

Abstract
During primary contact with susceptible hosts, microorganisms face an array of barriers that thwart their invasion process. Passage through the basement membrane (BM), a 50–100-nm-thick crucial barrier underlying epithelia and endothelia, is a prerequisite for successful host invasion. Such passage allows pathogens to reach nerve endings or blood vessels in the stroma and to facilitate spread to internal organs. During evolution, several pathogens have developed different mechanisms to cross this dense matrix of sheet-like proteins. To breach the BM, some microorganisms have developed independent mechanisms, others hijack host cells that are able to transverse the BM (e.g. leukocytes and dendritic cells) and oncogenic microorganisms might even trigger metastatic processes in epithelial cells to penetrate the underlying BM.
A crucial barrier to breach
A first step during infection is the attachment of microorganisms to various host proteins, which could include basement membrane (BM) proteins. Generally, adhesion of bacteria and fungi occurs through binding of adhesive molecules, called adhesins, to host proteins. Most adhesins are extensively described and some of the motifs participating in these processes have been identified. In comparison, knowledge about adhesion of viruses to BM components is scarce. On adhesion, microorganisms have evolved cunning techniques to overcome the BM barrier, which otherwise hampers their invasive process. Breakdown of the BM, via activation of microbial and/or host proteases, to cross the BM has been shown for many bacteria, fungi and, recently, some viruses. Pathogens might also gain access to the connective tissue by hijacking host cells, particularly local immune cells, to cross the BM. This review provides an overview of recent insights into different pathogen–BM interactions during host invasion, discusses these findings and projects how they could contribute to the design of novel strategies to interfere with microbial invasions and pathology.
The BM: a specialized extracellular matrix possessing unique properties
The BM provides a subtle interface between epithelial or endothelial cells and mesenchymal cells present in connective tissue. Besides providing tissue structure, the BM plays an important role in structural integrity, cell behavior and signaling [1]. In proximity to epithelial cells, BM components such as collagen XVII and laminin 5, 6 and 10 are linked to extracellular parts of different molecules, mainly belonging to the integrin family (e.g. α6β4, α5β1 and α9β1) and fibronectin network, present on basolateral cell surfaces. At the cellular–matrix interface, located underneath the cell-neighboring elements collagen IV, VII and XVIII, laminin 1, entactin/nidogen, BM-40/osteonectin, fibulins and proteoglycans such as bamacan, agrin, perlecan are abundant. Towards the lamina propria, collagen I, III, V and VI appear next to proteoglycans. Overall, the BM represents a firm network of the main components, collagen and laminin, bridged by various other components (Figure 1 ). Importantly, the average pore size in the collagen–laminin network is approximately 50 nm, which allows only very small particles to diffuse across this thin but firm barrier 2, 3. This implies that microorganisms have devised ways to cross the BM.
Figure 1.
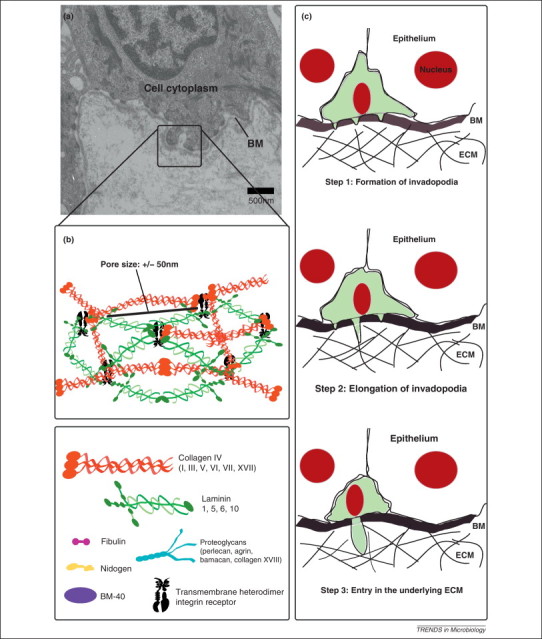
Immune cell trafficking through the basement membrane (BM) network. (a) Transmission electron micrograph of the BM zone. (b) Schematic illustration of a BM network and its main components. Laminin polymerization is believed to initiate the BM scaffold organization. Deposition of this polymer leads to association with a type IV collagen network. The other components of the BM interact with the laminin polymer and the type IV collagen network to organize a functional BM on the basolateral aspect of cells. (c) Despite the small pore size of the BM, immune cells scanning for signs of infection routinely traverse it. The BM transmigration program is a conserved mechanism. First, immune cells adhere to the matrix in an integrin-mediated manner. Subsequently, proteases degrade the BM before actin polymerization extends cell protrusions through the hole. Finally, the cell body moves behind the actin-rich protrusion.
Adhesion of pathogens to BM components
A crucial initial event in the pathogenesis of many microorganisms is adhesion to host tissues. Major players during these early steps in infection are microbial adhesive cell-surface molecules, termed adhesins [4]. Microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) are adhesins that attach to extracellular matrix (ECM) including the BM. Many MSCRAMMs are capable of binding more than one ECM component, and a single strain often possesses several different proteins that bind the same host component. Although current data about viral ECM adhesion and adhesins are still scarce, their importance in virulence has been shown for most bacteria and many fungi. For example, in many animal models of staphylococcal infections, CNA, a collagen-binding adhesin of pathogenic Staphylococcus aureus, increases disease severity. It has been suggested that the ability to interact with collagen grants these bacteria a clear advantage in pathogenesis [5]. For bacteria, adhesins are roughly subdivided according to the appendage morphology: there are fimbrial or non-flagellar adhesins (chaperone-usher pili, curli, type IV pili, type III secretion needle and type IV secretion pili) and non-fimbrial adhesins (autotransporters; outer membrane, secreted and biofilm-associated adhesins). Pili are further classified according to physical properties, antigenic determinants, adhesion characteristics, and characteristics of the major protein subunits or assembly pathways 6, 7. Although adhesins form a heterogeneous group with diverse architecture, domain content and binding mechanism, some do possess similarity in structure, ECM-binding domain organization and function 8, 9. ECM-binding MSCRAMMs of many pathogenic Gram-positive species are cell-wall-anchored surface proteins (CWPs). Common features of CWPs are an N-terminal signal peptide followed by a so-called A region or domain, segments of repeated domains and a characteristic C-terminal sorting signal. The sorting signal contains an important cell-wall-anchoring site or LPXTG motif, which covalently binds to the cell wall 10, 11. However, it has recently been shown that the collagen-binding adhesin Slr of Gram-positive Streptococcus pyogenes lacks the LPXTG motif, but uses a cell-wall-anchoring TLIA lipobox instead [12]. The structure and organization of viral and fungal adhesins to ECM are less well documented at present, although there is proof of diversity in fungal adhesins 8, 13. Some fungal adhesins show structural and/or functional similarities to bacterial adhesins 8, 13. Agglutinin-like sequence (Als) adhesins of Candida albicans are composed of a signal peptide, an N-terminal region, a nonrepeat Thr-rich (TR) region, a central region with a variable number of repeats, and a Ser/Thr/Asn-rich C-terminal domain that anchors the CWP via a glycophosphatidyl inositol (GPI) remnant 13, 14. In many cases, ECM-binding domains recognize carbohydrate residues or oligosaccharides, but not exclusively, because many of them also bind host protein-binding sites 10, 15, 16. Besides this direct microbial adhesin-ligand binding, microorganisms have developed other interesting indirect adhesive approaches. Haemophilus influenzae, Moraxella catarrhalis and Shigella spp. prevent cell detachment of infected cells from the BM through stabilization of focal adhesions. This strategy counteracts rapid exfoliation, which is an effective intrinsic defensive system of intestinal epithelium [17]. C. albicans promotes laminin 5 and type IV collagen protein secretion to enhance binding to the BM [18]. In the following paragraphs, adhesion of bacteria, fungi and viruses to the major BM components collagen, laminin, proteoglycans, entactin/nidogen, BM-40 and fibulin is discussed in more detail. Bacteria, fungi and viruses also show binding capacity to fibronectin, a minor component of the BM zone, that is extensively described elsewhere [19].
Collagen, the superhelix
Three collagen polypeptide chains, called α chains, are rich in proline and glycine and together constitute a long, stiff, rope-like superhelix known as the typical collagen molecule 1, 2, 3, 20. Numerous bacteria are able to interact with collagens via their proper adhesins 20, 21, 22. For many bacterial species, the ability to interact with collagen in the BM is a prerequisite for invasiveness. For example, PilA from Streptococcus agalactiae, causing meningitis in newborns, binds collagen I, which promotes interaction with integrins and subsequent penetration of the blood–brain barrier (BBB) [23].
Some fungal pathogens also interact with collagens during host invasion using distinct fungal receptors. Aspergillus fumigatus contains a sialic acid-specific lectin that interacts with collagen types I and IV [24]. Similarly, the Als3p glycoprotein, a major player in C. albicans pathogenesis, is responsible for binding collagen IV [25]. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and glycoprotein gp43 of Paracoccidioides brasiliensis bind type I collagen. Gp43 also binds type IV collagens and laminin, probably via a sialic acid recognition system, similar to A. fumigatus [24]. However, for most fungi, adhesins and adhesive mechanisms are still largely unknown 13, 14.
So far, viral binding to collagen has not been described.
The multidomain glycoprotein laminin
Three laminin chains (α,β,γ) form an asymmetric cross-like subunit via disulfide bonds. This non-collagenous protein contains multiple binding domains for interactions with ECM components (domains III, IV, V and VI) and with cellular receptors (G domain) 1, 2, 3. As described for collagen, many different bacteria and fungi are able to interact with laminins via adhesive proteins 6, 26, 27, 28, 29. Most adhesins recognize multiple molecules. However, there are adhesins that bind laminin but not collagen. Lsa24 and Lsa27 of Leptospira interrogans [26], Lmb of Streptococcus agalactiae [27] and others act as specific laminin-binding adhesins. Recently, ErpX of Borellia burgdorferi was found to have a unique mode of laminin binding through a hydrophobic segment at the center of the bacterial protein. This protein motif has not been identified yet in other bacterial laminin adhesins [29].
For human papillomaviruses (HPV), recent work has proposed laminin 5 as a possible basal ECM receptor. This interaction localizes virus particles to the basal surface of epithelial cells where they can reach their entry receptor, integrin α6β4, the physiological binding partner of laminin 5 [30]. However, another recent publication demonstrated that different HPV types show different binding characteristics (Figure 2 ) [31]. The nonstructural protein NSP4 of rotavirus plays a key role in the development of severe gastroenteritis by binding ECM proteins laminin β3 and fibronectin. Moreover, rotavirus induces phosphatidylinositol 3-kinase (PI3K) activation in intestinal cells, causing upregulation of integrin expression, prolonged attachment of infected cells to collagen and increased virus production 32, 33.
Figure 2.
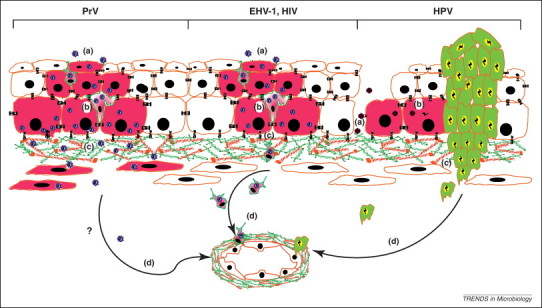
Different viral interactions with the BM. (a) Before entry into cells, viruses either attach directly to cell surface receptors (e.g. herpesviruses) or via intermediate binding to an exposed BM component in epithelial microlesions (e.g. human papillomavirus, HPV). (b) Viral replication and local dissemination (infected cells are in pink). Local immune cells may be infected (e.g. herpesviruses and HIV). (c) Viruses gain access to the stroma by breaching the BM. This may happen in a protease-mediated way (e.g. pseudorabies virus, PrV), via hijacking of immune cells to transverse the BM [e.g. equine herpesvirus 1 (EHV-1) and HIV] or via viral-driven metastasis out of a viral-induced tumor (e.g. HPV, green cells). (d) Finally, viruses may spread in the host by reaching blood or lymph vessels.
The heterogeneous molecule: proteoglycan
To be typed as a proteoglycan, at least one of the sugar side chains of a molecule has to be a glycosaminoglycan (GAG). All GAGs are covalently bound via a tetrasaccharide link to a serine amino acid in the core protein, the central polypeptide chain of proteoglycans 1, 2, 3. The ability to interact with proteoglycans, often heparan sulfate proteoglycans, is widespread in viruses and bacteria 34, 35. A clear distinction has to be made in this context between cell-surface proteoglycans, ECM-associated proteoglycans in general and specific proteoglycans residing inside the BM. Despite the numerous reports on pathogens interacting with ECM-associated proteoglycans in general 34, 35, no specific pathogen BM agrin or pathogen BM perlecan interactions have been described so far.
Currently, only one specific BM proteoglycan–pathogen interaction is known. The proteoglycan bamacan is a cellular ligand of vaccinia virus neurovirulence factor N1L. This interaction promotes viral growth and might contribute to virulence of the virus [36].
Entactin/nidogen, BM-40/osteonectin and fibulins: versatile ECM proteins
Members of the nidogen family are composed of a series of sulfated monomeric glycoproteins. Three globules, G1, G2 and G3, and one rod-like part, possessing different domains, make up the typical triglobular shape of nidogen 1, 2, 3. BM-40 is a glycoprotein of the ECM that binds calcium, collagen and hydroxyapatite and regulates the cell–matrix interaction 1, 2, 3. All fibulins contain epidermal growth factor-like repeats and a unique fibulin-type module at the C terminus that define this family [37]. To date, only one bacterium that uses nidogen as a potential ligand for ECM binding has been reported. SgrA of Enterococcus faecium has been identified as a bacterial receptor for nidogen and fibronectin [38]. The opportunistic bacterium Finegoldia magna depends on BM-40 interaction for colonization and survival [39]. Serum opacity factor is a streptococcal receptor for fibulin-1 [37].
No other bacteria, fungi or viruses are known to bind nidogen/entactin, BM-40 or fibulins during host invasion.
Pathogen-driven breakdown of the BM
Disruption of the BM in disease states often involves proteolytic enzymes [40] and an overview of the general characteristics of the different protease types is given elsewhere [41]. Many pathogens possess the ability to produce or modulate ECM-degrading enzymes. Regulation of ECM-degrading enzymes aids pathogens in invading deeper tissues, thereby enhancing dissemination throughout the host. Besides this direct effect of pathogens on BM-degrading enzymes, pathogens might also indirectly affect such enzymes. Indeed, during inflammation of infected tissues, local immune cells produce an array of these proteolytic enzymes. This indirect activation of proteases is beyond the scope of this review.
Several bacteria encode or modulate BM-degrading proteases, either directly or by engaging host-derived systems.
Various bacterial pathogens including Bacteroides fragilis and Clostridium perfringens [42] encode or modulate matrix metalloproteinases (MMPs). Other bacteria degrade the BM barrier by encoding or modulating serine proteases. Indeed, many bacterial pathogens, such as Enterobacteriaceae, Fusobacteriaceae, Helicobacteriaceae, Legionellaceae, Mycobacteriaceae, Neisseriaceae, Pasteurellaceae, Peptostreptococcaceae, Porphyromonadaceae, Pseudomonadaceae, Spirochaetaceae, Staphylococcaceae and Streptococcaceae, modulate the plasminogen (Plg)–plasmin system [43] and the structural and functional aspects of this system are described elsewhere [44]. In brief, through the activity of the two main physiological plasminogen activators, urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), plasminogen is converted to plasmin, which degrades laminin and fibronectin and activates precursors of MMPs. Several bacterial pathogens express plasminogen receptors, thereby recruiting plasminogen to the bacterial surface, which leads to enhanced plasminogen activation. In addition, some bacteria bind and/or induce uPA or tPA 43, 45 and/or express bacterial plasminogen activators. Moreover, some bacteria inactivate plasmin inhibitors [46].
Some bacterial species modulate multiple proteases to cross the BM. In addition to modulating the Plg system, Streptococcus pyogenes also expresses a cysteine protease [47]. Fusobacterium nucleatum can invade the BM in a strain-dependent manner by binding plasminogen [48] and pro-MMP9 and stimulating MMP9 and MMP13 secretion [49]. Helicobacter pylori induces expression of the cysteine protease cathepsin X in gastric epithelial cells and macrophages. In epithelial cells, H. pylori induces morphological and motility changes, partly via MMP9 [50] and probably also via other proteases. The mechanisms by which H. pylori induces expression of proteases in epithelial cells and macrophages is unknown. It has been suggested that proteolytic activities play a role in gastric tumorigenesis [50]. H. pylori also increases expression of the uPA system in gastric epithelial cells [51]. Mycobacterium tuberculosis 18, 52 and Neisseria meningitidis 45, 53 modulate both MMPs (MMP8 and MMP9, respectively) and the plasminogen–plasmin system. Both morphotypes of Peptostreptococcus micros possess plasminogen receptors. For P. micros, both bacterial (streptokinase) and human plasminogen activators (uPA) can activate plasminogen to plasmin. Rough morphotypes also possess chymotrypsin-like and gelatinase serine proteases [54]. Porphyromonas gingivalis upregulates MMPs, modulates the plasminogen–plasmin system and expresses cysteine protease gingipains. Gingipains contribute to BM penetration, either directly or by modulating MMP2 and MMP9 [55]. Pseudomonas aeruginosa expresses an MMP and modulates the plasminogen–plasmin system [56]. Treponema pallidum expresses an MMP-like and serine protease [57], induces MMPs and modulates the plasminogen–plasmin system [43]. Vibrio spp. express both MMPs and serine proteases [58].
Protease activity has also been implicated in tissue penetration by pathogenic fungi. Different fungi have been associated with multiple BM-degrading proteases. However, knowledge on substrate specificities and their contribution to virulence and pathogenesis is rather poor. During Aspergillus spp. infection, a serine protease, MMP and aspartic protease have been identified [59]. Candida spp. activate host MMP9, decrease tissue inhibitor of metalloproteinase 2 (TIMP2) secretion [18], and secrete aspartic proteases and unidentified MMP and serine protease activity [60]. Cryptococcus neoformans expresses a serine protease [61]. A total of 53 cDNAs encoding proteases were shown in Paracoccidioides brasiliensis including one unidentified gelatinase (collagenolytic protease) and an extracellular serine-thiol protease [62]. P. brasiliensis also induces MMP9 [63]. Nectriaceae, Saccharomycetaceae and Trichocomaceae also induce host MMPs [64].
It has also been reported that viruses modulate host-derived proteolytic activity to alter barrier properties of BMs, thereby enhancing viral dissemination (Figure 2). Most viral-induced alterations of BMs involve MMPs. The latent membrane protein-1 (LMP1) of Epstein–Barr virus (EBV) induces MMP9 and uPA [65]. Hepatitis B virus x protein (HBx) drives MMP14 expression [66]. It has been reported that HPV induces MMP2, MMP9 and MMP14 [67]. The glycoprotein K1 of Kaposi's sarcoma-associated herpesvirus (KSHV) modulates the production of MMP1, MMP2 and MMP9 [68]. Apart from the involvement of viral-induced proteolytic activities in EBV-, hepatitis B virus-, HPV- and KSHV-induced metastasis, it is unclear at present whether this induction plays a role in viral pathogenesis. A similar mechanism is observed in human T-cell leukemia/lymphoma virus type I (HTLV-1)-associated adult T-cell leukemia/lymphoma, in which MMP9 expression is increased in HTLV-1-infected malignant cells 69, 70. HTLV-I encephalitis is associated with MMP2 and MMP9 [70]. Other viral infections have also been implicated in neurological conditions because of their involvement in BBB impairment by damaging the vascular BM. Human cytomegalovirus (HCMV) infection of human microvascular endothelial cells (HMEC) induces collagenase type IV secretion, which may lead to BM degradation and subsequent release of infected endothelial cells into the circulation and access into the CNS [71]. Viral hijacking of immune cells might also modulate vascular permeability. Bunyaviridae (Andes virus, ANDV), Dengue virus, HIV and West Nile virus (WNV) enhance dendritic cell (DC) maturation, MMP9 expression and plasma vascular leakage [72]. MMP9 is also upregulated in both the periphery and brain on WNV infection and is partly localized to brain blood vessels. WNV may enter the brain directly through the BBB or may be carried within infected leukocytes (described in the next section). WNV also upregulates MMP1 and MMP3 [73]. Coronaviruses can also induce MMPs in susceptible cells and have been associated with multiple sclerosis-like disease in rodents. However, the role of MMPs in coronavirus CNS infection is unknown [74]. Finally, it has been postulated that bovine herpesvirus 5 (BHV-5) entry the CNS is facilitated by leukocytes and MMP9. However, induction of MMP9 expression by BHV-5 has not been directly demonstrated so far [75].
Reports describing the involvement of serine protease activity in viral-induced BM distortion are scarce. As described above, EBV also induces uPA [65]. An unidentified trypsin-like serine protease is involved in BM crossing by the porcine pseudorabies virus (PRV) in porcine nasal respiratory explants [76].
In summary, several bacteria, fungi and viruses enhance invasion through the BM barrier by (mis)using proteolytic systems by encoding and/or modulating host-derived proteases.
Hitchhiking across the BM
During physiological processes, such as development and immune surveillance, and during the pathology of many diseases, such as metastatic cancer, cells frequently traverse the BM barrier. Transmigration across the BM is a three-stage process (Figure 1). First, invadopodia-like protrusions perforate the BM. Then these protrusions elongate in the degraded zone and infiltrate the underlying compartment. It is believed that the rod-like shape of invadopodia allows for focal delivery of proteases to restrictive areas of the BM [77]. Although the primary function of immune cells is to sample pathogens to initiate an immune response, over time, several pathogens have developed mechanisms to use these cells as Trojan horses to cross the BM barrier and disseminate throughout the host. Mechanisms of intracellular survival involving inhibition of immune cell activation via alteration of their phenotype and function contribute to cell migration according to direct and indirect evidence [78].
Several bacteria survive in polymorphonuclear neutrophil granulocytes (PMN) or neutrophils: Anaplasma phagocytophilum, Bordetella pertussis, Brucella abortus, Chlamydia psittaci, Chlamydia trachomatis, Escherichia coli, Francisella tularensis, Mycobacterium leprae, Neisseria gonorrhoeae, Salmonella enterica serovar Typhi, Salmonella enterica serovar Typhimurium, S. aureus, S. pyogenes and Yersinia enterolytica. This may provide a mechanism by which bacteria infect distant sites, although this needs further experimental confirmation. Burkholderia pseudomallei, Chlamydia pneumoniae, Haemophilus somnus and Legionella pneumophila not only survive but also multiply in PMN. Afterwards, pathogens may enter through the uptake of infected apoptotic PMN, survive and multiply in macrophages, which subsequently may transport bacteria throughout the body 79, 80.
The Trojan horse hypothesis is also valid for DCs. Coxiella burnetii survives in DCs during infection [81]. DCs also transport intracellular Listeria monocytogenes, M. tuberculosis and S. Typhimurium from mucosal areas towards the draining lymph nodes [82].
Mononuclear phagocytes (monocytes and macrophages) can also be misused by intracellular bacteria to disseminate throughout the host. This has been reported or suggested for uropathogenic E. coli [83], L. pneumophila, Salmonella enterica [84], S. Typhimurium [78] and Yersinia pestis [85]. Furthermore, H. influenzae has been found within mononuclear cells both above and below the BM [86]. Although Bacillus anthracis is not an intracellular pathogen, it can use alveolar macrophages to cross airway mucosal barriers [87]. Mononuclear phagocyte-facilitated entry into CNS across the BBB has been described for Brucella spp., Ehrlichia chaffeensis, L. monocytogenes, M. tuberculosis and Streptococcus suis type II in swine [78]. L. monocytogenes invades both directly and when carried within infected leukocytes [82].
Several pathogens, such as Mycobacterium avium [88] and Shigella flexneri [89], predominantly exploit M cells to cross the epithelial barrier into the subepithelial lamina propria and subsequently invade macrophages. As described earlier, H. pylori is associated with metastatic processes and the spread of malignant cells throughout the body [50].
Some fungi also exploit immune cells to cross host barriers. Histoplasma capsulatun causes systemic mycosis, mainly in immunosuppressed patients, and can survive and multiply in PMN [80]. It has been demonstrated that the facultative intracellular pathogen C. neoformans crosses the BBB via mononuclear phagocytes (monocytes and macrophages), together with other mechanisms involving free yeasts [90].
Several viruses hijack immune cells to transverse the BM (Figure 2). DCs and Langerhans cells (LCs), an epidermal DC subtype, are located at mucosal or epidermal sites of entry for many viruses, such as herpesviruses, immunodeficiency viruses and HPV, and are thus of key importance in infections with these viruses. Initial infection of epidermal cells with herpes simplex virus type 1 (HSV-1) results in infection of resident LCs. After infection, a decrease in epidermal LC density and a corresponding increase in the number of langerin-positive cells in the underlying dermis has been noted, arguing for HSV-induced LC migration from the epidermal layer [91]. During primary infection with varicella zoster virus (VZV), another herpesvirus, DCs of the respiratory mucosa can transport VZV to human tonsillar CD4+ T lymphocytes, followed by T lymphocyte-mediated dissemination in the host [92]. HIV might use LCs for trans-epithelial transport of HIV to susceptible CD4+ T cells [93]. Measles virus (MV) is another virus that uses DCs to gain access to its main target cells in lymphoid tissues [94]. As described earlier, ANDV, Dengue virus, HIV and WNV hijack DCs [72].
HIV may use not only use LCs but also mononuclear phagocytes (monocytes/macrophages) for BM passage, which could contribute to their ability to invade the brain. Early in the course of infection, HIV-1 can enter the CNS. HIV encephalitis is characterized by HIV-laden monocytes and macrophage infiltration into CNS parenchyma. Local inflammation and HIV products such as gp120, Nef and Tat, which upregulate MMP2 and/or MMP9, lead to breaches in the BBB late in HIV CNS disease. This enables free virions to enter the brain 95, 96. In addition, CMV-infected monocytes can enter the CNS in a Trojan horse model [78]. For WNV, LCs support initial viral replication, followed by replication in lymphoid tissues and dissemination to organs and the CNS [97]; WNV may enter the brain through the BBB either directly or carried within infected monocytes or macrophages (as described above) [73].
Recently, it has been shown that equine herpesvirus 1 (EHV-1) might use monocytes, macrophages and lymphocytes as Trojan horses to transport the virus through the BM in nasal mucosae. EHV-1-infected monocytes, macrophages and lymphocytes were found in connective tissue below the BM in close proximity to epithelial plaques, suggestive of leukocyte-mediated viral passage through the BM [98]. The closely related EHV-4 did not efficiently infect these local immune cells, which might be the reason why EHV-4-induced viremia is rare [98]. It has been postulated that BHV-5 might use a similar invasive mechanism as EHV-1 and that BHV-5 entry into the CNS is facilitated by leukocytes and MMP9 [75]. Lymphocyte-mediated viral entry into the brain has also been demonstrated for HTLV-1 [70]. The association with MMP activity was described earlier.
Some oncogenic viruses such as EBV, hepatitis B virus, HPV, HTLV-1 and KSHV drive tumor invasiveness and metastasis by modulating proteases (Figure 2). However, the role of the spread of viral-infected malignant cells in the pathogenesis of these viruses is unclear at present.
In conclusion, different bacteria, fungi and viruses exploit host cells, particularly immune cells, to cross the BM barrier and disseminate throughout the host.
Concluding remarks and future perspectives
The BM represents a formidable barrier of the body against the outside world. However, it is clear that a wide array of pathogens have developed mechanisms to cross the BM and invade the host. There are several important outstanding questions about this still largely unexplored topic (Box 1 ). One aspect of BM passage that we have not discussed here is the role of local immunity in breaking down ECM during a microbial infection. Indeed, immune cells produce a large amount of proteases on stimulation at sites of inflammation and this might rupture important barriers such as the BM, allowing pathogens access to deeper tissues. However, inflammation increases the risk that pathogens will be neutralized by the immune system.
Box 1. Outstanding questions.
-
•
For what reason did certain pathogens, which might even belong to the same family, evolve different mechanisms of invasion? Did they co-evolve with their host?
-
•
Does tissue type play a role in the particular mechanism utilized for pathogen invasion? Alternatively, is tissue tropism driven by the invasion skills of a pathogen? How different are the BM composition and barrier function in different tissues of different species?
-
•
To what extent does inflammation really play a role in aiding pathogen invasion through the BM? Does immune evasion provide better and more rapid BM crossing?
-
•
What intrinsic capacities do viruses possess for host invasion?
-
•
Are the proteases involved in viral invasion produced by the virus or cellular proteases that are upregulated by the virus?
The BM represents one of the first barriers encountered by the pathogen, so dissection of pathogen interactions with and mechanisms to cross the BM may provide interesting leads towards the development of novel antimicrobial drugs. However, it is important to keep in mind that current detailed in vitro knowledge on this topic does not always translate to the in vivo situation as axiomatic truth. Improved in vitro models that better reflect the in vivo environment will provide excellent tools for the identification and characterization of putative adhesion and invasion mechanisms before progressing to the use of animal models.
An obvious target for preventing pathogen infiltration is the adhesion step to the BM. Moreover, it is important to know that different microbial invasive strategies might have synergistic effects on one another. Indeed, interactions between adhesion and proteolytic activity-mediating mechanisms to improve and enhance pathogen invasion have been described [99]. In some bacteria, production of proteases might even depend on quorum sensing [100]. Taking this possibility into account, hampering of bacterial adhesion might also influence proteolytic activity. Mechanisms underpinning binding to and breakdown of the BM are generally better understood for bacteria and fungi than for viruses. Hence, it will be interesting to identify possible viral factors that are required for efficient penetration through the BM and ECM. If protease activity plays a role, as described recently for a herpesvirus [76], then the use of protease inhibitors might be a useful strategy. However, to further develop the potential of proteases as antimicrobial targets, there is a need for identification of all signals, factors and domains involved during microbial invasion. Compounds that interfere with pathogen hijacking of migratory cells or limit interactions of metastatic cells with ECM elements could, if locally applied, provide an interesting way to prevent or delay further microbial invasion.
In conclusion, several bacteria, fungi and viruses have evolved different finely tuned techniques to adhere to, break down and/or hitchhike across the BM. Fundamental insights into these invasion mechanisms of pathogens could be a promising road towards new therapeutic approaches against these different types of pathogens.
Acknowledgments
We would like to thank Prof. Dr Gerlinde Van de Walle and Prof. Dr Greg Smith for their insightful comments. This work was supported by research grants from the Agency for Innovation by Science and Technology (IWT). We apologize for not being able to cite all the valuable work that has been done on this topic because of space limitations.
References
- 1.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer. 2003;3:422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 2.Rowe R.G., Weiss S.J. Breaching the basement membrane: who, when and how? Trends Cell Biol. 2008;18:560–574. doi: 10.1016/j.tcb.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 3.LeBleu V.S. Structure and function of basement membranes. Exp. Biol. Med. (Maywood) 2007;232:1121–1129. doi: 10.3181/0703-MR-72. [DOI] [PubMed] [Google Scholar]
- 4.Kline K.A. Bacterial adhesins in host–microbe interactions. Cell Host Microbe. 2009;5:580–592. doi: 10.1016/j.chom.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 5.Zong J. A ‘Collagen Hug’ model for Staphylococcus aureus CNA binding to collagen. EMBO J. 2005;24:4224–4236. doi: 10.1038/sj.emboj.7600888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amano A. Bacterial adhesins to host components in periodontitis. Periodontology. 2000;52:12–37. doi: 10.1111/j.1600-0757.2009.00307.x. [DOI] [PubMed] [Google Scholar]
- 7.Ilaria F., Giusti F. EM reconstruction of adhesins: future prospects. Adv. Exp. Med. Biol. 2011;715:271–284. doi: 10.1007/978-94-007-0940-9_17. [DOI] [PubMed] [Google Scholar]
- 8.Salgado P.S. Structural basis for the broad specificity to host-cell ligands by the pathogenic fungus Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 2011;108:15775–15779. doi: 10.1073/pnas.1103496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Q. The Enterococcus faecalis MSCRAMM ACE binds its ligand by the Collagen Hug model. J. Biol. Chem. 2007;282:19629–19637. doi: 10.1074/jbc.M611137200. [DOI] [PubMed] [Google Scholar]
- 10.Pieters R.J. Carbohydrate mediated bacterial adhesion. Adv. Exp. Med. Biol. 2011;715:227–240. doi: 10.1007/978-94-007-0940-9_14. [DOI] [PubMed] [Google Scholar]
- 11.Navarre W.W., Schneewind O. Surface proteins of Gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 1999;63:174–229. doi: 10.1128/mmbr.63.1.174-229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bober M. The membrane bound LRR lipoprotein Slr, and the cell wall-anchored M1 protein from Streptococcus pyogenes both interact with type I collagen. PLoS ONE. 2011;6:e20345. doi: 10.1371/journal.pone.0020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie X., Lipke P.N. On the evolution of fungal and yeast cell walls. Yeast. 2010;27:479–488. doi: 10.1002/yea.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaffin W.L. Candida albicans cell wall proteins. Microbiol. Mol. Biol. Rev. 2008;72:495–544. doi: 10.1128/MMBR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Greve H. Combining sites of bacterial fimbriae. Curr. Opin. Struct. Biol. 2007;17:506–512. doi: 10.1016/j.sbi.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Nobbs A.H. Streptococcus adherence and colonization. Microbiol. Mol. Biol. Rev. 2009;73:407–450. doi: 10.1128/MMBR.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim M. Reinforcement of epithelial cell adhesion to basement membrane by a bacterial pathogen as a new infectious stratagem. Virulence. 2010;1:52–55. doi: 10.4161/viru.1.1.10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Claveau I. Basement membrane protein and matrix metalloproteinase deregulation in engineered human oral mucosa following infection with Candida albicans. Matrix Biol. 2004;23:477–486. doi: 10.1016/j.matbio.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Henderson B. Fibronectin: a multidomain host adhesin targeted by bacterial fibronectin-binding proteins. FEMS Microbiol. Rev. 2010;35:147–200. doi: 10.1111/j.1574-6976.2010.00243.x. [DOI] [PubMed] [Google Scholar]
- 20.Krishnan V., Narayana S.V.L. Crystallography of Gram-positive bacteria. Adv. Exp. Med. Biol. 2011;715:175–195. doi: 10.1007/978-94-007-0940-9_11. [DOI] [PubMed] [Google Scholar]
- 21.Singh K.V. Importance of the collagen adhesin ace in pathogenesis and protection against Enterococcus faecalis experimental endocarditis. PLoS Pathog. 2010;6:e1000716. doi: 10.1371/journal.ppat.1000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hallström T. Complement regulator-acquiring surface protein 1 of Borrelia burgdorferi binds to human bone morphogenic protein 2, several extracellular matrix proteins, and plasminogen. J. Infect. Dis. 2010;202:490–498. doi: 10.1086/653825. [DOI] [PubMed] [Google Scholar]
- 23.Banerjee A. Bacterial pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat. Commun. 2011;2:462. doi: 10.1038/ncomms1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tronchin G. Adherence mechanisms in human pathogenic fungi. Med. Mycol. 2008;46:749–772. doi: 10.1080/13693780802206435. [DOI] [PubMed] [Google Scholar]
- 25.Nobbs A.H. Heterologous expression of Candida albicans cell wall-associated adhesins in Saccharomyces cerevisiae reveals differential specificities in adherence and biofilm formation and in binding oral Streptococcus gordonii. Eukaryot. Cell. 2010;9:1622–1634. doi: 10.1128/EC.00103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vieira M.L. Lsa63, a newly identified surface protein of Leptospira interrogans binds laminin and collagen IV. J. Infect. 2010;60:52–64. doi: 10.1016/j.jinf.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 27.Ragunathan P. Structure of laminin-binding adhesin (Lmb) from Streptococcus agalactiae. Acta. Crystallogr. D: Biol. Crystallogr. 2009;65:1262–1269. doi: 10.1107/S0907444909038359. [DOI] [PubMed] [Google Scholar]
- 28.Hallström T. Haemophilus influenzae protein E binds to the extracellular matrix by concurrently interacting with laminin and vitronectin. J. Infect. Dis. 2011;204:1065–1074. doi: 10.1093/infdis/jir459. [DOI] [PubMed] [Google Scholar]
- 29.Brissette C.A. The Borrelia burgdorferi outer-surface protein ErpX binds mammalian laminin. Microbiology. 2009;155:863–872. doi: 10.1099/mic.0.024604-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kines R.C. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. U.S.A. 2009;106:20458–20463. doi: 10.1073/pnas.0908502106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broutian T.R. Differential binding patterns to host cells associated with particles of several human alphapapillomavirus types. J. Gen. Virol. 2010;91:531–540. doi: 10.1099/vir.0.012732-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boshuizen J.A. Rotavirus enterotoxin NSP4 binds to the extracellular matrix proteins laminin-beta3 and fibronectin. J. Virol. 2004;78:10045–10053. doi: 10.1128/JVI.78.18.10045-10053.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halasz P. Rotavirus replication in intestinal cells differentially regulates integrin expression by a phosphatidylinositol 3-kinase-dependent pathway, resulting in increased cell adhesion and virus yield. J. Virol. 2008;82:148–160. doi: 10.1128/JVI.01980-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y. Microbial subversion of heparan sulfate proteoglycans. Mol. Cells. 2008;26:415–426. [PubMed] [Google Scholar]
- 35.Bartlett A.H., Park P.W. Heparan sulfate proteoglycans in infection. In: Pavão M.S.G., editor. Glycans in Diseases and Therapeutics. Springer; 2011. pp. 3–62. [Google Scholar]
- 36.Mohan K.V. The proteoglycan bamacan is a host cellular ligand of vaccinia virus neurovirulence factor N1L. J. Neuroviol. 2009;15:229–237. doi: 10.1080/13550280902913636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Courtney H.S. Serum opacity factor is a streptococcal receptor for the extracellular matrix protein fibulin-1. J. Biol. Chem. 2009;284:12966–12971. doi: 10.1074/jbc.M901143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hendrickx A.P. SgrA, a nidogen-binding LPXTG surface adhesin implicated in biofilm formation, and EcbA, a collagen binding MSCRAMM, are two novel adhesins of hospital-acquired Enterococcus faecium. Infect. Immun. 2009;77:5097–5106. doi: 10.1128/IAI.00275-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frick I.M. Identification of a novel protein promoting the colonization and survival of Finegoldia magna, a bacterial commensal and opportunistic pathogen. Mol. Microbiol. 2008;70:695–708. doi: 10.1111/j.1365-2958.2008.06439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buttle D.J. Factors controlling matrix turnover in health and disease. Biochem. Soc. Trans. 2007;35:643–646. doi: 10.1042/BST0350643. [DOI] [PubMed] [Google Scholar]
- 41.Barrett A.J. Managing peptidases in the genomic era. Biol. Chem. 2003;384:873–882. doi: 10.1515/BC.2003.098. [DOI] [PubMed] [Google Scholar]
- 42.Pruteanu M. Degradation of the extracellular matrix components by bacterial-derived metalloproteases: implications for inflammatory bowel diseases. Inflamm. Bowel Dis. 2011;17:1189–1200. doi: 10.1002/ibd.21475. [DOI] [PubMed] [Google Scholar]
- 43.Bergmann S., Hammerschmidt S. Fibrinolysis and host response in bacterial infections. Thromb. Haemost. 2007;98:512–520. [PubMed] [Google Scholar]
- 44.Schaller J., Gerber S.S. The plasmin–antiplasmin system: structural and functional aspects. Cell. Mol. Life Sci. 2011;68:785–801. doi: 10.1007/s00018-010-0566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eberhard T. Surface bound plasmin promotes migration of Streptococcus pneumoniae through reconstituted basement membranes. Microb. Pathog. 1999;26:175–181. doi: 10.1006/mpat.1998.0262. [DOI] [PubMed] [Google Scholar]
- 46.Lähteenmäki K. Bacterial plasminogen activators and receptors. FEMS Microbiol. Rev. 2001;25:531–552. doi: 10.1111/j.1574-6976.2001.tb00590.x. [DOI] [PubMed] [Google Scholar]
- 47.Burns E.H., Jr. Activation of a 66-kilodalton human endothelial cell matrix metalloprotease by Streptococcus pyogenes extracellular cysteine protease. Infect. Immun. 1996;64:4744–4750. doi: 10.1128/iai.64.11.4744-4750.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Darenfed H. Acquisition of plasmin activity by Fusobacterium nuclaeatum subsp. nucleatum and potential contribution to tissue destruction during periodontitis. Infect. Immun. 1999;67:6439–6444. doi: 10.1128/iai.67.12.6439-6444.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gursoy U.K. Stimulation of epithelial cell-matrix metalloproteinase (MMP-2, -9, -13) and interleukin-8 secretion by fusobacteria. Oral Microbiol. Immunol. 2008;23:432–434. doi: 10.1111/j.1399-302X.2008.00453.x. [DOI] [PubMed] [Google Scholar]
- 50.Krueger S. Up-regulation of cathepsin X in Helicobacter pylori gastritis and gastric cancer. J. Pathol. 2005;207:32–42. doi: 10.1002/path.1820. [DOI] [PubMed] [Google Scholar]
- 51.Kenny S. Increased expression of the urokinase plasminogen activator system by Helicobacter pylori in gastric epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;295:G431–G441. doi: 10.1152/ajpgi.90283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Monroy A. Binding and activation of human plasminogen by Mycobacterium tuberculosis. Infect. Immun. 2000;68:4327–4330. doi: 10.1128/iai.68.7.4327-4330.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schubert-Unkmeir A. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PloS Pathog. 2010;6:e1000874. doi: 10.1371/journal.ppat.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grenier D., Bouclin R. Contribution of proteases and plasmin-acquired activity in migration of Peptostreptococcus micros through a reconstituted basement membrane. Oral Microbiol. Immunol. 2006;21:319–325. doi: 10.1111/j.1399-302X.2006.00298.x. [DOI] [PubMed] [Google Scholar]
- 55.Andrian E. Regulation of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases by Porphyromonas gingivalis in an engineered human oral mucosa model. J. Cell. Physiol. 2006;211:56–62. doi: 10.1002/jcp.20894. [DOI] [PubMed] [Google Scholar]
- 56.da Silva C.M. Binding of plasminogen to Pseudomonas aeruginosa results in formation of surface-associated plasmin and enhanced bacterial invasiveness. Microb. Pathog. 2004;36:59–66. doi: 10.1016/j.micpath.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 57.Houston S. Bifunctional role of the Treponema pallidum extracellular matrix binding adhesin Tp0751. Infect. Immun. 2011;79:1386–1398. doi: 10.1128/IAI.01083-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shinoda S., Miyoshi S. Proteases produced by vibrios. Biocontrol. Sci. 2011;16:1–11. doi: 10.4265/bio.16.1. [DOI] [PubMed] [Google Scholar]
- 59.Lee J.D., Kolattukudy P.E. Molecular cloning of the cDNA and gene for an elastinolytic aspartic proteinase from Aspergillus fumigatus and evidence of its secretion by the fungus during invasion of the host lung. Infect. Immun. 1995;63:3796–3803. doi: 10.1128/iai.63.10.3796-3803.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pärnänen P. Human laminin-332 degradation by Candida proteinases. J. Oral Pathol. Med. 2008;37:329–335. doi: 10.1111/j.1600-0714.2008.00638.x. [DOI] [PubMed] [Google Scholar]
- 61.Rodrigues M.L. Cleavage of human fibronectin and other basement membrane-associated proteins by a Cryptococcus neoformans serine proteinase. Microb. Pathog. 2003;34:65–71. doi: 10.1016/s0882-4010(02)00195-x. [DOI] [PubMed] [Google Scholar]
- 62.Puccia R. Exocellular proteolytic activity of Paracoccidioidies brasiliensis: cleavage of components associated with the basement membrane. Med. Mycol. 1998;36:345–348. [PubMed] [Google Scholar]
- 63.Nishikaku A.S. Matrix metalloproteinases with gelatinolytic activity induced by Paracoccidioides brasiliensis infection. Int. J. Exp. Pathol. 2009;90:527–537. doi: 10.1111/j.1365-2613.2009.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dong X. Roles of adherence and matrix metalloproteinases in growth patterns of fungal pathogens in cornea. Curr. Eye Res. 2005;30:613–620. doi: 10.1080/02713680590968196. [DOI] [PubMed] [Google Scholar]
- 65.Yoshizaki T. Promotion of metastasis in nasapharyngeal carcinoma by Epstein–Barr virus latent membrane protein-1. Histol. Histopathol. 2002;17:845–850. doi: 10.14670/HH-17.845. [DOI] [PubMed] [Google Scholar]
- 66.Larra-Pezzi E. The hepatitis B virus X protein promotes tumor cell invasion by inducing membrane-type matrix metalloproteinase-1 and cyclooxygenase-2 expression. J. Clin. Invest. 2002;110:1831–1838. doi: 10.1172/JCI200215887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.da Silva C.M. Higher expression and activity of metalloproteinases in human cervical carcinoma cell lines is associated with HPV presence. Biochem. Cell Biol. 2006;84:713–719. doi: 10.1139/o06-084. [DOI] [PubMed] [Google Scholar]
- 68.Qian L.W. Kaposi's sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J. Virol. 2007;81:7001–7010. doi: 10.1128/JVI.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mori N. Human T-cell leukemia virus type I Tax transactivates the matrix metalloproteinase-9 gene: potential role in mediating adult T-cell leukemia invasiveness. Blood. 2002;99:1341–1349. doi: 10.1182/blood.v99.4.1341. [DOI] [PubMed] [Google Scholar]
- 70.Bazarbachi A. Human T-cell lymphotropic virus type I-infected cells extravasate through the endothelial barrier by a local angiogenesis-like mechanism. Cancer Res. 2004;64:2039–2046. doi: 10.1158/0008-5472.can-03-2390. [DOI] [PubMed] [Google Scholar]
- 71.Poland S.D. Cytomegalovirus-caused release of collagenase IV from human cerebral microvascular endothelial cells. Clin. Diagn. Virol. 1995;4:301–309. doi: 10.1016/0928-0197(95)00023-2. [DOI] [PubMed] [Google Scholar]
- 72.Marsac D. Infection of human monocyte-derived dendritic cells by ANDES hantavirus enhances pro-inflammatory state, the secretion of active MMP-9 and indirectly enhances endothelial permeability. Virol. J. 2011;8:223. doi: 10.1186/1743-422X-8-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verma S. Reversal of West Nile virus-induced blood–brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology. 2010;397:130–138. doi: 10.1016/j.virol.2009.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Edwards J.A. Activation of glial cells by human coronavirus OC43 infection. J. Neuroimmunol. 2000;108:73–81. doi: 10.1016/S0165-5728(00)00266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cardoso T.C. Immunohistochemical approach to the pathogenesis of clinical cases of Bovine Herpesvirus type 5 infections. Diagn. Pathol. 2010;5:57. doi: 10.1186/1746-1596-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Glorieux S. A trypsin-like serine protease is involved in pseudorabies virus invasion through the basement membrane barrier of porcine nasal respiratory mucosa. Vet. Res. 2011;42:58. doi: 10.1186/1297-9716-42-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schoumacher M. Cytoskeleton networks in basement membrane transmigration. Eur. J. Cell Biol. 2011;90:93–99. doi: 10.1016/j.ejcb.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 78.Drevets D.A., Leenen P.J.M. Leukocyte-facilitated entry of intracellular pathogens into the central nervous system. Microbes Infect. 2000;2:1609–1618. doi: 10.1016/s1286-4579(00)01317-4. [DOI] [PubMed] [Google Scholar]
- 79.Staali L. Streptococcus pyogenes expressing M and M-like surface proteins are phagocytosed but survive inside human neutrophils. Cell. Microbiol. 2003;5:253–265. doi: 10.1046/j.1462-5822.2003.00272.x. [DOI] [PubMed] [Google Scholar]
- 80.Laskay T. Neutrophil granulocytes as host cells and transport vehicles for intracellular pathogens: apoptosis as infection-promoting factor. Immunobiology. 2008;213:183–191. doi: 10.1016/j.imbio.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 81.Broederdorf L.J., Voth D.E. Cheating death: a Coxiella effector prevents apoptosis. Front. Microbiol. 2011;2:43–44. doi: 10.3389/fmicb.2011.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Drevets D.A. The Ly-6Chigh monocyte subpopulation transports Listeria monocytogenes into the brain during systemic infection of mice. J. Immunol. 2004;172:4418–4424. doi: 10.4049/jimmunol.172.7.4418. [DOI] [PubMed] [Google Scholar]
- 83.Bokil N.J. Intramacrophage survival of uropathogenic Escherichia coli: differences between diverse clinical isolates and between mouse and human macrophages. Immunobiology. 2011;216:1164–1171. doi: 10.1016/j.imbio.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 84.Vranckx L. Legionella pneumophila exhibits plasminogen activator activity. Microbiology. 2007;153:3757–3765. doi: 10.1099/mic.0.2007/010116-0. [DOI] [PubMed] [Google Scholar]
- 85.Fukuto H.S. Global gene expression profiling of Yersinia pestis replicating inside macrophages reveals the roles of a putative stress-induced operon in regulating type III secretion and intracellular cell division. Infect. Immun. 2010;78:3700–3715. doi: 10.1128/IAI.00062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Farley M.M. Pathogenesis of IgA1 protease-producing and -nonproducing Haemophilus influenzae in human nasaopharyngeal organ cultures. J. Infect. Dis. 1986;154:752–759. doi: 10.1093/infdis/154.5.752. [DOI] [PubMed] [Google Scholar]
- 87.Guidi-Rontani C. The alveolar macrophage: the Trojan horse of Bacillus anthracis. Trends Microbiol. 2002;9:405–409. doi: 10.1016/s0966-842x(02)02422-8. [DOI] [PubMed] [Google Scholar]
- 88.Secott T.E. Mycobacterium avium subsp. paratuberculosis fibronectin attachment protein facilitates M-cell targeting and invasion through a fibronectin bridge with host integrins. Infect. Immun. 2004;72:3724–3732. doi: 10.1128/IAI.72.7.3724-3732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.d’Hauteville H. Two msbB genes encoding maximal acylation of lipid A are required for invasive Shigella flexneri to mediate inflammatory rupture and destruction of the intestinal epithelium. J. Immunol. 2002;168:5240–5251. doi: 10.4049/jimmunol.168.10.5240. [DOI] [PubMed] [Google Scholar]
- 90.Casadevall A. Cryptococci at the brain gate: break and enter or use a Trojan horse? J. Clin. Invest. 2010;120:1389–1392. doi: 10.1172/JCI42949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eidsmo L. Differential migration of epidermal and dermal dendritic cells during skin infection. J. Immunol. 2009;182:3165–3172. doi: 10.4049/jimmunol.0802950. [DOI] [PubMed] [Google Scholar]
- 92.Abendroth A. Varicella zoster virus immune evasion strategies. Curr. Top. Microbiol. Immunol. 2010;342:155–171. doi: 10.1007/82_2010_41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cunningham A.L. Viruses and Langerhans cells. Immunol. Cell Biol. 2010;88:416–423. doi: 10.1038/icb.2010.42. [DOI] [PubMed] [Google Scholar]
- 94.Ludlow M. Systemic spread of measles virus: overcoming the epithelial and endothelial barriers. Thromb. Haemost. 2009;102:1050–1056. doi: 10.1160/TH09-03-0202. [DOI] [PubMed] [Google Scholar]
- 95.Gras G., Kaul M. Molecular mechanisms of neuroinvasion by monocytes-macrophages in HIV-1 infection. Retrovirology. 2010;7:30. doi: 10.1186/1742-4690-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Louboutin J.P. HIV-1 gp120-induced injury to the blood–brain barrier: role of metalloproteinases 2 and 9 and relationship to oxidative stress. J. Neuropathol. Exp. Neurol. 2010;69:801–816. doi: 10.1097/NEN.0b013e3181e8c96f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rios M. Monocytes–macrophages as a potential target in human infection with West Nile virus through blood transfusion. Transfusion. 2006;46:659–667. doi: 10.1111/j.1537-2995.2006.00769.x. [DOI] [PubMed] [Google Scholar]
- 98.Vandekerckhove A.P. Equine alphaherpesviruses (EHV-1 and EHV-4) differ in their efficiency to infect mononuclear cells during early steps of infection in nasal mucosal explants. Vet. Microbiol. 2011;152:21–28. doi: 10.1016/j.vetmic.2011.03.038. [DOI] [PubMed] [Google Scholar]
- 99.Lähteenmäki K. Expression of plasminogen activator Pla of Yersinia pestis enhances bacterial attachment to the mammalian extracellular matrix. Infect. Immun. 1998;66:5755–5762. doi: 10.1128/iai.66.12.5755-5762.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kievit T.R., Iglewski B.H. Bacterial quorum-sensing in pathogenic relationships. Infect. Immun. 2000;68:4839–4849. doi: 10.1128/iai.68.9.4839-4849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]