

Graphical abstract
Keywords: 2-Deoxy-d-ribose-1-phosphate, Carbasugar, Phosphoramidate, Prodrugs, Antiviral, Anticancer
Abstract
2-Deoxy-α-d-ribose-1-phosphate is of great interest as it is involved in the biosynthesis and/or catabolic degradation of several nucleoside analogues of biological and therapeutic relevance. However due to the lack of a stabilising group at its 2-position, it is difficult to synthesize stable prodrugs of this compound. In order to overcome this lack of stability, the synthesis of carbasugar analogues of 2-deoxyribose-1-phosphate was envisioned. Herein the preparation of a series of prodrugs of two carbocyclic analogues of 2-deoxyribose-1-phosphate using the phosphoramidate ProTide technology, along with their biological evaluation against HIV and cancer cell proliferation, is reported.
1. Introduction
Glycosyl-1-phosphates are essential constituents of larger biomolecules and play a diverse and important role in many physiological processes.1 In particular they are key intermediates in the metabolism of carbohydrates, critical in their transformation into nucleosides.2, 3 Among them, 2-deoxy-α-d-ribose-1-phosphate 1 (Fig. 1 ) is a catabolic product of thymidine phosphorylase (TP, EC 2.4.2.4), a nucleoside phosphorylase (NP) enzyme involved in the pyrimidine nucleoside salvage pathway.3 However, TP was also shown to be responsible for promoting angiogenesis.4 Increased TP expression levels, found in many solid tumours, are often correlated with neovascularisation, onset of metastasis and poor prognosis.5
Figure 1.
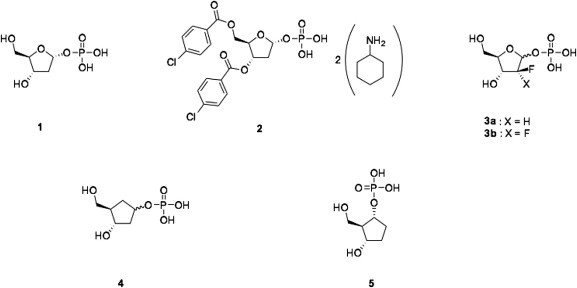
Structures of 2-deoxy-α-d-ribose-1-phosphate (1); 3,5-di-p-chlorobenzoyl-2-deoxy-d-ribose-1-phosphate (2); 2-fluoro-2-deoxyribose-1-phosphate (3a); 2,2-difluoro-2-deoxyribose-1-phosphate (3b); (3S,4R)-3-hydroxy-4-(hydroxymethyl)cyclopentyl-1-phosphate (4) and (1R,2R,3S)-3-hydroxy-2-(hydroxymethyl)cyclopentyl-1-phosphate (5).
During previous studies, aimed at preparing novel inhibitors of NPs as anticancer agents, we have identified 3,5-di-p-chlorobenzoyl-2-deoxy-d-ribose-1-phosphate 2 (Fig. 1), which was found to inhibit a variety of pyrimidine and purine NPs with preference for uridine- and inosine-hydrolyzing enzymes.6 This compound efficiently prevented the enzymatic breakdown of therapeutic analogues such as 5-fluoro-2′-deoxyuridine (FdUrd). In these studies we also demonstrated the difficulty in synthesizing phosphoramidate prodrugs at the anomeric position of 2-deoxyribose as they were found unstable. To overcome this instability we have shown that introduction of fluorine atoms on the 2-position of 2-deoxyribose enabled the synthesis of phosphoramidate prodrugs of 2-fluoro and 2,2-difluoro-2-deoxyribose-1-phosphate7 (compounds 3a and 3b, respectively, Fig. 1). Within this context we have more recently reported on the synthesis of a series of phosphonamidate prodrugs of another stable analogue of 2-deoxy-d-ribose-1-phosphate, in which the anomeric oxygen has been replaced by a methylene group.8
As a continuation of our interest in preparing prodrugs of stable analogues of 2-deoxy-d-ribose-1-phosphate we report here the application of the ProTide approach to two carbasugar analogues of 2-deoxyribose. The term ‘carbasugar’ refers to a family of compounds in which the oxygen atom of the furanose sugar ring has been replaced by a methylene group.9, 10, 11 Carbasugars are chemically more stable towards degradation than their sugar analogues12 but at the same time, due to their resemblance to natural sugars, they may be still recognized by the same enzymes.12 It is well known that the therapeutic potential of drugs bearing a phosphate moiety is decreased because of the negative charges of the phosphate group at physiological pH.13 To overcome this problem, the ProTide technology has been successfully applied in the past to various nucleoside14 and sugar analogues.15 With this in mind we herein report the synthesis of a series of prodrugs of (3S,4R)-3-hydroxy-4-(hydroxymethyl)cyclopentyl-1-phosphate 4 and (1R,2R,3S)-3-hydroxy-2-(hydroxymethyl)cyclopentyl-1-phosphate 5, as potential antiviral and anticancer agents (Fig. 1).
2. Results and Discussion
2.1. Chemistry
The synthesis of (3S,4R)-4-(hydroxymethyl)cyclopentane-1,3-diol 14 and (1R,2R,3S)-2-(hydroxymethyl)cyclopentane-1,3-diol 15 is depicted in Scheme 1 . Compound 8 was prepared in two steps from (1R,4S)-1-acetoxy-4-hydroxycyclopent-2-ene 6 according to a previously reported procedure.16, 17, 18, 19 In order to confirm the formation of compound 7, we further protected its hydroxyl groups as benzyl ethers obtaining the protected carbocycle 8. NMR analyses of 8 were in agreement with data reported in the literature.20 In addition, we prepared the Cbz-protected analogue 9, which we envisaged as a better intermediate for the synthesis of nucleoside aryloxyphosphoramidate prodrugs. The benzyloxycarbonyl group can be easily introduced into a nucleoside and most importantly, as reported in a recent paper by Schinazi et al.,21 it can be removed in neutral conditions, perhaps without affecting the phosphoramidate moiety. Hydroboration–oxidation reaction of 8 was investigated using different borane reagents such as 9-BBN,16, 20, 22 dicyclohexylborane23, 24 or BH3·THF.23 The best yield was obtained with BH3·THF. As expected in addition to the carbocycle 10, formation of its regio-isomer 12 was also observed. Only the α-epimer of compound 12 was isolated whereas both alpha and beta epimers of compound 10 were observed in a 1:1 ratio. NMR spectra of compounds 10 and 12 were in agreement with the literature.20, 23 We then adapted these optimized hydroboration conditions to compound 9 obtaining carbocycle 11 (α-epimer) and its regio-isomer 13 (mixture of epimers in a 1:0.5 ratio) in 26% and 28% yield, respectively.
Scheme 1.
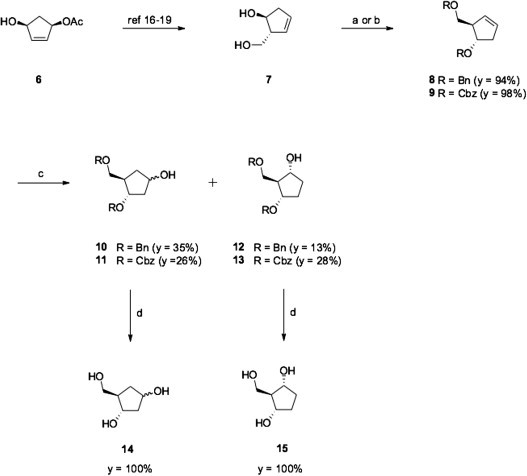
Synthesis of the carbocyclic intermediates 10–15. Reagents and conditions: (a) NaH, BnBr, THF, reflux, 3 h. (b) CbzCl, DMAP, CH2Cl2, rt, 19 h. (c) BH3·THF, 0 °C, 24 h then NaOH, H2O2, rt, 15 h. (d) H2, Pd/C, EtOH, rt, 1–2 h.
In order to biologically evaluate the parent carbasugars and to eventually compare their activities with those of the corresponding phosphoramidate prodrugs, we also prepared triol 14 and its regio-isomer 15 by hydrogenation reaction of compounds 11 and 13. NMR data of compound 15 showed that it is symmetrical meso system, which confirmed that we have prepared only the α-epimer of 15.25
With both carbocyclic intermediates 10 and 11 in hand we then studied the reaction of an appropriate phosphorochloridate with compound 11 (Scheme 2 and Table 1 ) in the presence of different bases (tBuMgCl,26, 27 NMI26, 27 pK a = 7, Et3N pK a = 10.6, n-BuLi pK a ca. 50) and different solvents (THF, toluene). As shown in Table 1 the best results were obtained when Et3N and NMI were used (entry 4).28 In particular, NMI (2.5% mol) was first added to a solution of carbocycle 11 in toluene. The reaction mixture was then cooled at 0 °C, followed by addition of Et3N (3 equiv) and the desired phosphorochloridate (3 equiv). The reaction was monitored by 31P NMR analysis until formation of the desired phosphorylated compounds was completed (signals between 1 and 3 ppm by 31P NMR). The crude was purified by column chromatography to yield the Cbz-protected prodrug, which was submitted to a hydrogenation reaction to afford the final phosphoramidate prodrug 16.
Scheme 2.
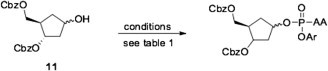
General reaction scheme for phosphorylation of compound 11.
Table 1.
Phosphorylation studies of compound 11
| Entry | Conditions | Yield (%) |
|---|---|---|
| 1 | t-BuMgCl (1.3 equiv) phosphorochloridatea (2.5 equiv) THF, rt, 20 h | 5 |
| 2 | t-BuMgCl (1.3 equiv) phosphorochloridateb (2.5 equiv) THF, rt, 20 h | <1 |
| 3 | t-BuMgCl (1.3 equiv) phosphorochloridatec (2.5 equiv) THF, rt, 20 h | No reaction |
| 4 | NMI (2.5% mol) Et3N (3 equiv + 3 equiv) phosphorochloridated (3 equiv + 3 equiv) toluene, rt, 19 h | 13 |
| 5 | Et3N (2 equiv + 3 equiv) phosphorochloridatec (2 equiv + 3 equiv) THF, rt, 2 days | No reaction |
| 6 | BuLi (1.1 equiv) phosphorochloridatee (2 equiv) THF, rt, 14 h | No reaction |
| 7 | BuLi (3 equiv) phosphorochloridatee (3 equiv) THF, rt, 18 h | <1 |
| 8 | NMI (10 equiv) phosphorochloridatea (4 equiv) THF, rt, 40 h | No reaction |
Ar = Naphthyl, AA ester = l-Ala-neopentyl.
Ar = Naphthyl, AA ester = l-Ala-cyclohexyl.
Ar = Naphthyl, AA ester = l-Ala-methyl.
Ar = Phenyl, AA ester = l-Ala-methyl.
Ar = Naphthyl, AA ester = l-Ala-neopentyl.
Following this methodology, six prodrugs (16a–f) of the carbasugar 14 were synthesized with yields ranging from 2% to 13% over two steps (Scheme 3 ). This strategy was then applied to the regio-isomer 13 affording the two prodrugs 17a (2% yield) and 17b (3% yield).
Scheme 3.
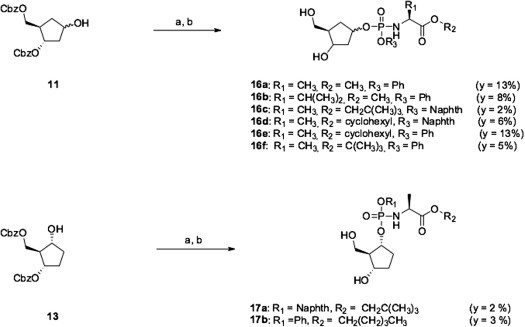
Synthesis of prodrugs 16a–f and 17a–b. Reagents and conditions: (a) appropriate phosphorochloridate, NMI, Et3N, toluene, rt, 24 h. (b) H2, Pd/C, EtOH, rt, 1–2 h.
2.2. Biological evaluation
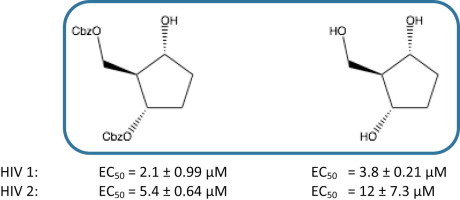
Prodrugs 16a–f were subjected to biological evaluation as antiviral and antiproliferative agents. None of the prodrugs showed any anti-HIV-1 or anti-HIV-2 activity or any cytostatic effect in CEM cells (Table 2 ). The parent carbocyclic analogue 14 and its Cbz-protected derivative 11 were not active either. However the regio-isomer 15 and its Cbz-protected derivative 13 showed pronounced anti-HIV activity in CEM cell cultures (2–12 μM range) when compared to the corresponding C1-regio-isomers in CEM cells (Table 2). When 15 was evaluated against HIV in MT-4 cell cultures, the antiviral activity against both HIV-1 and HIV-2 was confirmed at low micromolar concentrations without marked cytotoxicity (CC50: 90 μM). Unfortunately the corresponding prodrugs 17a and 17b were devoid of any significant antiviral activity (Table 3 ). When assessed for the antiproliferative activity 17a and 17b did not show pronounced inhibition of the proliferation of murine leukemia cells (L1210) and human T-lymphocyte cells (CEM) or cervix carcinoma (HeLa) cells (Table 4 ).
Table 2.
Anti-HIV-1 and -HIV-2 activity and cytostatic properties of the compounds 16a–f, 11, 13, 14 and 15 in human T-lymphocyte (CEM) cells
| Compound | EC50 (μM) |
CC50 (μM) | ||
|---|---|---|---|---|
| CEM |
CEM-TK− | |||
| HIV-1 | HIV-2 | HIV-2 | ||
| 16a | >250 | >250 | >250 | >250 |
| 16b | >250 | >250 | >250 | >250 |
| 16d | >50 | >50 | >50 | >250 |
| 16e | >250 | >250 | >250 | >250 |
| 16f | >250 | >250 | >250 | >250 |
| 13 | 2.1 ± 0.99 | 5.4 ± 0.64 | nd | 94 ± 4.2 |
| 15 | 3.8 ± 0.21 | 12 ± 7.3 | nd | >250 |
| 11 | >50 | >50 | nd | 110 ± 4.2 |
| 14 | >250 | >250 | nd | >250 |
EC50 = 50%-effective concentration or concentration required to protect CEM cells against the cytopathogenicity of HIV by 50%.
CC50 = 50%-cytotoxic concentration or concentration required to reduce CEM cell proliferation by 50%.
nd: not done.
Table 3.
Anti-HIV-1 and -HIV-2 activity and cytostatic properties of the compounds 15, 17a and 17b in MT-4 cell cultures
| Compound | HIV-1 (NL4.3) | HIV-2 (ROD) | MT-4 |
|---|---|---|---|
| EC50a (μM) | EC50a (μM) | CC50b (μM) | |
| 17a | >50 | >50 | 112 |
| 17b | >250 | 145 ± 30 | >250 |
| 15 | 6.7 ± 2.3 | 7.5 ± 1.5 | 90 |
nd: not done.
50% effective concentration or compound concentration required to inhibit HIV-induced cytopathic effect in MT-4 cell cultures.
50% cytostatic concentration in MT-4 cell cultures.
Table 4.
Inhibitory effects of compounds 15, 17a and 17b on the proliferation of murine leukemia cells (L1210) and human T-lymphocyte cells (CEM) and cervix carcinoma (HeLa) cells
| Compound | IC50⁎ (μM) |
||
|---|---|---|---|
| L1210 | CEM | HeLa | |
| 17a | 70 ± 5 | 64 ± 14 | 94 ± 13 |
| 17b | 116 ± 5 | 106 ± 3 | 166 ± 18 |
| 15 | 55 ± 15 | ⩾250 | 175 ± 42 |
50% inhibitory concentration.
To probe this result, incubation of compound 16d with carboxypeptidase Y in d 6-acetone and Trizma buffer was performed and the assay was followed by 31P NMR (Fig. 2 ). Compound 16d was slowly metabolized and after 13 h the starting material was still present. This slow processing may explain the lack of activity of this kind of prodrugs.
Figure 2.
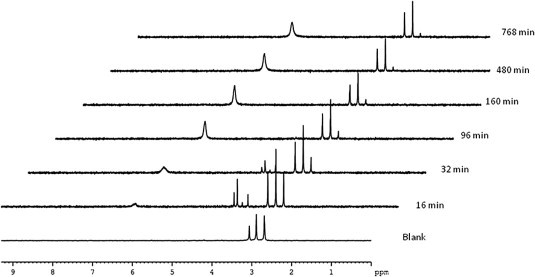
Carboxypeptidase Y-mediated processing of compound 16d, monitored by 31P NMR.
The anti-HIV activity of 13 and 15 at low micromolar concentrations is surprising and worth further exploring with respect to its molecular mechanism of antiviral action. Indeed, it should be noticed that compound 15 did not show any measurable activity at 100 μM against a wide variety of other viruses including herpes simplex virus type 1 (HSV-1), HSV-2 and vaccinia virus in HEL cell cultures, vesicular stomatitis virus (VSV), Coxsackie virus B4 and respiratory syncytial virus (RSV) in HeLa cell cultures, parainfluenza virus-3, reovirus-1, Sindbis virus and Punta Toro virus in Vero cell cultures, influenza virus A (H1N1, H3N2) and influenza virus B in MDCK cell cultures, and feline corona virus and feline herpesvirus in CrFK cell cultures. These findings make the compounds 13 and 15 highly selective for HIV. Reverse transcriptase has been excluded as a direct target since the compounds were found inactive against this enzyme (data not shown).
3. Conclusion
In summary, we have prepared several prodrugs of two carbasugar analogues of 2-deoxyribose-1-phosphate. Biological evaluation of these prodrugs showed that they had no inhibitory activity against HIV and cancer cell proliferation. However we note that parent carbocycle 15 and its synthetic intermediate 13 showed micromolar activity against HIV. Further investigations to elucidate the underlying mechanism by which 13 and 15 exert their antiviral activity against HIV will be performed. In particular, the synthetic route to (1R,2R,3S)-3-hydroxy-2-(hydroxymethyl)cyclopentyl-1-phosphate 5 is under investigation in order to confirm whether this compound can indeed act as an inhibitor against NPs. Application of other prodrug approaches is also under consideration.
4. Experimental part
4.1. Chemistry
Solvents and reagents: The following anhydrous solvents were bought from Aldrich with subaseal stopper: chloroform (CHCl3), dichloromethane (DCM), diethyl ether (Et2O), N,N-dimethylformamide (DMF), N-methylimidazole (NMI), pyridine, tetra-hydrofuran (THF), triethylamine (TEA). All reagents commercially available were used without further purification. Thin Layer Chromatography (TLC): Precoated, aluminium backed plates (60 F 254, 0.2 mm thickness, Merck) were visualized under both short and long wave ultraviolet light (254 nm and 366 nm). Preparative TLC plates (20 × 20 cm, 500–2000 μm) were purchased from Merck. Column Chromatography (CC): Column chromatography processes were carried out using silica gel supplied by Fisher (60A, 35–70 μm). Glass columns were slurry packed using the appropriate eluent and samples were applied either as a concentrated solution in the same eluent or pre-adsorbed on silica gel. High Performance Liquid Chromatography (HPLC): Analytical and semi-preparative reversed phase HPLC were conducted by Varian Prostar (LC Work Station-Varian prostar 335 LC detector, Varian fraction collector (model 701), pro-star 210 solvent delivery system, using Varian Polaris C18-A (10 μm) as an analytic column and Varian Polaris C18-A (10 μm) as a semi-preparative column. The software used was Galaxie Chromatography Data System. Nuclear Magnetic Resonance (NMR): 1H NMR (500 MHz), 13C NMR (125 MHz), 31P NMR (202 MHz) were recorded on a Bruker Avance 500 MHz spectrometer at 25 °C. Spectra were calibrated to the residual signal of the deuterated solvent used. Chemical shifts are given in parts per million (ppm) and coupling constants (J) in Hertz. The following abbreviations are used in the assignment of NMR signals: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br s (broad singlet), dd (doublet of doublet), dt (doublet of triplet).
4.1.1. Synthesis of (1S,2R)-2-(hydroxymethyl)cyclopent-3-enol (7)
To a suspension of Mg (893 mg, 36.7 mmol, 5.2 equiv) in THF (4 mL) was added 50 μL of dibromoethane and a solution of ClCH2SiMe2 iPr (6.4 mL, 35.3 mmol, 5 equiv) in THF (20 mL) in a quick dropwise (over 20 min) in order to keep a gently exothermic reaction. The reaction mixture was then heated at reflux for 30 min until no more solid Mg was observed and was then cooled down to 0 °C. To a slurry of CuCN (3.479 mg, 38.8 mmol, 5.5 equiv) in THF (40 mL) at −18 °C was slowly cannulated the fleshly prepared Grignard reaction. After 20 min of stirring at −18 °C was added to this organocopper reaction mixture a solution of compound 6 (1.004 g, 7.06 mmol) in THF (8 mL) at −18 °C. The reaction mixture was stirred at −18 °C and was slowly allowed to warm up until room temperature. After 19 h of reaction, the reaction was quenched by addition at 0 °C of 150 mL of NH4OH 10% saturated with NH4Cl and the reaction mixture was stirred at room temperature for 25 min. Phases were separated and the aqueous phase was extracted with AcOEt (3 * 100 mL). The combined organic phases were dried over MgSO4 and solvents were evaporated to dryness. Purification of the crude by column chromatography (eluent: Hexane/AcOEt 95:5 to 92:8) in order to obtain (1S,2S)-2-((isopropoxydimethylsilyl)methyl)cyclopent-3-enol (1.290 g, 6.01 mmol, 85%) as a pale yellow oil. 1H NMR (500 MHz, CDCl3): δ 5.59 (m, 1H, H3), 5.52–5.49 (m, 1H, H4), 4.08–4.01 (m, 2H, H1, CH(CH3)2), 3.79 (d, J = 3.0 Hz, 1H, OH), 2.68–2.62 (m, 2H, H2, H5a), 2.32 (m, 1H, H5b), 1.20 (s, 3H, CHCH 3), 1.19 (s, 3H, CHCH 3), 0.78 (dd, J = 14.9, 4.4 Hz, 1H, SiCH2a), 0.68 (dd, J = 14.8, 10.8 Hz, 1H, SiCH2b), 0.19 (s, 3H, SiCH3), 0.18 (s, 3H, SiCH3). 13C NMR (126 MHz, CDCl3): δ 136.2 (C3), 126.9 (C4), 81.1 (C1), 65.6 (CH(CH3)2), 49.1 (C2), 40.0 (C5), 25.6 (CH(CH 3)2), 25.5 (CH(CH 3)2), 22.22 (SiCH2), −1.0 (SiCH3), −1.2 (SiCH3).
A suspension of (1S,2S)-2-((isopropoxydimethylsilyl)methyl)cyclopent-3-enol (5.28 g, 24.6 mmol), potassium fluoride (7.25 g, 124.9 mmol, 5.07 equiv), KHCO3 (7.42 g, 74.1 mmol, 3.01 equiv) and hydrogen peroxide 30% (27 mL) in methanol/THF 1:1 (154 mL) was heated at reflux for 15 h. The reaction mixture was diluted with AcOEt (800 mL) and sodium thiosulfate (1.87 g) and magnesium sulfate (6.34 g) were added. The reaction mixture was stirred at room temperature for 30 min before filtration though a pad of Celite. The filtrate was then evaporated to dryness. The crude was purified by column chromatography (Eluent Hexane/AcOEt 5:95 to 0:100) in order to give compound 7 (2.689 g, 23.6 mmol, 96%) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 5.72 (dddd, J = 2.0, 2.0, 2.0, 6.0 Hz, 1H, H3), 5.54 (m, 1H, H4), 4.28 (ddd, J = 3.5, 3.5, 7.0 Hz, 1H, H1), 4.05 (br s, 1H, OH), 3.92–3.89 (br s, 1H, OH), 3.62 (dd, J = 10.7, 5.0 Hz, 1H, CH 2 aOH), 3.36 (dd, J = 10.7, 8.2 Hz, 1H, CH 2 bOH), 2.73–2.65 (m, 2H, H2, H5a), 2.28–2.23 (m, 1H, H5b). 13C NMR (126 MHz, CDCl3): δ 130.9 (C3), 128.9 (C4), 75.7 (C1), 64.4 (CH2OH), 57.5 (C2), 41.9 (C5). MS (ES+) m/z: 137.0 (M+Na+).
4.1.2. Synthesis of (1S,2R)-1-benzyloxy-2-benzyloxymethylcyclopent-3-ene (8)
To a solution of compound 7 (150 mg, 1.31 mmol) in THF (4.7 mL) at 0 °C was added NaH (60% slurry, 152 mg, 1.87 mmol, 2.8 equiv). The reaction mixture was stirred at room temperature for 1 h before addition of BnBr (560 μL, 4.72 mmol, 3.6 equiv). The reaction mixture was heated at reflux for 3 h before addition of crushed ice. The reaction mixture was stirred for 30 min and was then diluted with AcOEt (20 mL). The organic phase was washed with H20 (2 * 20 mL) and brine (2 * 20 mL) and dried over MgSO4. Solvents were evaporated to dryness. Purification by column chromatography (eluent: PE/AcOEt 100:0 to 100:4) led to compound 8 (364 mg, 1.24 mmol, 94%) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.40–7.28 (m, 10H, ArH), 5.77 (J = 2.0, 2.0, 2.0, 6.0 Hz, 1H, H3), 5.67 (J = 2.1, 2.1, 2.1, 5.9 Hz, 1H, H4), 4.56–4.53 (m, 4H, 2* CH 2Ph), 4.10 (ddd, J = 3.5, 3.5, 7.0 Hz, 1H, H1), 3.46 (dd, J = 9.3, 5.7 Hz, 1H), 3.35 (dd, J = 9.2, 7.3 Hz, 1H), 3.09–3.07 (m, 1H), 2.73–2.67 (m, 1H), 2.46–2.42 (m, 1H). 13C NMR (126 MHz, CDCl3): δ 138.8 (Cq), 138.5 (Cq), 130.0 (C3), 129.9 (C4), 128.6, 128.3, 128.3, 129.0, 127.7, 127.5, 127.5, 127.4 (CAr), 81.4 (C1), 73.0 (CH2Ph), 71.6 (OCH2), 70.8 (CH2Ph), 53.0 (C2), 39.1 (C5). MS (ES+) m/z: 317.1 (M+Na+).
4.1.3. Synthesis of (1S,2R)-1-benzyloxycarbonyl-2-benzyloxycarbonylmethylcyclopent-3-ene (9)
To a solution of compound 7 (1.983 g, 17.4 mmol) and DMAP (12.735 g, 104.2 mmol, 6 equiv) in CH2Cl2 (100 mL) at 0 °C was added dropwise CbzCl (11.02 mL, 78.2 mmol, 4.5 equiv). The reaction mixture was stirred at room temperature for 4 h and was then diluted with CH2Cl2 (100 mL) and washed with HCl 1 N (30 mL) and H2O (50 mL). The organic phase was dried over MgSO4 and solvents were evaporated to dryness. Purification of the crude by column chromatography (eluent: PE/AcOEt 98:2 to 90:10) gave compound 9 (6.54 g, 17.1 mmol, 98%) as a colorless oil. 1H NMR (500 MHz, CDCl3): δ 7.41–7.32 (m, 10H, ArH), 5.79 (dddd, J = 2.0, 2.0, 2.0, 6.0, Hz, 1H, H3), 5.63 (dddd, J = 2.5, 2.5, 2.5, 6.5 Hz, 1H, H4, 2* CH 2Ph), 5.13 (ddd, J = 3.0, 3.0, 6.0 Hz, 1H, H1), 4.20 (s, 1H, OCH2a), 4.19 (s, 1H, OCH2b), 3.15–3.11 (m, 1H, H2), 2.87 (ddddd, J = 2.5, 2.5, 2.5, 7.0 18.0 Hz, 1H, H5a), 2.49–2.43 (m, 1H, H5b). 13C NMR (126 MHz, CDCl3): δ 155.1 (C O), 154.8 (C O), 135.2 (Cq), 135.1 (Cq), 130.7 (C6), 128.6, 128.6 (CAr), 128.5 (C1), 128.3, 128.3 (CAr), 79.9 (C1), 69.7 (CH 2Ph), 69.6 (CH 2Ph), 68.0 (OCH2), 52.0 (C2), 39.0 (C5). MS (ES+) m/z: 405.1 (M+Na+).
4.1.4. Synthesis of (3S,4R)-3-(benzyloxy)-4-(benzyloxymethyl)cyclopentenol (10) and (2S,3S)-3-(benzyloxy)-2-(benzyloxymethyl)cyclopentenol (12)
To a solution of compound 8 (141 mg, 0.48 mmol) in THF (2.4 mL) at 0 °C was added dropwise a 1 M solution of BH3·THF (960 μL, 0.96 mmol, 2 equiv). The reaction mixture was stirred at 0–5 °C for 24 h and then at room temperature for 19 h before addition at 0 °C of 120 μL of NaOH 3 N and 120 μL of H2O2 30%. The reaction mixture was stirred at room temperature for 24 h. The reaction mixture was diluted in H2O (10 mL) and AcOEt (10 mL). The aqueous phase was extracted with AcOEt (3 * 10 mL) and the combined organic phases were dried over MgSO4. Solvents were evaporated to dryness. Purification of the crude by column chromatography (eluent: PE/AcOEt 9:1 to 7:3) gave compound 10 (46 mg, 0.15 mmol, 31%) and compound 12 (18 mg, 0.6 mmol, 12%) as colorless oils.
4.1.4.1. (3S,4R)-3-(Benzyloxy)-4-(benzyloxymethyl)cyclopentenol (10). Epimeric mixture α/β 1:1
1H NMR (500 MHz, CDCl3): δ 7.38–7.28 (m, 20H, ArH), 4.56–4.46 (m, 8H, CH 2Ph), 4.35–4.34 (m, 1H, H1-β), 4.29 (m, 1H, H1-α), 4.10 (ddd, J = 4.5, 6.5, 6.5 Hz, 1H, H3-β), 3.99 (ddd, J = 1.5, 1.5, 4.5 Hz, 1H, H3-α), 3.53 (dd, J = 9.0, 4.3 Hz, 1H, OCH2a-β), 3.49 (dd, J = 9.0, 4.3 Hz, 1H, OCH2b-β), 3.43 (dd, J = 9.3, 5.5 Hz, 1H, OCH2a-α), 3.29 (dd, J = 9.3, 7.5 Hz, 1H, OCH2b-α), 2.78 (br s, 2H, OH-α, OH-β), 2.69 (m, 1H, H4-α), 2.38–2.27 (m, 2H, H4-β, H5a-β), 2.10–1.99 (m, 3H, H2a-β, H5a-α, H2a-α), 1.92–1.85 (m, 2H, H2b-β, H2b-α), 1.57 (ddd, J = 13.7, 7.8, 5.6 Hz, 1H, H5b-α), 1.54–1.49 (m, 1H, H5b-β). 13C NMR (126 MHz, CDCl3): δ 138.6 (Cq), 138.4 (Cq), 138.3 (Cq), 137.8 (Cq), 128.4, 128.3, 128.2, 127.7, 127.7, 127.6, 127.5, 127.4 (CAr), 83.0 (C3-β), 81.9 (C3-α), 73.6 (C1-α), 73.3 (CH 2Ph), 72.9 (CH 2Ph), 72.3 (OCH2), 72.14 (OCH2), 72.13 (C1-β), 71.3 (CH 2Ph), 70.7 (CH 2Ph), 44.5 (C4-β), 44.0 (C4-α), 42.6 (C2-α), 40.3 (C2-β), 37.9 (C5-α), 37.3 (C5-β). MS (ES+) m/z: 335.1 (M+Na+).
4.1.4.2. (1R,2S,3S)-3-(Benzyloxy)-2-(benzyloxymethyl)cyclopentanol (12), α-epimer
1H NMR (500 MHz, CDCl3): δ 7.37–7.28 (m, 10H, ArH), 4.56–4.44 (m, 4H, 2* CH 2Ph), 4.03–4.00 (m, 1H, H1), 3.75–3.72 (m, 1H, H3), 3.62 (dd, J = 9.1, 5.7 Hz, 1H, OCH2a), 3.46 (dd, J = 9.1, 8.0 Hz, 1H, OCH2b), 2.46 (br s, 1H, OH), 2.28 (m, 1H, H2), 1.97–1.79 (m, 4H, H5, H4). 13C NMR (126 MHz, CDCl3): δ 138.5 (Cq), 138.1 (Cq), 128.4, 128.3, 127.7, 127.6, 127.6, 127.5 (CAr), 80.7 (C3), 75.7 (C1), 73.3 (CH 2Ph), 71.1 (CH 2Ph), 70.9 (OCH2), 54.0 (C2), 31.9 (C4), 28.9 (C5). MS (ES+) m/z: 335.1 (M+Na+).
4.1.5. Synthesis of (3S,4R)-3-(benzyloxycarbonyl)-4-(benzyloxycarbonylmethyl)cyclopentenol (11) and (2S,3S)-3-(benzyloxycarbonyl)-2-(benzyloxycarbonylmethyl)cyclopentenol (13)
To a solution of compound 9 (3.1 g, 8.1 mmol) in THF (40 mL) at 0 °C was added dropwise a 1 M solution of BH3·THF (16.2 mL, 16.2 mmol, 2 equiv). The reaction mixture was stirred at 0–5 °C for 18 h before addition at 0 °C of 5.05 mL of NaOH 3 N and 5.05 mL of H2O2 30%. The reaction mixture was stirred at room temperature for 24 h. The reaction mixture was diluted in H2O (100 mL) and AcOEt (100 mL). The aqueous phase was extracted with AcOEt (3 * 100 mL) and the combined organic phases were dried over MgSO4. Solvents were evaporated to dryness. Purification of the crude by column chromatography (eluent: PE/AcOEt 9:1 to 75:25) gave compound 11 (822 mg, 2.05 mmol, 26%) and compound 13 (904 mg, 2.25 mmol, 28%) as colorless oils.
4.1.5.1. (3S,4R)-3-(Benzyloxycarbonyl)-4-(benzyloxycarbonylmethyl)cyclopentenol (11)
Epimeric ratio 1:0.5 configuration of the epimeric stereocenter was not assigned. 1H NMR (500 MHz, CDCl3): δ 7.43–7.31 (m, 15H, ArH), 5.17–5.13 (m, 6H, 2* CH 2Ph of both epimers), 5.06 (ddd, J = 5.0, 5.0, 7.0 Hz, 1H, H3 of major epimer), 4.96–4.93 (m, 0.5H, H3 of minor epimer), 4.44–4.41 (m, 1H, H1 of major epimer), 4.34 (m, 0.5H, H1 of minor epimer), 4.31–4.19 (m, 3H, OCH2 of both epimers), 2.74 (dddd, J = 5.5, 5.5, 10.5, 10.5 Hz, 0.5H, H4 of minor epimer), 2.47–2.41 (m, 1H, H4 of major epimer), 2.35–2.24 (m, 1.5H, H2a of minor epimer, H5a of major epimer), 2.14–1.97 (m, 2.5H, H2 of major epimer, H5a of minor epimer), 1.90–1.83 (m, 0.5H, H2b of minor epimer), 1.68 (ddd, J = 5.5, 9.5, 14.5 Hz, 0.5H, H5b of minor epimer), 1.50–1.45 (m, 1H, H5b of major epimer). 13C NMR (126 MHz, CDCl3): δ 155.0, 155.0, 154.7, 155.5, 135.1, 135.0 (Cq), 128.6, 128.6, 128.5, 128.4, 128.3, 128.3 (CAr), 80.4 (C3 of minor epimer), 80.0 (C3 of major epimer), 71.6 (C1 of minor epimer), 71.2 (C1 of minor epimer), 69.7, 69.7, 69.6, 69.6 (CH 2Ph of both epimers), 69.1 (OCH2 of major epimer), 68.4 (OCH2 of minor epimer), 43.3 (C4 of major epimer), 43.1 (C4 of minor epimer), 41.5 (C2 of minor epimer), 41.4 (C2 of major epimer), 37.1 (C5 of minor epimer), 36.4 (C5 of major epimer). MS (ES+) m/z: 423.1 (M+Na+).
4.1.5.2. (1R,2S,3S)-3-(Benzyloxycarbonyl)-2-(benzyloxycarbonylmethyl)cyclopentanol (13), α-epimer
1H NMR (500 MHz, CDCl3): δ 7.38 (m, 10H, ArH), 5.17–5.15 (m, 4H, 2 × CH2Ph), 4.86–4.82 (m, 1H, H3), 4.41 (dd, J = 11.1, 4.8 Hz, 1H, OCH2a), 4.25 (dd, J = 11.1, 5.8 Hz, 1H, OCH2b), 4.05–4.00 (m, 1H, H1), 2.27–2.23 (m, 1H, H2), 2.06–1.89 (m, 4H, 5a, H4+OH), 1.83–1.79 (m, 1H, H5b). 13C NMR (126 MHz, CDCl3): δ 155.3 (C O), 154.8 (C O), 135.1 (Cq), 135.0 (Cq), 128.6, 128.6, 128.5, 128.4, 128.3 (CAr), 79.1 (C3), 73.5 (C1), 69.9 (CH 2Ph), 69.7 (CH 2Ph), 66.6 (OCH2), 53.1 (C2), 32.0 (C4), 29.1 (C5). MS (ES+) m/z: 423.1 (M+Na+).
4.1.6. Synthesis of (3S,4R)-3-hydroxy-4-hydroxymethyl cyclopentanol (14), epimeric ratio 1:0.15
To a solution of 11 in EtOH (C SM = 0.014 M) was added Pd/C 10% (1 mg/mmol of 11) and the reaction mixture was stirred at room temperature under H2 atmosphere until all 11 was consumed (1–2 h). Pd was filtrated through a pad of Celite and solvents were evaporated to dryness. To afford the desired final compounds 14 in quantitative yield.
1H NMR (500 MHz, CD3OD): δ 4.36–4.33 (m, 1H, H1, major epimer), 4.22–4.18 (m, 1H, H1, minor epimer), 4.09 (dt, J = 6.0, 6.5 Hz, 1H, H4, major epimer), 3.90 (dt, J = 6.5, 7.05 Hz, 1H, H4, minor epimer), 3.66 (dd, J = 6.0, 10.5 Hz, 1H, CH2aOH, major epimer), 3.62 (dd, J = 5.5, 10.5 Hz, 1H, CH2aOH, minor epimer), 3.56 (dd, J = 6.5, 10.5 Hz, 1H, CH2bOH, major epimer), 3.50 (dd, J = 6.5, 10.5 Hz, 1H, CH2bOH, minor epimer) 2.26–2.22 (m, 2H, CH2a), 1.98–1.92 (m, 2H, H3), 1.91–1.82 (m, 2H, CH2-5 major epimer), 1.69–1.59 (m, 2H, CH2-5), 1.37–1.32 (m, 2H, CH2b). 13C NMR (125 MHz, CD3OD): δ 74.88 (C1, minor epimer), 74.58 (C1, major epimer), 71.82 (C4 major epimer), 71.63 (C4, minor epimer), 65.44 (CH2OH major epimer), 64.66 (minor epimer), 50.18 (C3), 44.83 (CH2-5), 37.92 (CH2-2, major epimer), 37.82 (CH2-2, minor epimer). MS (ES+) m/z: 155.1 (M+Na+). HRMS TOF MS ES+ for C6H12O3Na: calculated: 155.0684, found: 155.0672.
4.1.7. Synthesis of 2-hydroxymethyl-3-hydroxycyclopentanol (15), α-epimer
To a solution of 13 in EtOH (C SM = 0.014 M) was added Pd/C 10% (1 mg/mmol of 13) and the reaction mixture was stirred at room temperature under H2 atmosphere until all 13 was consumed (1–2 h). Pd was filtrated through a pad of Celite and solvents were evaporated to dryness. To afford the desired final compounds 15 in quantitative yield.
1H NMR (500 MHz, CD3OD): δ 3.95 (dt, J = 6.0, 5.5 Hz, 2H, H1 and H3), 3.68 (d, J = 5.0 Hz, 2H, CH2OH), 1.88–1.71 (m, 5H, 2× CH2, H2). 13C NMR (125 MHz, CD3OD): δ 74.42 (C1, C3) 62.25 (CH2OH), 59.39 (C2), 32.98 (C4, C5). MS (ES+) m/z: 155.1 (M+Na+). HRMS TOF MS ES+ for C6H12O3Na: calculated: 155.0684, found: 155.0674.
4.1.8. General method for the condensation of the desired phosphorochloridate on carbocycle 11 followed by hydrogenation
To a solution of carbocycle 11 (200 mg, 0.5 mmol) in toluene (C SM = 0.14 M) was added dropwise 2.5% of NMI (1 μL). The reaction mixture was then cooled down to 0 °C before addition of 3 equiv of Et3N dropwise followed by 3 equiv of the desired phosphorochloridate. The reaction was then monitored by 31P NMR until no more evolution was observed (addition of more equivalents of Et3N and phosphorochloridate was sometimes necessary). The crude was then purified by column chromatography (Eluent: PE/AcOEt 7:3 to 5:5) and the fractions containing signals between 1 and 3 ppm (31P NMR) were combined and are called intermediate A.
To a solution of intermediate A in EtOH (C SM = 0.014 M) was added Pd/C 10% (1 mg/mmol of A) and the reaction mixture was stirred at room temperature under H2 atmosphere until all the intermediate A was consumed (1–2 h). Pd was filtrated through a pad of Celite and solvents were evaporated to dryness. Purification of the crude by preparative TLC (Eluent: AcOEt/MeOH 95:5) gave the desired final compounds 16a–f with yields comprised between 2% and 13%.
4.1.8.1. (3S,4R)-3-Hydroxy-4-(hydroxymethyl)cyclopentyl-1-O-phenyl-(methoxy-l-alaninyl)-phosphate (16a)
This compound has been synthesized according to the method described above and was obtained with 13% yield as a α/β/P S/P R mixture (4 diastereoisomers). 1H NMR (500 MHz, MeOD): δ 7.37–7.34 (m, 2H, ArH), 7.24–7.16 (m, 3H, ArH), 5.06–4.87 (m, 1H, H1), 4.12–4.08 (ddd, J = 6.0, 6.0, 12.5 Hz, 0.2H, H3), 4.04 (ddd, J = 6.2, 6.2, 12.5 Hz, 0.3H, H3 of one diastereoisomer), 3.98–3.90 (m, 1.5H, CH Ala+H3 of one diastereoisomer), 3.69 (s, OCH3 of one diastereoisomer), 3.68 (s, OCH3 of one diastereoisomer), 3.68 (s, OCH3 of one diastereoisomer), 3.67 (s, OCH3 of one diastereoisomer), 3.65–3.60 (m, 1H, CH 2OH), 3.57–3.47 (m, 1H, CH 2OH), 2.43–2.30 (m, 1H, H5, H2), 2.25–2.14 (m, 0.8H, H5, H4, H2), 2.12–2.05 (m, 0.5H, H5, H2), 2.00–1.90 (m, 1H, H5, H4), 1.85 (m, 0.5H, H5), 1.78–1.70 (m, 0.6H, H2), 1.66–1.61 (m, 0.6H, H2), 1.35 (dd, J = 7.1, 1.1 Hz, CH3Ala of one diastereoisomer), 1.34 (dd, J = 7.1, 1.1 Hz, CH3Ala of one diastereoisomer), 1.31 (d, J = 7.1 Hz, CH3Ala of one diastereoisomer), 1.30 (d, J = 7.0 Hz, CH3Ala of one diastereoisomer). 13C NMR (126 MHz, MeOD): δ 175.5 (C O), 175.5 (C O), 175.4 (C O), 175.4 (C O), 152.4–152.3 (Cq), 130.7, 126.0, 125.9, 121.5, 121.5, 121.5, 121.4 (CAr), 79.3 (m, C1 of four diastereoisomers), 74.1 (C3 of one diastereoisomer), 74.1 (C3 of one diastereoisomer), 74. (C3 of one diastereoisomer), 73.9 (C3 of one diastereoisomer), 64.9 (CH 2OH of one diastereoisomer), 64.8 (CH 2OH of one diastereoisomer), 64.0 (CH 2OH of one diastereoisomer), 63.9 (CH 2OH of one diastereoisomer), 52.7 (OCH3 of two diastereoisomers), 52.7 (OCH3 of two diastereoisomers), 51.6 (CH Ala of one diastereoisomer), 51.6 (d, J = 1.6 Hz, CH Ala of one diastereoisomer), 51.5 (CH Ala of one diastereoisomer), 51.4 (CH Ala of one diastereoisomer), 49.8 (C4 of one diastereoisomer), 49.7 (of one diastereoisomer), 49.6 (C4 of one diastereoisomer), 49.5 (C4 of one diastereoisomer), 43.7 (d, J = 4.5 Hz, C2 of one diastereoisomer), 43.6 (d, J = 5.1 Hz, C2 of one diastereoisomer), 43.4 (d, J = 3.9 Hz, C2 of one diastereoisomer), 43.3 (d, J = 5.3 Hz, C2 of one diastereoisomer), 36.7 (d, J = 4.5 Hz, C5 of one diastereoisomer), 36.6 (d, J = 5.4 Hz, C5 of one diastereoisomer), 36.2 (d, J = 4.5 Hz, C5 of one diastereoisomer), 36.1 (d, J = 5.5 Hz, C5 of one diastereoisomer), 20.5 (CH3Ala of one diastereoisomer), 20.5 (CH3Ala of one diastereoisomer), 20.4 (d, J = 4.5 Hz, CH3Ala of one diastereoisomer), 20.3 (t, J = 4.4 Hz, CH3Ala of one diastereoisomer). 31P NMR (202 MHz, MeOD): δ 2.70, 2.64, 2.48, 2.34. MS (ES+) m/z: 396.1 (M+Na+). HRMS TOF MS ES+ for C16H24NO7NaP: calculated: 396.1188, found: 396.1178. Reverse HPLC eluting with H2O/MeOH from 90:10 to 0:100 in 25 min: t R = 14.04, 14.24, 14.47, 14.63 min (94%).
4.1.8.2. (3S,4R)-3-Hydroxy-4-(hydroxymethyl)cyclopentyl-1-O-phenyl-(methoxy-l-valinyl)-phosphate (16b)
This compound has been synthesized according to the method described above and was obtained with 8% yield as an α-epimers mixture according the NOESY experiments (no signal between H1 and H4 on NOESY experiments). The ratio P S/P R is 1:0.5 but we do not know which one is P S and which one is P R. 1H NMR (500 MHz, MeOD): δ 7.34 (dd, J = 8.1, 7.5 Hz, 2H, ArH), 7.23–7.20 (m, 2H, ArH), 7.18–7.15 (m, 1H, ArH), 4.89 (m, 1H, H1 of both diastereoisomers), 3.96–3.91 (m, 1H, H3, of both diastereoisomers), 3.66 (s, OCH3 of one diastereoisomer), 3.66 (s, OCH3 of one diastereoisomer), 3.63 (m, 2H, NCH, CH 2OH), 3.52 (m, 1H, CH 2OH), 2.42–2.35 (m, 1H, H2), 2.22–2.05 (m, 2H, H4, H5), 2.03–1.96 (m, 1H, H4, CH(CH3)2), 1.85 (m, 1H, H2), 1.79–1.69 (m, 1H, H5), 0.93 (d, J = 6.8 Hz, CH(CH 3)2 of one diastereoisomer), 0.92 (d, J = 6.8 Hz, CH(CH 3)2 of one diastereoisomer), 0.90 (d, J = 6.8 Hz, CH(CH 3)2 of one diastereoisomer), 0.88 (d, J = 6.8 Hz, CH(CH 3)2 of one diastereoisomer). 13C NMR (126 MHz, MeOD): δ 174.8 (d, J = 3.3 Hz, C O of one diastereoisomer), 174.6 (d, J = 3.5 Hz, C-O of one diastereoisomer), 152.4 (Cq) of one diastereoisomer, 152.3 (Cq of one diastereoisomer), 130.6, 125.9, 121.5, 121.4 (CAr), 79.4 (d, J = 6.2 Hz, C1 of one diastereoisomer), 79.2 (d, J = 6.3 Hz, C1 of one diastereoisomer), 74.1 (C3 of both diastereoisomers), 64.0 (CH 2OH of one diastereoisomer), 63.9 (CH 2OH of one diastereoisomer), 61.9 (NCH of one diastereoisomer), 61.8 (NCH of one diastereoisomer), 52.4 (OCH3 of one diastereoisomer), 52.3 (OCH3 of one diastereoisomer), 49.6 (C4 of one diastereoisomer), 49.5 (C4 of one diastereoisomer), 43.7 (d, J = 4.1 Hz, C2 of one diastereoisomer), 43.6 (d, J = 5.1 Hz, C2 of one diastereoisomer), 36.7 (d, J = 4.6 Hz, C5 of one diastereoisomer), 36.7 (d, J = 45.9 Hz, C5 of one diastereoisomer), 33.3 (d, J = 6.7 Hz, CH(CH3)2 of one diastereoisomer), 33.2 (d, J = 7.1 Hz, CH(CH3)2 of one diastereoisomer), 19.5 (CH(CH 3)2 of one diastereoisomer), 19.5 (CH(CH 3)2 of one diastereoisomer), 18.4 (CH(CH 3)2 of one diastereoisomer), 18.3 (CH(CH 3)2 of one diastereoisomer). 31P NMR (202 MHz, MeOD): δ 3.39, 3.36. MS (ES+) m/z: 424 (M+Na+); HRMS TOF MS ES+ for C18H28NO7NaPNa: calculated: 424.1501, found: 424.1499. Reverse HPLC eluting with H2O/MeOH from 90:10 to 0:100 in 25 min: t R = 16.91, 17.29 min (96%).
4.1.8.3. (3S,4R)-3-Hydroxy-4-(hydroxymethyl)cyclopentyl-1-O-naphthyl-(neopentoxy-l-alaninyl)-phosphate (16c)
This compound has been synthesized according to the method described above and was obtained with 2% yield as a mixture of two diastereoisomers. 1H NMR (500 MHz, MeOD): δ 8.18–8.16 (m, 1H, ArH), 7.90–7.88 (m, 1H, ArH), 7.70 (d, J = 8.3 Hz, 1H, ArH), 7.55 (m, 2H, ArH), 7.49–7.41 (m, 2H, ArH), 5.11–5.03 (m, 1H, H1), 4.08–3.97 (m, 2H, H3, NCH), 3.81, 3.79, 3.77, 3.71 (two AB systems, J = 10.5 Hz, 2H, CH2neopentyl), 3.66–3.34 (m, 2H, CH 2OH), 2.35–2.31 (m, 1H, H5), 2.24–2.17 (m, 0.5H, H4, H2), 2.07 (m, 0.5H, H2), 1.99–1.82 (m, 2H, H4, H2), 1.68–1.58 (m, 1H, H5), 1.36 (d, J = 7.5 Hz, CH3Ala of one diastereoisomer), 1.34 (d, J = 7.5 Hz, CH3Ala of one diastereoisomer), 0.93, 0.90 (two s, 9H, C(CH3)3 of both diastereoisomers). 13C NMR (126 MHz, MeOD): δ 175.1 (d, J = 4.6 Hz, C O of one diastereoisomer), 175.0 (d, J = 4.6 Hz, C O of one diastereoisomer), 148.2 (d, J = 2.7 Hz, Cq of one diastereoisomer), 148.1 (d, J = 2.8 Hz, Cq of one diastereoisomer), 136.32 (Cq), 128.9, 128.8 (CAr), 128.0 (d, J = 2.8 Hz, Cq), 128.0 (d, J = 3.5 Hz, Cq), 127.7, 127.7, 127.4, 127.3, 126.5, 126.5, 125.8, 122.9, 122.8, 116.36 (CAr), 116.3 (d, J = 3.4 Hz, CAr of one diastereoisomer), 116.2 (d, J = 3.0 Hz, CAr of one diastereoisomer), 79.5 (d, J = 2.5 Hz, C1 of one diastereoisomer), 79.4 (d, J = 2.5 Hz, C1 of one diastereoisomer), 75.4 (CH2neopentyl of one diastereoisomer), 75.3 (CH2neopentyl of one diastereoisomer), 74.0 (C3 of one diastereoisomer), 73.9 (C3 of one diastereoisomer), 64.9 (CH2OH of one diastereoisomer), 64.9 (CH2OH of one diastereoisomer), 51.8 (NCH), 49.8 (C4 of one diastereoisomer), 49.7 (C4 of one diastereoisomer), 43.5 (d, J = 3.8 Hz, C2 of one diastereoisomer), 43.4 (d, J = 5.0 Hz, C2 of one diastereoisomer), 36.3 (d, J = 3.8 Hz, C5 of one diastereoisomer), 36.2 (d, J = 6.0 Hz, C5 of one diastereoisomer), 32.3 (Cq of one diastereoisomer), 32.3 (Cq of one diastereoisomer) 26.7 (C(CH 3)3 of one diastereoisomer), 26.7 (C(CH 3)3 of one diastereoisomer), 20.7 (d, J = 6.9 Hz, CH3Ala of one diastereoisomer), 20.6 (d, J = 7.1 Hz, CH3Ala of one diastereoisomer). 31P NMR (202 MHz, MeOD): δ 3.03, 2.84. MS (ES+) m/z: 480.21 (M+H+), HRMS TOF MS ES+ for C24H34NO7NaP: calculated: 480.2151, found: 480.2141 Reverse HPLC eluting with H2O/MeOH from 90:10 to 0:100 in 25 min: t R = 22.72 min (94%).
4.1.8.4. (3S,4R)-3-Hydroxy-4-(hydroxymethyl)cyclopentyl-1-O-naphthyl-(cyclohexoxy-l-alaninyl)-phosphate (16d)
This compound has been synthesized according to the method described above and was obtained with 2% yield as a α/β/P S/P R mixture (4 diastereoisomers). 1H NMR (500 MHz, MeOD): δ 8.20–8.13 (m, 1H, ArH), 7.92–7.86 (m, 1H, ArH), 7.73–7.68 (m, 1H, ArH), 7.61–7.41 (m, 4H, ArH), 5.13–4.91 (m, 1H, H1 of all diastereoisomers), 4.77–4.63 (m, 1H, H3 of all diastereoisomers), 4.08–3.92 (m, 2H, CH Ala, CH cyclohexyl), 3.67–3.38 (m, 2H, CH 2OH), 2.43–2.30 (m, 1H, H5), 2.24–2.05 (m, 1.4H, H2, H4), 2.00–1.86 (m, 2H, H2, H4), 1.85–1.46 (m, 5.6H, CH2cyclohexyl, H5), 1.44–1.21 (m, 8H, CH2cyclohexyl, CH3Ala). 13C NMR (126 MHz, MeOD): δ 174.6 (d, J = 3.2 Hz, C O of one diastereoisomer), 174.5 (d, J = 3.3 Hz, C O of one diastereoisomer), 174.3, (C O), 148.2 (d, J = 2.7 Hz, Cq of one diastereoisomer), 148.1 (d, J = 2.8 Hz, Cq of one diastereoisomer), 136.4 (Cq), 128.9, 128.9 (CAr), 128.0, 128.0 (Cq), 127.8, 127.7, 127.7, 127.7, 127.4, 127.4, 127.3, 126.7, 126.6, 126.5, 125.8, 125.8, 125.7, 122.9, 122.9, 122.81 (CAr), 116.3 (d, J = 3.2 Hz, CAr), 116.3 (d, J = 3.5 Hz, CAr), 116.2 (d, J = 3.5 Hz, CAr), 79.6 (C1 of one diastereoisomer), 79.5 (d, J = 3.4 Hz, C1 of one diastereoisomer), 79.4 (d, J = 5.7 Hz, C1 of one diastereoisomer), 74.9 (C3 of one diastereoisomer), 74.9 (C3 of one diastereoisomer), 74.8 (C3 of one diastereoisomer), 74.7 (C3 of one diastereoisomer), 74.1 (CH cyclohexyl of one diastereoisomer), 74.1 (CH cyclohexyl of one diastereoisomer), 74.1 (CH cyclohexyl of one diastereoisomer), 73.9 (CH cyclohexyl of one diastereoisomer), 64.9 (CH2OH of one diastereoisomer), 64.9 (CH2OH of one diastereoisomer), 64.0 (CH2OH of one diastereoisomer), 63.9 (CH2OH of one diastereoisomer), 51.9 (CH Ala of all diastereoisomers), 49.9 (C4 of one diastereoisomer), 49.8 (C4 of one diastereoisomer), 49.8 (C4 of one diastereoisomer), 49.7 (C4 of one diastereoisomer), 43.8 (d, J = 5.0 Hz, C2 of one diastereoisomer), 43.6 (d, J = 4.7 Hz, C2 of one diastereoisomer), 43.5 (d, J = 3.8 Hz, C2 of one diastereoisomer), 43.5 (d, J = 4.9 Hz, C2 of one diastereoisomer), 36.8 (d, J = 5.5 Hz, C5 of one diastereoisomer), 36.7 (d, J = 5.5 Hz, C5 of one diastereoisomer), 36.3 (d, J = 4.3 Hz, C5 of one diastereoisomer), 36.2 (d, J = 5.9 Hz, C5 of one diastereoisomer), 32.5 (CH2cyclohexyl), 32.5 (CH2cyclohexyl), 32.4 (CH2cyclohexyl), 32.4 (CH2cyclohexyl), 26.5 (CH2cyclohexyl), 26.4 (CH2cyclohexyl), 24.6 (CH2cyclohexyl), 24.5 (CH2cyclohexyl), 20.78–20.54 (m, CH3Ala of all diastereoisomers). 31P NMR (202 MHz, MeOD): δ 3.04, 3.02, 3.00, 2.90. MS (ES+) m/z: 514 (M+Na+); Reverse HPLC eluting with H2O/MeOH from 90:10 to 0:100 in 25 min: t R = 22.91 min (95%).
4.1.8.5. (3S,4R)-3-Hydroxy-4-(hydroxymethyl)cyclopentyl-1-O-phenyl-(cyclohexoxy-l-alaninyl)-phosphate (16e)
This compound has been synthesized according to the method described above and was obtained with 13% yield as a α/β/P S/P R mixture (4 diastereoisomers, ratio 0.7:1:0.7:0.7). 1H NMR (500 MHz, MeOD): δ 7.35 (m, 2H, ArH), 7.24–7.16 (m, 3H, ArH), 5.06–4.86 (m, 1H, H1 of all diastereoisomers), 4.78–4.71 (m, 1H, H3), 4.12–4.08 (m, 0.4H, CH cyclohexyl of one diastereoisomer), 4.04 (q, J = 6.2 Hz, 0.4H, CH cyclohexyl of one diastereoisomer), 3.96–3.91 (m, 1.4H, CH cyclohexyl, CH Ala), 3.67–3.59 (m, 1H, CH 2OH), 3.55–3.47 (m, 1H, CH 2OH), 2.43–2.30 (m, 1H, H5), 2.23–2.05 (m, 1.5H, H2, H4), 1.95 (m, 1H, H2, H4), 1.88–1.70 (m, 5H, CH2cyclohexyl, H5), 1.66–1.61 (m, 0.5H, H5), 1.57–1.50 (m, 1H, CH2cyclohexyl), 1.50–1.27 (m, 8H, CH2cyclohexyl, CH3Ala). 13C NMR (126 MHz, MeOD): δ 174.6 (d, J = 5.7 Hz, C O of one diastereoisomer), 174.5 (d, J = 4.9 Hz, C O of one diastereoisomer), 174.5 (d, J = 3.5 Hz, C O of one diastereoisomer), 174.4 (d, J = 3.9 Hz, C O of one diastereoisomer), 152.4, 152.4, 152.4, 152.3 (Cq), 130.7, 126.0, 125.9, 121.5, 121.5, 121.5, 121.5, 121.4, 121.4 (CAr), 79.2 (d, J = 6.0 Hz, C1 of all diastereoisomers), 74.82–74.78 (C3 of all diastereoisomers), 74.1 (CH cyclohexyl of one diastereoisomer), 74.0 (CH cyclohexyl of one diastereoisomer), 73.9 (CH cyclohexyl of one diastereoisomer), 73.9 (CH cyclohexyl of one diastereoisomer), 64.9 (CH2OH of one diastereoisomer), 64.9 (CH2OH of one diastereoisomer), 63.9 (CH2OH of one diastereoisomer), 63.8 (CH2OH of one diastereoisomer), 51.81 (CH Ala of one diastereoisomer), 51.8 (CH Ala of one diastereoisomer), 51.7 (CH Ala of one diastereoisomer), 51.7 (CH Ala of one diastereoisomer), 49.9 (C4 of one diastereoisomer), 49.8 (C4 of one diastereoisomer), 49.7 (C4 of one diastereoisomer), 49.6 (C4 of one diastereoisomer), 43.7 (d, J = 4.5 Hz, C2 of one diastereoisomer), 43.6 (d, J = 5.3 Hz, C2 of one diastereoisomer), 43.4 (d, J = 3.5 Hz, C2 of one diastereoisomer), 43.4 (d, J = 4.7 Hz, C2 of one diastereoisomer), 36.7 (d, J = 4.5 Hz, C6 of one diastereoisomer), 36.6 (d, J = 5.4 Hz, C6 of one diastereoisomer), 36.2 (d, J = 5.2 Hz, C6 of one diastereoisomer), 36.2 (d, J = 6.3 Hz, C6 of one diastereoisomer), 32.5 (CH2cyclohexyl), 32.4 (CH2cyclohexyl), 32.4 (CH2cyclohexyl), 26.4 (CH2cyclohexyl), 24.6 (CH2cyclohexyl), 20.7 (d, J = 6.7 Hz, CH3Ala of one diastereoisomer), 20.6 (d, J = 6.7 Hz, CH3Ala of one diastereoisomer), 20.6 (d, J = 6.9 Hz, CH3Ala of one diastereoisomer), 20.5 (d, J = 6.8 Hz, CH3Ala of one diastereoisomer). 31P NMR (202 MHz, MeOD): δ 2.75, 2.68, 2.58, 2.42. MS (ES+) m/z: 464. HRMS TOF MS ES+ for C21H32NO7NaPNa: calculated: 464.1814, found: 464.1798. Reverse HPLC eluting with H2O/MeOH from 90/10 to 0/100 in 25 min: t R = 19.08, 21.33 min (89%).
4.1.8.6. (3S,4R)-3-Hydroxy-4-(hydroxymethyl)cyclopentyl-1-O-phenyl-(tert-butoxy-l-alaninyl)-phosphate (16f)
This compound has been synthesized according to the method described above and was obtained with 5% yield as a α/β/P S/P R mixture (4 diastereoisomers). 1H NMR (500 MHz, MeOD): δ 7.37–7.33 (m, 2H, ArH), 7.24–7.16 (m, 3H, ArH), 5.04–4.85 (m, 1H, H1), 4.10 (q, J = 6.2 Hz, 0.4H, H3 of one diastereoisomer), 4.05 (q, J = 6.1 Hz, 0.4H, H3 of one diastereoisomer), 3.94 (q, J = 6.8 Hz, 0.2H, H3 of two diastereoisomers), 3.83–3.75 (m, 1H, CH Ala), 3.61 (m, 1H, CH 2OH), 3.52–3.48 (m, 1H, CH 2OH), 2.42–2.30 (m, 1H, H5, H2), 2.20–2.08 (m, 1.2H, H2, H4, H5), 1.99–1.90 (m, 1.5H, 2H, H4, H5), 1.89–1.71 (m, 0.5H, H5, H2), 1.67–1.61 (m, 0.8H, H2), 1.46, 1.44, 1.44 (three s, 9H, C(CH 3)3), 1.31 (dd, J = 0.9, 7.1 Hz, CH3Ala), 1.29 (dd, J = 0.9, 7.1 Hz, CH3Ala). 13C NMR (126 MHz, MeOD): δ 174.5 (d, J = 5.5 Hz, C O), 174.4 (d, J = 5.5 Hz, C O), 174.3 (d, J = 5.5 Hz, C O), 174.2 (d, J = 5.5 Hz, C O), 152.5 (Cq), 152.5 (Cq), 152.4 (Cq), 152.4 (Cq), 130.7, 130.7, 125.6.0 126.0, 125.9, 125.9, 121.6, 121.6, 121.5, 121.5, 121.5, 121.4 (CAr), 82.6, 82.6, 82.6, 82.6 (C(CH3)3 of all diastereoisomers), 79.2 (d, J = 5.9 Hz, C1), 79.2 (d, J = 6.2 Hz, C1), 74.1 (C3 of one diastereoisomer), 74.1 (C3 of one diastereoisomer), 74.0 (C3 of one diastereoisomer), 73.9 (C3 of one diastereoisomer), 64.9 (CH2OH of one diastereoisomer), 64.9 (CH2OH of one diastereoisomer), 64.0 (CH2OH of one diastereoisomer), 63.9 (CH2OH of one diastereoisomer), 52.3 (d, J = 1.1 Hz, CH Ala of one diastereoisomer), 52.2 (CH Ala of one diastereoisomer), 52.2 (CH Ala of one diastereoisomer), 52.2 (CH Ala of one diastereoisomer), 49.8 (C4 of one diastereoisomer), 49.8 (C4 of one diastereoisomer), 49.7 (C4 of one diastereoisomer), 49.5 (C4 of one diastereoisomer), 43.7 (d, J = 4.6 Hz, C2 of one diastereoisomer), 43.7 (d, J = 5.3 Hz, C2 of one diastereoisomer), 43.5 (d, J = 3.6 Hz, C2 of one diastereoisomer), 43.4 (d, J = 3.6 Hz, C2 of one diastereoisomer), 36.7 (d, J = 4.3 Hz, C5 of one diastereoisomer), 36.7 (d, J = 4.7 Hz, C5 of one diastereoisomer), 36.3 (d, J = 4.3 Hz, C5 of one diastereoisomer), 36.2 (d, J = 5.5 Hz, C5 of one diastereoisomer), 28.3 (C(CH 3)3), 28.3 (C(CH 3)3), 20.8 (d, J = 6.3 Hz, CH3Ala of one diastereoisomer), 20.8 (d, J = 6.3 Hz, CH3Ala of one diastereoisomer), 20.6 (d, J = 6.3 Hz, CH3Ala of one diastereoisomer), 20.6 (d, J = 6.6 Hz, CH3Ala of one diastereoisomer). 31P NMR (202 MHz, MeOD): δ 2.78, 2.71, 2.67, 2.52. MS (ES+) m/z: 438.16 (M+Na+), HRMS TOF MS ES+ for C19H30NO7NaPNa: calculated: 438.1658, found: 438.1641. Reverse HPLC eluting with H2O/MeOH from 90:10 to 0:100 in 25 min: t R = 17.67, 19.51 min (91%).
4.1.8.7. (1R,2R,3S)-3-Hydroxy-2-(hydroxymethyl)cyclopentyl-1-O-naphthyl-(neopentyl-l-alaninyl)-phosphate (17a), α-epimer
This compound has been synthesized from compound 13 according to the method described above and it was obtained with 2% yield over two steps as a mixture of two diastereoisomers. 1H NMR (500 MHz, CD3OD): 8.23–8.17 (m, 1H, naph), 7.90–7.89 (m, 1H, naph), 7.74–7.70 (m, 1H, naph), 7.57–7.44 (m, 4H, naph), 4.80 (m, 1H, CHOP, under the residual H2O solvent peak), 4.09–4.05 (m, 1H, CHCH3), 3.99–3.95 (m, 1H, CHOH), 3.88–3.62 (m, 4H, CH 2OH, OCH 2C(CH3)3), 2.11–1.68 (m, 4H, 2× CH2), 1.38–1.33 (m, 3H, CHCH 3), 0.93, 0.92 (2s, 9H, 2× C(CH3)3). 13C NMR (125 MHz, CD3OD): 175.36 (d, J cp = 4.6 Hz, C O), 175.36 (d, J cp = 6.25 Hz, C O), 148.26 (d, J cp = 3.75 Hz, Cq), 148.21 (d, J cp = 3.62 Hz, Cq), 136.33 (Cq), 136.24 (C naph), 128.86, 128.62 (C naph), 128.05 (d, J cp = 6.25 Hz, Cq naph), 128.98 (d, J cp = 6.25 Hz, Cq naph), 127.75, 127.39, 127.37, 126.56, 125.85, 125.79, 122.93, 122.88 (CH, naph), 116.39 (d, J cp = 3.6 Hz, C naph), 116.20 (d, J cp = 3.6 Hz, C naph), 81.69 (d, J cp = 6.25 Hz, CHOP), 81.62 (d, J cp = 6.25 Hz, CHOP), 73.96, 73.94 (CH2OH), 58.47 (d, J cp = 5.4 Hz, CHCHOP), 58.40 (d, J cp = 6.25 Hz, CHCHOP), 51.89, 51.84 (CHCH3), 31.96 (d, J cp = 4.25 Hz, CH2CHOP), 32.03 (d J = 2.9 Hz, CH2CHOP), 33.19, 33.12 (CH2), 29.23, 29.14 (C(CH3)3), 26.76, 26.75 (C(CH3)3), 20.83 (d, J cp = 6.75 Hz, CH3), 20.68 (d, J cp = 7.0 Hz, CH3). 31P NMR (202 MHz, MeOD): δ 3.29, 3.04. MS (ES+) m/z: 502.19 (M+Na+).
4.1.8.8. (1R,2R,3S)-3-Hydroxy-2-(hydroxymethyl)cyclopentyl-1-O-phenyl-(pentoxy-l-alaninyl)-phosphate (17b), α-epimer
This compound has been synthesized from compound 13 according to the method described above and was obtained with 3% yield over two steps as a mixture of two diastereoisomers. 1H NMR (500 MHz, MeOD): δ 7.43–7.36 (m, 2H, ArH), 7.30–7.21 (m, 3H, ArH), 4.79–4.70 (m, 1H, H1), 4.15–4.09 (m, 2H, OCH 2 ester), 4.01–3.94 (m, 2H, CHCH3, H3), 3.75–3.08 (m, 2H, CH 2OH), 2.10–1.88 (m, 4H, H2, 2× H5, H4), 1.81–1.73 (m, 1H, H4), 1.66–1.61 (m, 2H, CH 2 ester), 1.40–1.34 (m, 7H, 2× CH 2 ester, CHCH 3), 0.95–0.91 (m, 3H, CH 3 ester). 13C NMR (126 MHz, MeOD): δ 175.4 (d, J = 5.5 Hz, C O), 175.1 (d, J = 5.5 Hz, C O), 152.4 (d, J = 6.2, Cq), 131.0, 130.7, 129.9, 126.8, 126.0, 125.9, 121.6, 121.5, 121.4, 121.2 (CHAr), 81.4 (d, J = 6.2 Hz, C1), 81.3 (d, J = 6.2 Hz, C1), 74.0 (C3), 66.4 (OCH2), 61.6 (CH2OH of one diastereoisomer), 61.3 (CH2OH of one diastereoisomer), 58.5 (d, J = 4.0 Hz, C2 of one diastereoisomer), 58.4 (d, J = 4.0 Hz, C2 of one diastereoisomer), 51.73 (CH Ala of one diastereoisomer), 51.70 (CH Ala of one diastereoisomer), 33.2 (C4 of one diastereoisomer), 33.1 (C4 of one diastereoisomer), 32.0 (C5 of one diastereoisomer), 31.9 (C5 of one diastereoisomer), 29.4, 29.2, 29.1, 23.4 (CH2 ester), 20.6 (d, J = 6.5 Hz, CH3Ala of one diastereoisomer), 20.5 (d, J = 6.5 Hz, CH3Ala of one diastereoisomer), 14.3 (CH3 ester). 31P NMR (202 MHz, MeOD): δ 2.96, 2.67. MS (ES+) m/z: 452.20 (M+Na+). Reverse HPLC eluting with H2O/MeOH from 90:10 to 0:100 in 25 min: t R = 18.20, 19.07.
4.2. Biology
4.2.1. Anti-HIV activity assays
Inhibition of HIV-1(IIIB)- and HIV-2(ROD)-induced cytopathicity in CEM and MT-4 cell cultures was measured in microtiter 96-well plates containing ∼3 × 105 cells/mL infected with 100 CCID50 of HIV per milliliter and containing appropriate dilutions of the test compounds. After 4–5 days of incubation at 37 °C in a CO2-controlled humidified atmosphere, CEM giant (syncytium) cell formation was examined microscopically. Viable MT-4 cells were estimated by trypan blue dye exclusion. The EC50 (50% effective concentration) was defined as the compound concentration required to inhibit HIV-induced cytopathic effect by 50%.
4.2.2. Cytostatic activity assays
All assays were performed in 96-well microtiter plates. To each well were added (5–7.5) × 104 tumour cells and a given amount of the test compound. The cells were allowed to proliferate for 48 h (murine leukaemia L1210 cells) or 72 h (human lymphocyte CEM cells) or 96 h (human cervix carcinoma HeLa cells) at 37 °C in a humidified CO2-controlled atmosphere. At the end of the incubation period, the cells were counted in a Coulter counter. The IC50 (50% inhibitory concentration) was defined as the concentration of the compound that inhibited cell proliferation by 50%.
4.3. Carboxypeptidase Y (EC 3.4.16.1) assay
The experiment was carried out by dissolving compound 16d (5.8 mg) in acetone-d 6 (0.15 mL) followed by addition of 0.30 mL of Trizma buffer (pH 7.6). After recording the control 31P NMR at 25 °C, a previously defrosted carboxypeptidase Y (0.1 mg dissolved in 0.15 mL of Trizma) was added to the sample, which was then immediately submitted to the 31P NMR experiments (at 25 °C). The spectra were recorded every 16 min over 13 h. 31P NMR recorded data were processed and analyzed with the Bruker Topspin 2.1 program.
Acknowledgments
We are grateful to Leentje Persoons, Frieda De Meyer, Leen Ingels and Lizette van Berckelaer for excellent technical assistance. N.H. is grateful to Cardiff University for support. The biological work was supported by the KU Leuven (GOA 10/014).
References and notes
- 1.Dwek R.A. Chem. Rev. 1996;96:683. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 2.Heidlas J.E., Williams K.W., Whitesides G.M. Acc. Chem. Res. 1992;25:307. [Google Scholar]
- 3.Heidlas J.E., Lees W.J., Whitesides G.M. J. Org. Chem. 1992;57:152. [Google Scholar]
- 4.Griffiths L., Stratford I.J. Br. J. Cancer. 1997;76:689. doi: 10.1038/bjc.1997.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bronckaers A., Gago F., Balzarini J., Liekens S. Med. Res. Rev. 2009;29:903. doi: 10.1002/med.20159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vande Voorde J., Quintiliani M., McGuigan C., Liekens S., Balzarini J. Biochem. Pharmacol. 2012;83:1358. doi: 10.1016/j.bcp.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Hamon N., Quintiliani M., Balzarini J., McGuigan C. Bioorg. Med. Chem. Lett. 2013;23:2555. doi: 10.1016/j.bmcl.2013.02.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quintiliani M., Balzarini J., McGuigan C. Tetrahedron. 2013;69:9111. [Google Scholar]
- 9.McCasland G.E., Furuta S., Durham L.J. J. Org. Chem. 1966;31:1516. [Google Scholar]
- 10.McCasland G.E., Furuta S., Durham L.J. J. Org. Chem. 1968;33:2835. doi: 10.1021/jo01271a049. [DOI] [PubMed] [Google Scholar]
- 11.McCasland G.E., Furuta S., Durham L.J. J. Org. Chem. 1968;33:2841. doi: 10.1021/jo01271a050. [DOI] [PubMed] [Google Scholar]
- 12.Arjona O., Gómez A.M., López J.C., Plumet J. Chem. Rev. 2007;107:1919. doi: 10.1021/cr0203701. [DOI] [PubMed] [Google Scholar]
- 13.He G.-X., Krise J.P., Oliyai R. In: Stella V.J., Borchardt R.T., Hageman M.J., Oliyai R., Maag H., Tilley J.W., editors. Springer; New York: 2007. Prodrugs; pp. 923–964. (Biotechnology: Pharmaceutical Aspects). [Google Scholar]
- 14.Mehellou Y., Balzarini J., McGuigan C. ChemMedChem. 2009;4:1779. doi: 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- 15.Serpi M., Bibbo R., Rat S., Roberts H., Hughes C., Caterson B., Alcaraz M.J., Gibert A.T., Verson C.R.A., McGuigan C. J. Med. Chem. 2012;55:4629. doi: 10.1021/jm300074y. [DOI] [PubMed] [Google Scholar]
- 16.Huang H., Greenberg M.M. J. Org. Chem. 2008;73:2695. doi: 10.1021/jo702614p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamao K., Ishida N., Ito Y., Kumada M. Org. Synth. 1993:315. [Google Scholar]
- 18.Tamao K., Ishida N., Ito Y., Kumada M. Org. Synth. 1990:96. [Google Scholar]
- 19.Matsuumi M., Ito M., Kobayashi Y. Synlett. 2002:1508. [Google Scholar]
- 20.Ludek O.R., Meier C. Synthesis. 2003:2101. [Google Scholar]
- 21.Cho J.H., Amblard F., Coats S.J., Schinazi R.F. Tetrahedron. 2011;67:5487. doi: 10.1016/j.tet.2011.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jessel S., Meier C. Eur. J. Org. Chem. 2011;2011:1702. [Google Scholar]
- 23.Ludek O.R., Kramer T., Balzarini J., Meier C. Synthesis. 2006:1313. [Google Scholar]
- 24.Brown H.C., Singaram B. J. Org. Chem. 1984;49:945. [Google Scholar]
- 25.See Section 4.
- 26.Derudas M., Carta D., Brancale A., Vanpouille C., Lisco A., Margolis L., Balzarini J., McGuigan C. J. Med. Chem. 2009;52:5520. doi: 10.1021/jm9007856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGuigan C., Gilles A., Madela K., Aljarah M., Holl S., Jones S., Vernachio J., Hutchins J., Ames B., Bryant K.D., Gorovits E., Ganguly B., Hunley D., Hall A., Kolykhalov A., Liu Y., Muhammad J., Raja N., Walters R., Wang J., Chamberlain S., Henson G. J. Med. Chem. 2010;53:4949. doi: 10.1021/jm1003792. [DOI] [PubMed] [Google Scholar]
- 28.Miller, S.; Sculimbrene, B.; Morgan, S. J. Patent US 20,050,267,291 A1, 2005.