >100 μg/mL). Considering the chemical structure of those nucleoside derivatives, compounds 1–4 may exhibit antibacterial, analgesic, sedative, and cardiac depressant activities,15 however, further biological evaluations could not be performed due to paucity of materials.
Acknowledgment
This study was supported by the National Research Foundation of Korea (Grant No. 20090083538).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2013.05.097.
Supplementary data
Supplementary data.
References and notes
- 1.Verbist J. F. J. Pharm. Belg. 1995;50:98. [PubMed] [Google Scholar]
- 2.Watters D.J., van den Brenk A.L. Toxicon. 1993;31:1349. doi: 10.1016/0041-0101(93)90202-t. [DOI] [PubMed] [Google Scholar]
- 3.Li J.L., Han S.C., Yoo E.S., Shin S., Hong J., Cui Z., Li H., Jung J.H. J. Nat. Prod. 2011;74:1792. doi: 10.1021/np200397g. [DOI] [PubMed] [Google Scholar]
- 4.Li J.L., Xiao B., Park M., Yoo E.S., Shin S., Hong J., Chung H.Y., Kim H.S., Jung J.H. J. Nat. Prod. 2012;75:2082. doi: 10.1021/np300401g. [DOI] [PubMed] [Google Scholar]
- 5.Ochi M., Kataoka K., Ariki S., Iwatsuki C., Kodama M., Fukuyama Y. J. Nat. Prod. 1998;61:1043. doi: 10.1021/np980097r. [DOI] [PubMed] [Google Scholar]
- 6.Santalova E.A., Denisenko V.A., Berdyshev D.V., Aminin D.L., Sanamyan K.E. Nat. Prod. Commun. 2008;3:1617. [Google Scholar]
- 7.García A., Vázquez M.J., Quiñoá E., Riguera R., Debitus C. J. Nat. Prod. 1996;59:782. [Google Scholar]
- 8.Carroll A.R., Avery V.M. J. Nat. Prod. 2009;72:696. doi: 10.1021/np800831z. [DOI] [PubMed] [Google Scholar]
- 9.Davidson B.S. Chem. Rev. 1993;93:1771. [Google Scholar]
- 10.Extraction and isolation: Frozen ascidians (0.4 kg) were chopped into small pieces and extracted with MeOH and CH2Cl2 at room temperature. The combined extract was partitioned between CH2Cl2 and H2O. The H2O fraction was further extracted with n-BuOH, and the BuOH fraction (6.0 g) was subjected to reversed-phase MPLC column chromatography (YMC gel ODS-A, 60 Å, 230 mesh) using a step gradient solvent system from 30% to 100% MeOH in H2O to afford 21 fractions. Fraction 8 (136.9 mg) was first purified on a Shodex-packed NH-5E column using 85% MeOH as eluent and further purified on a YMC-packed C-8 column (250 × 10 mm, 5 μm, 12 nm) using 35% MeOH as eluent to afford four components (1–4). However, 1 and 2 were interconvertible and 1.6 mg of an equilibrium mixture was obtained. Likewise, 3 and 4 were interconvertible and 2.0 mg of an equilibrium mixture was obtained.Momusine A/B (1/2): White powder; (c 0.025, MeOH); UV (MeOH) λmax (log ε) 261 (1.62), 290 (0.87) nm; CD (c 1.4 × 10−4 M, MeOH) Δ∊ (nm) 0.31 (200); −0.28 (220); −0.04 (246); −0.10 (257); 1H and 13C NMR (see Table 1, Table 2); (+)-ESIMS m/z 575/573 [M+Na]+; (+)-FABMS m/z 554/552 [M+2H]+; (+)-FABMS m/z 555/553 [M+3H]+; (+)-HRFABMS m/z 552.0418 (calcd for C20H2179BrN6O6S, 552.0427).epi-Momusine A/B (3/4): White powder; (c 0.05, MeOH); UV (MeOH) λmax (log ε) 263 (2.34), 291 (1.17) nm; CD (c 3.1 × 10−4 M) Δ∊ (nm) 0.56 (200); −0.77 (220); −0.14 (247); −0.20 (256); 1H and 13C NMR (see Table 1, Table 2); (+)-ESIMS m/z 575/573 [M+Na]+; (−)-ESIMS m/z 551/549 [M−H]−; (+)-FABMS m/z 554/552 [M+2H]+; (+)-FABMS m/z 555/553 [M+3H]+; (+)-HRFABMS m/z 552.0413 (calcd for C20H2179BrN6O6S, 552.0427).
- 11.Cerny R.L., Gross M.L. Anal. Chem. 1985;57:1160. [Google Scholar]
- 12.Ostroswki H., Maurizot J., Adeline M., Fourrey J., Clivio P. J. Org. Chem. 2003:6502. doi: 10.1021/jo030086p. [DOI] [PubMed] [Google Scholar]
- 13.Ohigashi H., Kaji M., Sakaki M., Koshimuzi K. Phytochemistry. 1989;28:1365. [Google Scholar]
- 14.Jiao P., Mudur S.V., Gloer J.B., Wicklow D.T. J. Nat. Prod. 2007;70:1308. doi: 10.1021/np070241l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kehraus S., Gorzalka S., Hallmen C., Iqbal J., Müller C.E., Wright A.D., Wiese M., König G.M. J. Med. Chem. 2004;47:2243. doi: 10.1021/jm031092g. [DOI] [PubMed] [Google Scholar]
- 16.Kawagishi H., Fukuhara F., Sazuka M., Kawashima A., Mrrsubori T., Tomita T. Phytochemistry. 1993;32:239. [Google Scholar]
- 17.Jackson M.D., Denu J.M. J. Biol. Chem. 2002;277:18535. doi: 10.1074/jbc.M200671200. [DOI] [PubMed] [Google Scholar]
- 18.Corcoran O., Mortensen R.W., Hansen S.H., Troke J., Nicholson J.K. Chem. Res. Toxicol. 2001;14:1363. doi: 10.1021/tx010015q. [DOI] [PubMed] [Google Scholar]
- 19.Hasegawa H., Akira K., Shinohara Y., Kasuya Y., Hashimoto T. Biol. Pharm. Bull. 2001;24:852. doi: 10.1248/bpb.24.852. [DOI] [PubMed] [Google Scholar]
- 20.Mortensen R.W., Corcoran O., Cornett C., Sidelmann U.G., Lindon J.C., Nicholson J.K., Hansen S.H. Drug Metab. Dispos. 2001;29:375. [PubMed] [Google Scholar]
- 21.Jemnitz K., Levai F., Monostory K., Szatmari I., Vereczkey L. Eur. J. Drug Metab. Pharmacokinet. 2000;25:153. doi: 10.1007/BF03192307. [DOI] [PubMed] [Google Scholar]
- 22.Morimitsu Y., Kawakishi S. Phytochemistry. 1990;29:3435. [Google Scholar]
- 23.Oxidation of compounds 1–4 and RP HPLC analysis: Hydrogen peroxide (10 μL, 30% w/w solution in H2O) was added to a compound 1/2 mix (0.2 mg) or a compound 3/4 mix (0.2 mg) in buffer (300 μL, 50 nM sodium phosphate, pH 7.4, DMSO 10 μL). TLC analysis showed that the starting materials were completely converted to two major products after 48 h at room temperature. The reaction mixture was then treated with MeOH (50 μL) and centrifuged at 1000 rpm for 2 min. MeOH soluble portions were subjected to RP HPLC (YMC-packed C-18 column, 250 × 4.6 mm, S-5 μm, 12 nm) using a linear gradient from 5% MeOH (0.1% TFA) to 90% MeOH (0.1% TFA) over 60 min (flow rate 0.5 mL/min; with UV detection at 220 nm).
- 24.Morimitsu Y., Kawakishi S. Agric. Biol. Chem. 1991;55:889. [Google Scholar]
- 25.Bayer T., Breu W., Seligmann O., Wray V., Wagner H. Phytochemistry. 1989;28:2373. [Google Scholar]
- 26.Block E. The Lore of the Science. Eng., Royal Society of Chemistry; Cambridge: 2009. Garlic and other alliums; p. 100. [Google Scholar]
- 27.Staford S.A., Fascione M.A., Sasindran S.J., Chatterjee D., Dhandayuthapani S., Turnbull W.B. Chem. Commun. 2009:110. doi: 10.1039/b817483k. [DOI] [PubMed] [Google Scholar]
- 28.MsrA reduction experiments: The buffer used for all NMR experiments was 50 mM sodium phosphate in D2O (pD 7.4). In each case, reactions with MsrA and compounds were performed in an NMR tube for 2 days at 37 °C before 1H NMR spectroscopy. Reaction mixes were as follows: Compounds 1/2 (mixture) (0.48 mg, 0.87 μmol), DTT (1.0 mg, 4.7 μmol), MsrA (30 μg, in a solution of 50 nM Tris containing 150 nM NaCl), and NMR buffer (400 μL); and Compounds 3/4 (mixture) (0.50 mg, 0.91 μmol), DTT (0.9 mg, 4.2 μmol), MsrA (30 μg, in a solution of 50 nM Tris containing 150 nM NaCl) and NMR buffer (400 μL).
- 29.Diwan A.R., Robins R.K., Prusoff W.H. Experientia. 1969;25:98. doi: 10.1007/BF01903922. [DOI] [PubMed] [Google Scholar]
- 30.Leneva I.A., Russell R.J., Boriskin Y.S., Hay A.J. Antiviral Res. 2009;81:132. doi: 10.1016/j.antiviral.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Brooks M.J., Sasadeusz J.J., Tannock G.A. Curr. Opin. Pulm. Med. 2004;10:197. doi: 10.1097/00063198-200405000-00009. [DOI] [PubMed] [Google Scholar]
- 32.Chai H., Zhao Y., Zhao C., Gong P. Bioorg. Med. Chem. 2006;14:911. doi: 10.1016/j.bmc.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 33.Boriskin Y.S., Pécheur E.I., Polyak S.J. Virol. J. 2006;3:56. doi: 10.1186/1743-422X-3-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antiviral activity: Antiviral assays were performed at the Korea Research Institute of Chemical Technology using the virus-induced CPE (cytopathic effect) inhibition assay. 50 μL aliquots of prepared samples (20 mg/mL in DMSO) diluted with MEM/2% FBS were applied to confluent HeLa cells grown in 96 well microplates in duplicate. Virus solution (50 μl) at an MOI (multiplicity of infection) of 200 CCID50 (50% cell culture inhibitory dose) was added to each well, and cells were incubated in CO2 incubator. Antiviral activities in virus-infected cell cultures and the cytotoxic effects of drugs on mock-infected cells were determined using an MTT assay and by microscopic observation. 50% cytotoxic concentration (CC50) was defined as the concentration of a compound that reduced the absorbance of mock-infected control by 50%. The concentrations of the compounds that protected 50% of cells against the cytotoxic effects of the virus strains were defined as the 50% effective concentration values (EC50).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data.