

Graphical abstract
This is first report to describe the specific and selective inhibition effects of tanshinones against SARS-CoV 3CLpro and PLpro with deubiquitination. In detail kinetic mechanism, tanshinones manifest a slow-binding inhibition profile.
Abbreviations: IC50, the inhibitor concentration leading to 50% activity loss; Ki, inhibition constant; Kiapp, apparent Ki; k, rate constant; Vmax, maximum velocity; Km, Michaelis-Menten constant; kobs, apparent first-order rate constant for the transition from vi to vs; vi, initial velocity; vs, steady-state rate; SARS, severe acute respiratory syndrome; CoV, coronavirus
Keywords: Tanshinone, SARS-CoV, 3CLpro, PLpro, Slow-binding inhibitor
Abstract
In the search for anti-SARS-CoV, tanshinones derived from Salvia miltiorrhiza were found to be specific and selective inhibitors for the SARS-CoV 3CLpro and PLpro, viral cysteine proteases. A literature search for studies involving the seven isolated tanshinone hits showed that at present, none have been identified as coronaviral protease inhibitors. We have identified that all of the isolated tanshinones are good inhibitors of both cysteine proteases. However, their activity was slightly affected by subtle changes in structure and targeting enzymes. All isolated compounds (1–7) act as time dependent inhibitors of PLpro, but no improved inhibition was observed following preincubation with the 3CLpro. In a detail kinetic mechanism study, all of the tanshinones except rosmariquinone (7) were identified as noncompetitive enzyme isomerization inhibitors. However, rosmariquinone (7) showed a different kinetic mechanism through mixed-type simple reversible slow-binding inhibition. Furthermore, tanshinone I (5) exhibited the most potent nanomolar level inhibitory activity toward deubiquitinating (IC50 = 0.7 μM). Additionally, the inhibition is selective because these compounds do not exert significant inhibitory effects against other proteases including chymotrysin, papain, and HIV protease. These findings provide potential inhibitors for SARS-CoV viral infection and replication.
1. Introduction
Severe acute respiratory syndrome (SARS) is a life-threatening form of atypical pneumonia that first occured in the Guandong province in China and rapidly spread through several countries in other parts of the world in 2003.1 The fatality rate of SARS is rather high. In the first wave of the SARS outbreak, approximately 8500 people worldwide were affected and over 900 died. The reemergence of SARS in south China was reported in December 2003 and again in the spring of 2004, while these instances are not necessarily indicative of another global outbreak, they to illustrate the need to continue to study the virus and develop appropriate therapeutics for its treatments.
SARS-CoV is a single-stranded positive-strand RNA virus, and its genome structure comprises both replicases and structural regions.2 It encodes two overlapping polyproteins, ppla (486 kDa) and pplab (790 kDa).3 The polypeptides are released from each polyprotein through extensive proteolytic processing, primarily performed by the virally encoded chymotrypsin-like protease 3CLpro (also called the main protease, Mpro) with additional cleavage performed by the viral papain-like protease PLpro.4 Because of their essential roles in viral replication, both proteases are recognized as attractive targets for the development of anti-SARS therapeutics.5 Not surprisingly, numerous studies related to the development of SARS-CoV 3CLpro inhibitors have already been reported. The active site of SARS-CoV 3CLpro contains Cys145 and His41, constituting a catalytic dyad in which cysteine functions as the common nucleophile in the proteolytic process.6 Untill now, all of the inhibitor studies have focused on the SARS-CoV 3CLpro and have utilized both synthetic peptidyl libraries and naturally derived libraries. Examples of the peptide-derive inhibitors include C2-symmetric diols,7 quinolinecarboxylic acids,8 and anilides,9 and the naturally derived inhibitors include quinone-methide triterpens,10 and biflavonoids amentoflavone,11 which are included in our screen of natural products with activity against 3CLpro. In contrast, structural and functional studies focused on PLpro are far less numerous but have established important roles for PLpro beyond viral peptide cleavage, including deubiquitination, deISGylation, and involvement in virus evasion of the innate response.12 It is located on polypeptide 1a and undergoes self-cleavage at three sites on the polypeptide. Its catalytic triad (Cys1651-His1812-Asp1826) has a broad range of optimum pH values, characteristic of papain-like cysteine protease.12, 13 To date, only a small number of inhibitors of PLpro have been studied, including molecules identified from high-throughput screening, such as thiopurine analogs,14 benzamide derivatives,15 and the noncovelent inhibitor GRL0617.16 Despite numerous biochemical, strucutral, and inhibitor-development studies directed at 3CLpro and PLpro, potent antiviral that directly target proteases have yet to be developed.
While searching for a SARS-CoV 3CLpro and PLpro inhibitor from natural sources, we found that the lipophilic fraction of a Salvia miltiorrhiza ethanol extract possessed inhibitory activity against both proteases. The Salvia miltiorrhiza root (Laminaceae) has been widly used in China, Korea, Japan, and other Asian countries to treat coronay heart disease, paricularly angina pectoris and myocardial infarction.17 There are two classes of major active compounds in S. miltiorrhiza, namely lipophilic tanshinones and hydrophilic salvianolic acids. Both compounds have been found to exhibit anti-oxidant, anti-inflammation, and anti-viral properties.18 Huang and coworkers isolated two potent, non-toxic HIV-1 intergrase inhibitors from this plant that were salvianolid acid derivatives: lithospermic acid and lithospermic acid B.19 To the best of our knowledge, their application to the inhibition of SARS-CoV’s main cysteine proteases has never been reported.
In this study, we conducted a biological evaluation of seven isolated tanshinones from S. miltiorrhiza and investigated the inhibition selectivity of its constituents on SARS-CoV 3CLpro and PLpro. For the study, we have synthesized and expressed in Escherichia coli (E. coli) the 3CLpro and PLpro as well as open reading frame (ORF) containing only the catalytic domains. The inhibitors were screened against the proteases using a fluorescent peptide substrate, and the steady-state inhibition parameters of the effective inhibitors were evaluated. The mode of action of the inhibitors was further investigated with a kinetic assay and the function of time exposed to the inhibitors. To our knowledge, this structure is the first to show SARS coronaviral cysteine proteases (3CLpro and PLpro) and deubiquitinating (DUB) enzyme activities. Further characterization of these quinoids suggests that they provide good scaffolds for further optimization and drug development against SARS.
2. Results and discussion
2.1. Preparation of SARS-CoV 3CLpro and PLpro for inhibition assay
The numerous functions and requisite roles of SARS-CoV cysteine proteases (3CLpro and PLpro) in viral replication and pathogenesis suggest that they may serve as an attractive target for antiviral drugs. Ultimately, our goal is to develop a naturally derived SARS viral cysteine protease inhibitor for use as an anti-SARS drug. As a first step toward this goal, we expressed SARS-CoV 3CLpro in E. coli BL21 (DE3) CodonPlus-RIL cells, purified the protein to homogeneity, and evaluated a FRET-based peptide cleavage assay to measure the kinetic activity of SARS-CoV 3CLpro. The purification consisted of a combination of hydrophobic interaction and anion exchange chromatography and resulted in a purified single protein with a weight of approximately 33 kDa. The peptide substrate utilized was KNSTLQSGLRKE labelled with Dabcyl and Edans. We also expressed and purified SARS-CoV PLpro (36 kDa) from E. coli BL21 (DE3) CodonPlus-RIL. PLpro, located within nsp3, cleaves at the nsp1/2, nsp2/3 and nsp3/4 boundaries using the consensus motif LXGG. Therefore, to identify potential inhibitors of PLpro, we used a sensitive fluorescence screen developed during our initial work, which used a commercially available peptide substrate to represent the C-terminal residues of LXGG with a C-terminal 7-amido-4-methylcoumarin (AMC) fluorogenic reporter group. In this study, the SARS-CoV cysteine proteases’ inhibitory activity was evaluated with the above expressed proteins and substrates following the general procedure previously described.11, 16 The inhibition of proteases slowly increased with increasing concentrations of the corresponding compounds. The IC50 value of the compounds, which expresses their ability to inhibit the catalytic activity of the protease, was calculated by fitting the dose–response curve to a logistic derivative equation.
2.2. Isolation and identification of tanshinones
In the preliminary study, we confirmed the SARS-CoV 3CLpro and PLpro inhibitory activities of an ethanol extract of Salvia miltiorrhiza. The ethanol extract exhibited 60% and 88% inhibition to 3CLpro and PLpro, respectively, at 30 μg/mL. The main constituents of this plant are a number of tanshinones, which include lipophilic compounds such as diterpene quinolones and hydrophilic phenolics such as salvianolic acid derivatives. For partition of crude extract, it fractionated with organic solvents (n-hexane, ethyl acetate, and H2O) of the different polarities. Among the partitioned fractions of the ethanolic extracts, the n-hexane-soluble fraction showed noticeable protease inhibition activity. Activity-guided fraction of the lipophilic n-hexane fraction yielded seven tanshinones (1–7), which were purified over silica gel, Sephadex LH-20, and octadecyl-functionalized silica gel. as delineated below. The isolated tanshinones (1–7) were identified as the known species tanshinone IIA (1), tanshinone IIB (2), methyl tanshinonate (3), cryptotanshinone (4), tanshinone I (5), dihydrotanshinone I (6), and rosmariquinone (7) by analysis of spectroscopic data and comparison with previous studies (Fig. 1 ).20
Figure 1.
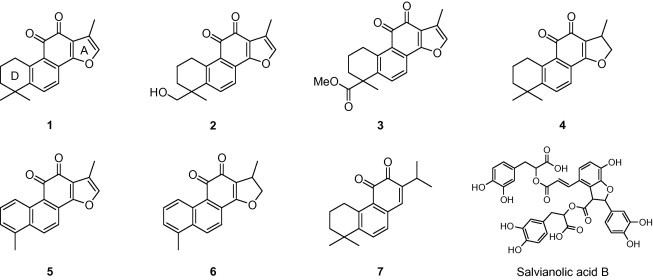
Chemical structures of isolated tanshinones from S. miltiorrhiza lipophilic fraction and representative hydrophilic compound, salvianolic acid B.
2.3. Effects of isolated tanshinones on the activity of SARS-CoV 3CLpro
The isolated tanshinones (1–7) were evaluated in-house for their SARS-CoV 3CLpro and PLpro inhibitory activities, which in turn allowed us to develop a new class inhibitor with increased selectivity. The results of the inhibition studies of both proteases are expresses as IC50 values and are reported in Table 1 . First, the inhibitory potencies and capacities of these tanshinones toward SARS-CoV 3CLpro were investigated. The SARS-CoV 3CLpro inhibitory activity was assayed by following the proteolysis of the fluorogenic substrate in the presence or absence of the test compounds, as described in our previous studies.11 As shown in Table 1, all isolated tanshinones examined, apart from cryptotanshinone (4), exhibited a dose-dependent inhibitory effect on 3CLpro (Fig. 2 A); however, the compounds did not have time-dependent inhibition profiles. No improved inhibitory activity was observed after preincubation of the inhibitors with 3CLpro. Salvianolic acid B, a representative bioactive compound in the hydrophilic fraction, did not show a significant inhibitory effect (30% at 200 μM). We found that the activity of these compounds ranged from 14.4 to 89.1 μM against 3CLpro. Importantly, dihydrotanshinone I (6) showed extremely high inhibition with an IC50 value of 14.4 μM. In addition, the activity was slightly affected by subtle changes in structure. To identify the active inhibitory pharmacophore, a structure–function relationship study was carried out. Initially, the presence of naphthalene in the diterpene quinolone backbone appeared to be important for activity in comparison to the presence of dimethyl tetranaphthalene. For example, 1 and 4 contain dimethyl tetranaphthalene and have IC50 values of 89.1 and 226.7 μM, respectively, while 5 and 6 contain naphthalene and showed IC50 values of 38.7 and 14.4 μM. Subsequently, when we compared a series of tanshinone II analogs (1–4), we found that the introduction of a dihydrofuran moiety on the A-ring to this group (4) decreases the inhibitory activity ∼2-fold, which is in contrast to the results obtained with inhibitors 1–3 that have furan moiety. The hydroxymethyl group on the D-ring derivative 2 exhibited improvement of enzyme inhibitory activity with an IC50 value of 24.8 μM. The corresponding methyl ester on the D-ring of derivative 3 also showed a similar potency enhancement (IC50 = 21.1 μM). In contrast, the dihydrofuran moiety of tanshinone I (6, IC50 = 14.4 μM) turned out to be more potent than furan (5, IC50 = 38.7 μM) within the series of tanshinone I analogs (5 and 6).
Table 1.
Inhibitory effects of isolated compounds 1–7 on SARS-CoV cysteine proteases
| Compound | 3CLpro | PLpro |
|---|---|---|
| IC50a (μM) | IC50a (μM) | |
| 1 | 89.1 ± 5.2 | 1.6 ± 0.5b (17.1 ± 5.0)c |
| 2 | 24.8 ± 0.8 | 10.7 ± 1.7 (18.0 ± 1.2) |
| 3 | 21.1 ± 0.8 | 9.2 ± 2.8 (10.6 ± 0.6) |
| 4 | 226.7 ± 6.2 | 0.8 ± 0.2 (10.1 ± 2.2) |
| 5 | 38.7 ± 8.2 | 8.8 ± 0.4 (11.5 ± 1.5) |
| 6 | 14.4 ± 0.7 | 4.9 ± 1.2 (10.7 ± 2.3) |
| 7 | 21.1 ± 0.8 | 30.0 ± 5.5 (78.9 ± 0.8) |
All compounds were examined in a set of triplicates experiment.
An IC50 value of inhibitor exposed by 30 min preincubation.
An IC50 value of inhibitor with no preincubation.
Figure 2.
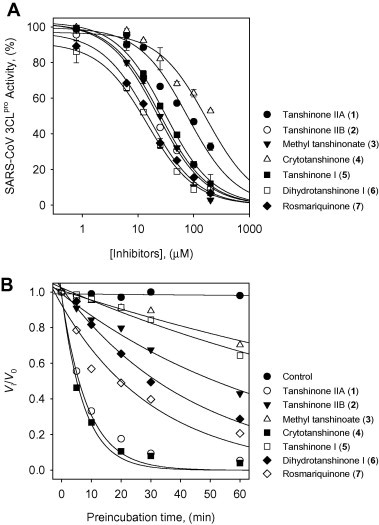
(A) Effects of compounds 1–7 on the activity of SARS-CoV 3CLpro for proteolysis of substrate. (B) SARS-CoV PLpro inhibition as a function of preincubation time for compounds 1–7 at IC50 value.
2.4. Effects of isolated tanshinones on the activity of SARS-CoV PLpro
Compounds 1–7 were tested against SARS-CoV PLpro using a continuous fluorometric assay during which an increase in fluorescence (λ ex 360 nm, λ em 460 nm) was monitored using a spectrofluorimeter. The initial rates were calculated from the first 5 min of the progress curves. Interestingly, all of the isolated compounds showed a time-dependent inhibitory effect on PLpro activity, which is the first report of naturally derived inhibitors of PLpro. As can be observed from the data presented in Table 1 and Figure 2B, when the isolated compounds from S. miltiorrhiza were preincubated for 60 min, they showed more inhibition than those without incubation. Most of these tanshinones show an increased inhibition of the enzymatic activity of PLpr°Compared to 3CLpro in the nanomolar or micromolar range. As evidenced from the PLpro inhibition, the dimethyl tetrahydronaphthalen structure showed a more potent inhibitory response than the naphthalene chymotype. Of the isolated tanshinones, cryptotanshinone (4) displayed the most potent inhibitory activity (IC50 = 0.8 μM). By comparing the series of tanshinone II derivatives, it was shown that the furan moiety on the A-ring does not show improved inhibition with PLpro. This result was unexpected because of the increased inhibition of 3CLpro with 1 compared to that of 4. Tanshinone IIA (1, IC50 = 1.6 μM) showed a two-fold lower inhibition effect than cryptotanshinone (4). Similar trends can also be observed with tanshinone I (5, IC50 = 8.8 μM) and dihydrotanshinone I (6, IC50 = 4.9 μM), which showed respectively similar inhibitory activities because they have similar A-ring structures. Other tanshinone II analogs 2 (IC50 = 10.7 μM) and 3 (IC50 = 9.2 μM) suggested that the presence of a hydroxymethyl and methyl ester on the D-ring has a weak effect on inhibition potency. Interestingly, the abietane analog, rosmariquinone (7), displayed significant activity against both SARS-CoV cysteine proteases with IC50 values of 21.1 and 30.0 μM. The abietane skeleton is structurally related to the tanshinones, and its activity is noteworthy, suggesting that further research on this important lead compound is needed.
2.5. Time-dependent inhibitory effects of tanshinones on SARS-CoV PLpro
To investigate the inhibition mechanism, the time dependence of the inhibition of the proteolysis of Arg-Leu-Arg-Gly-Gly-AMC (RLRGG-AMC) by these inhibitors was subsequently probed. Reaction progress curves in the presence of varied concentrations of the isolated tanshinones are illustrated. Taken as an ensemble, the residual enzyme activity decreases as a function PLpro/tanshinones incubation time in Figure 3 . Representatively, compound 4 show a clear time- and concentration-dependent approach to the steady-state because it curves downwards, which is characteristic of slow-binding inhibition kinetics (Fig. 3A and Fig. S50). All of the PLpro inhibitors (1–7) manifested the same relationship for the enzyme activity. Slow-binding inhibition mechanisms can be investigated by preincubation of the enzyme with the inhibitor, followed by measurement of the initial velocities for substrate proteolysis as a function of preincubation time. Increasing the concentration of compound 4 led to a decrease in both the initial velocity (v i) and the steady-state rate (v s). The progress curves obtained using various concentrations of the inhibitors were fit to (Eq. 1) to determine v i, v s, and k obs. The plot represents the relationship between k obs and [I] for all possible inhibition modes (simple reversible slow-binding, enzyme isomerization, and mechanism-based inhibition) to determine which kinetic profile was at play. The kinetic parameters (k 3, k 4, k 5, k 6, and K i app) were derived from the plots by fitting the results (Table 2 ). The results of all the inhibitors were fit to the slow-binding (Eq. 2), illustrating a hyperbolic dependence of kobs on the concentration of PLpro by tanshinones except for rosmariquinone (7). Based on the above kinetic observations, we believed that the reaction undergoes the following kinetic mechanism (Fig. 3B and Fig. S51). The hyperbolic dependence observed with the inhibitors suggests that they inhibit PLpro by rapid formation of an enzyme complex (E·I), which slowly isomerizes to from a modified enzyme complex (E∗·I) (Fig. 4 ). This is expected to sustain an inhibitory process longer than conventional inhibitors because k 5 is higher than k 6. Interestingly, the only abietane-containing structure, rosmariquinone (7), found in the isolated compounds was shown to be time dependent because k obs exhibited a linear dependence on the inhibitor concentration as depicted in Figure 3C. Thus, this result proves that the abietane type compound 7 from S. miltiorrhiza inhibits SARS-CoV PLpro by the simple reversible slow-binding model according to the data yielded by the analysis by Eqs. (3), (4) as listed in Table 2. This conclusion suggests that the four-ringed inhibitors may have a different binding mode than the three-ringed inhibitor. Additionally, these compounds represent a new class of SARS-CoV PLpro inhibitors and, to our knowledge, are the first naturally derived, potent and time-dependent inhibitors of this enzyme.
Figure 3.
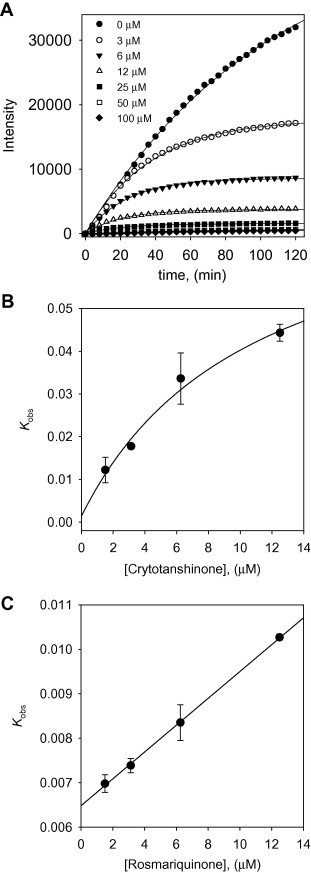
(A) Time course of substrate proteolyzed by PLpro in the presence of compound 4. (B) Dependence of the values for kobs on the concentrations of compound 4. (C) Dependence of the values for kobs on the concentrations of compound 7. The kobs values were fitted to Eqs. (2), (3).
Table 2.
Kinetic parameters for time dependent inhibition of PLpro by compounds 1–7a
| Compound | k5 (min−1) | k6 (min−1) | Kiapp (μM) |
|---|---|---|---|
| 1b | 0.04825 ± 0.006 | 0.008485 ± 0.0001 | 11.2 ± 0.1 |
| 2 | 0.02503 ± 0.003 | 0.004704 ± 0.0002 | 21.6 ± 2.0 |
| 3 | 0.01930 ± 0.001 | 0.002657 ± 0.0002 | 9.7 ± 0.7 |
| 4 | 0.08127 ± 0.001 | 0.001406 ± 0.0001 | 9.0 ± 0.2 |
| 5 | 0.03015 ± 0.003 | 0.003349 ± 0.0003 | 13.7 ± 0.1 |
| 6 | 0.03203 ± 0.001 | 0.003175 ± 0.0003 | 11.2 ± 0.0 |
| 7c | k3 (μM−1min-1) | k4 (min−1) | Kiapp (μM) |
| 0.00030 ± 0.00005 | 0.006483 ± 0.0004 | 21.5 ± 1.3 | |
Figure 4.
Scheme for time dependent enzyme inhibition. The upper part denoted the turnover of the enzyme in the absence of inhibition. The lower part illustrates the equilibrium for a slow-binding inhibition process. In simple reversible slow-binding inhibition process that the low values of k3 and k4 relative to enzyme turnover. In enzyme isomerisation, an initial binding of the inhibitor to the enzyme leads to formation of the EI complex, which undergoes an isomerisation of the enzyme to form the new complex E∗I.
2.6. Determination of the inhibition type of tanshinones on SARS-CoV 3CLpro and PLpro
Next, we studied the kinetic mechanism of the interaction of these inhibitors with the proteases (3CLpro and PLpro). The enzymatic activity was measured with a series of substrate concentrations and various inhibitor concentrations. The inhibition data were globally fit to all of the possible kinetic models. As a result of this kinetic analysis, we found that S. miltiorrhiza-derived proteases inhibitors could be divided into two classes based upon their mode of inhibition. These classes could also be divided on the basis of their structure for the targeted enzyme. The isolated tanshinones (1–3 and 5–7) exhibit noncompetitive inhibition characteristics toward 3CLpro (see Supplementary data Fig. S46 and Fig. S47). In case of PLpro kinetic plots, four-ringed tanshinones (1–6) also showed noncompetitive inhibition kinetics (see Supplementary data Fig. S48 and Fig. S49). The three-ringed compound 7 was the only exception to this rule. As illustrated representatively in Figure 5 A, the inhibition kinetics analyzed by Dixon plots shows that compound 4 has a noncompetitive mechanism because the Dixon plot of [I] versus 1/V results in a family of straight lines with the same x-axis intercept, as illustrated for the substrate concentrations. Compound 7 displayed mixed type inhibition against SARS-CoV PLpro, as shown in the Dixon plot (Fig. 5B). The Dixon plot of [I] versus 1/V resulted in a family of straight lines with an above the [I]-axis intercept. These kinetic mechanisms support the inferences drawn from the time-dependent assay, which reveals the important roles of the SARS-CoV PLpro inhibition modes. Notably, most slow-binding enzyme inhibitors act as competitive inhibitors, although it is possible for them to interact with the enzyme by competitive, noncompetitive, or uncompetitive inhibitioin pattens. Despite having similar molecular size and functionality, the diterpene quinolone tanshinone derivatives, 1–6, inhibit the SARS-CoV PLpro isomerization slow-binding mode, whereas the abietane type derivative, 7, is active against PLpro through the simple slow-binding mode.
Figure 5.
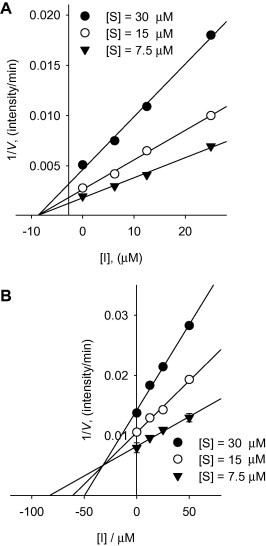
Dixon plots for inhibition of compounds 4 and 7 on SARS-CoV PLpro for the proteolysis of substrate. (A) Noncompetitive inhibition of PLpro by compound 4. (B) Mixed-type inhibition of PLpro by compound 7.
2.7. Effects of isolated tanshinones on the activity of deubiquitination and other proteases
PLpro, located within nsp3, cleaves at nsp1/2, nsp2/3 and nsp3/4 boundaries using the consensus motif LXGG, along with a consensus cleavage sequence of cellular deubiquitinating (DUB) enzymes.12 Modelling studies and crystal structures revealed a correlation between SARS-CoV PLpro and the herpes virus associated ubiquitin-specific protease, indicating potential DUB activity observed in in vitro cleavage assays.22 It has become clear that deubiquitination may have important implications on viral replication and pathogenesis. To experimentally confirm the predicted DUB activity of the isolated cysteine protease inhibitors, we observed the DUB activity for in vitro characterization. Table 3 lists the in vitro activity of compounds 1–7 against DUB. The IC50 values demonstrated that the presence of naphthalene in 5 (IC50 = 0.7 μM) and 6 (IC50 = 1.2 μM) provide a greater inhibitory effect than the other tanshinone derivatives. Although compound 4 was reported to be a potent inhibitor for SARS-CoV PLpro, 4 exhibited only a moderate inhibition effect against DUB activity (IC50 = 87.6 μM). To date, there is no potent and selective chemical compound available for the inhibition of DUB. Hence, the biological consequence of inhibiting DUB is not clear at the moment. Moreover, the function of the DUB activity of PLpro in vivo is unknown, as is the identity of its in vivo cellular substrate.14 Thus, further investigation is necessary to determine the specifics of the inhibition with the tanshinones and the PLpro-DUB interaction. Other studies are needed to measure the cleavage of the ubiquitin-like protein, interferon (IFN)-induced 15 kDa protein (ISG15), from an ISG15-fusion protein, suggesting de-ISGylation by PLpro as a mechanism by which SARS-CoV inactivates an IFN-α/β-induced innate immune response.22 Subsequently, compounds 1–7 were also evaluated against various proteases to study their selectivity and no detectable inhibition of any of the other proteases tested was observed (Table 3). Nevertheless, no detectable inhibition was observed at that concentration, indicating that tanshinones have an interesting selectivity for the viral proteases and could prove to be a viable lead for these proteolytic enzymes. Currently, there are many reports of rational designs and library-based screens that have identified several small molecules that act as potent inhibitors of the replication of SARS-CoV but are also HIV protease inhibitors. In contrast to this, our tanshinones-based viral protease inhibitors did not show a significant HIV protease inhibition effect.
Table 3.
Selective inhibition (IC50, μM) against various proteases by compounds 1–7a
| Compound | Ubb | Chymotrypsin | Papain | HIV-1 |
|---|---|---|---|---|
| 1 | NAc | 68.3 ± 2.0 | 122.0 ± 4.4 | 59.2 ± 1.6 |
| 2 | 52.0 ± 3.2 | NA | NA | NA |
| 3 | NA | 124.0 ± 6.2 | NA | 103.5 ± 2.2 |
| 4 | 87.6 ± 6.3 | 119.8 ± 1.1 | 89.8 ± 3.3 | 69.6 ± 0.6 |
| 5 | 0.7 ± 0.2 | 102.7 ± 3.6 | NA | 45.9 ± 3.0 |
| 6 | 1.2 ± 2.0 | NA | NA | 50.6 ± 9.1 |
| 7 | 60.2 ± 2.8 | 138.9 ± 5.1 | 88.0 ± 1.1 | 147.6 ± 1.9 |
All compounds were examined in a set of triplicates experiment; IC50 values of compounds represent the concentration that caused 50% enzyme activity loss.
Ub = deubiquitination activity.
NA = not activity at 200 μM.
3. Conclusion
SARS-CoV is the causative agent of severe acute respiratory syndrome (SARS). The virally encoded 3C-like protease (3CLpro) and papain-like protease (PLpro) have been presumed critical for the viral replication of SARS-CoV. In this study, we focused on naturally derived tanshinones for inhibitory activity against both proteases. These compounds are abundant in the lipophilic fraction of S. miltiorrhiza extract. To the best of our knowledge, this is the first report to describe the inhibitory effects of tanshinone against SARS-CoV main proteases. All of the isolated compounds 1–7 showed marked inhibitory activities to the 3CLpro and PLpro. The activity was significantly affected by subtle changes in structure. The IC50 values of these inhibitors, although higher than those of peptide-derived and small molecule viral cysteine protease inhibitors, are nonetheless in the low micromolar range. In particular, seven compounds exerted significant PLpro inhibitory activity (range from 0.8 to 30.0 μM) and inhibition was improved preincubation with the PLpro. In addition, our detailed kinetic mechanism analysis of these species has unveiled that they all exhibit slow-binding inhibition with enzyme isomerisation and noncompetitive inhibitors with the exception of compound 7. Interestingly, the introduction of three-ringed abietane analog, 7, showed simple reversible slow-binding inhibitor and mixed-type inhibition. Furthermore, the potential usage and development of other novel inhibitors for PLpro and cellular deubitquitination enzyme (DUB) based on the isolated compounds is discussed in this study. Out of the tanshinones, tanshinone I (5) showed the most potent DUB activity with an IC50 value of 0.7 μM. The results from this study warrant further investigation to examine the effect to these natural products on the inhibition of SARS-CoV. In an effort to advance this initiative, we sought to identify novel small molecules that specifically target SARS-CoV.
4. Materials and methods
4.1. General apparatus and chemicals
1H and 13C NMR data were obtained on 400 and 600 MHz FT-NMR spectrometer (Jeol Ltd, Japan) spectrometer in CDCl3 and tetramethylsilane (TMS) as internal standard. Melting points were measured on a Thomas Scientific Capillary Melting Point Apparatus (Electronthermal 9300, UK) and are uncorrected. Electrospray ionization (ESI) mass spectra were scanned using EIS in negative or positive mode with esquire 6000 mass spectrometer (Bruker Daltonik GmbH, Germany). All the reagent grade chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, USA). All purifications were monitored on commercially available glass-backed Merck precoated TLC plates and visualized under UV illumination at 254 nm and/or 366 nm or stained with 10% H2SO4 solution. Silica gel (230-400 mesh, Merck), RP-19 (ODS-A, 12 nm, S-150 mM, YMC), and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden) were used for column chromatography.
4.2. Extraction and isolation of tanshinones from Salvia miltiorrhiza
The dried roots of S. miltiorrhiza (2.5 kg) were extracted with 95% EtOH (10 L) for a week at room temperature. The crude extract was filtered and evaporated under reduced pressure. The extract (140 g) obtaining brown residue was suspended with distilled water (2 L). The suspended extract was partitioned by the organic solvents of the different polarities (n-hexane, ethyl acetate, and distilled water, each of 6 L, respectively). In consequence, we are to obtain four fractions which are n-hexane (20 g), ethyl acetate (31 g), and distilled water (80 g). Lipophilic (n-hexane) fraction (20 g) was subjected to silica gel column chromatography using a gradient of mixed solvent with n-hexane/ethyl acetate (from 10:1 to 1:10) and then these were achieved to five fractions (DH.1–DH.5). Fraction DH.3 (7 g) was chromatography by Sephadex LH-20 chromatography using a methanol solvent to give three sub-fractions (DH.3.1–DH.3.3). Sub-fraction DH.3.2 (3 g) was purified by preparative HPLC using an isocratic system of acetonitrile/distilled water (80:20) to give compound 6 (50 mg). Sub-fraction GH.3.1 (2 g) was purified by preparative HPLC using an isocratic system of acetonitrile/distilled water (90:10) to give compounds 1 (120 mg) and 5 (80 mg). Sub-fraction DH.3.3 (4 g) was subjected to C-18 reverse column chromatography using gradient of mixed solvent with methanol/distilled water (from 10:0 to 1:5) to afford compound 4 (10 mg). Fraction DH.4 (5 g) was chromatographed by silica gel column chromatography using a mixed solvent with n-hexane/ethyl acetate (from 10:0 to 0:10) to achieved to five sub-fractions (DH.4.1–DH.4.5). Compounds 2 (12 mg) and 7 (13 mg) were obtained by C-18 reverse column chromatography using a mixed solvent with methanol/distilled water (from 10:0 to 1:5) at sub-fraction DH.4.2 (1 g). Compound 3 (15 mg) which was chromatography by C-18 reverse column chromatography using a mixed solvent with methanol/distilled water (from 20:0 to 1:3) was purified at sub-fraction DH.4.4 (0.65 g).
4.2.1. Tanshinone IIA (1)
Red powder; mp 200–210 °C; ESI-MS m/z = 295 [M+H]+, 317 [M+Na]+, [calcd C19H18O3, 294]; 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 8.29 Hz, 1H, H-6), 7.52 (d, J = 8.29 Hz, 1H, H-7), 7.19 (s, 1H, H-16), 3.15 (t, J = 6.34 Hz, 2H, H-1), 2.23 (s, 3H, H-17), 1.78 (m, 2H, H-2), 1.63 (m, 2H, H-3), 1.28 (s, 6H, H-18, 19). 13C NMR (100 MHz, CDCl3) δ 183.8, 175.9, 161.9, 150.3, 144.6, 141.4, 133.6, 127.6, 126.7, 121.3, 120.4, 120.1, 38.1, 34.9, 32.0, 30.1, 19.3, 9.0.
4.2.2. Tanshinone IIB (2)
Red powder; ESI-MS m/z = 311 [M+H]+, 333 [M+Na]+, [calcd C19H18O4, 310]; 1H NMR (600 MHz, CDCl3) δ 7.62 (d, J = 8.25 Hz, 1H, H-6), 7.51 (d, J = 8.25 Hz, 1H, H-6), 7.19 (d, J = 1.37 Hz, 1H, H-16), 3.75 (d, J = 11.00 Hz, 1H, H-18b), 3.58 (d, J = 11.00 Hz, 1H, H-18a), 3.17 (m, 2H, H-1), 2.22 (s, 3H, H-17), 1.95 (m, 1H, H-3b), 1.85 (m, 1H, H-2b), 1.74 (m, 1H, H-2a), 1.54 (m, 1H, H-3a), 1.27 (s, 3H, H-19). 13C NMR (150 MHz, CDCl3) δ 183.6, 175.7, 161.6, 146.4, 146.1, 141.7, 133.9, 128.1, 126.8, 121.4, 120.5, 120.3, 71.7, 40.1, 32.4, 29.9, 26.8, 19.0, 9.0.
4.2.3. Methyl tanshinonate (3)
Red powder; mp 174–176 °C; ESI-MS m/z = 339 [M+H]+, 361 [M+Na]+, [calcd C20H18O5, 338]; 1H NMR (600 MHz, CDCl3) δ 7.56 (d, J = 8.25 Hz, 1H, H-6), 7.48 (d, J = 8.25 Hz, 1H, H-7), 7.23 (d, J = 1.37 Hz, 1H, H-16), 3.67 (s, 3H, H-20), 3.24 (m, 2H, H-1), 2.26 (m, 1H, H-3b) 2.26 (s, 3H, H-17), 1.82 (m, 2H, H-2), 1.75 (m, 1H, H-3a), 1.58 (s, 3H, H-18). 13C NMR (150 MHz, CDCl3) δ 183.6, 177.3, 175.8, 161.5, 144.6, 143.3, 141.8, 135.2, 128.8, 126.7, 125.3, 121.5, 120.5, 52.8, 47.4, 34.2, 29.2, 27.8, 19.4, 9.0.
4.2.4. Cryptotanshinone (4)
Red powder; mp 190–192 °C; ESI-MS m/z = 297 [M+H]+, 319 [M+Na]+, [calcd C19H20O3, 296]; 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 7.80 Hz, 1H, H-6), 7.45 (d, J = 8.29 Hz, 1H, H-7), 4.85 (t, J = 9.27 Hz, 1H, H-16b), 4.34 (dd, J = 9.27, 5.85 Hz, 1H, H-16a), 3.56 (m, 1H, H-15), 3.17 (t, J = 6.34 Hz, 1H, H-1), 1.75 (m, 2H, H-2), 1.63 (m, 2H, H-3), 1.32 (d, J = 6.83 Hz, 1H, H-17), 1.27 (s, 6H, H-18, 19). 13C NMR (100 MHz, CDCl3) δ 184.4, 175.8, 170.9, 152.5, 143.8, 132.7, 128.5, 126.4, 122.7, 118.4, 38.0, 35.0, 34.7, 32.1, 29.8, 19.2, 19.0.
4.2.5. Tanshinone I (5)
Dark brown powder; mp 223–234 °C; ESI-MS m/z = 277 [M+H]+, 299 [M+Na]+, [calcd C18H12O3, 276]; 1H NMR (400 MHz, CDCl3) δ 9.21 (d, J = 8.78 Hz, 1H, H-1), 8.27 (d, J = 8.78 Hz, 1H, H-6), 7.42 (d, J = 8.78 Hz, 1H, H-7), 7.52 (dd, J = 8.78, 6.83 Hz, 1H, H-2), 7.32 (d, J = 6.83 Hz, 1H, H-3), 7.27 (d, J = 1.46 Hz, 1H, H-16), 3.16 (s, 3H, H-18), 3.02 (s, 3H, H-17). 13C NMR (100 MHz, CDCl3) δ 183.9, 175.9, 161.4, 142.3, 135.4, 133.9, 133.1, 133.0, 130.9, 129.9, 128.6, 125.0, 123.4, 122.0, 120.7, 119.0, 20.1, 9.0.
4.2.6. Dihydrotanshinone I (6)
Dark brown powder; mp 197–202 °C; ESI-MS m/z = 279 [M+H]+, 301 [M+Na]+, [calcd C18H14O3, 278]; 1H NMR (500 MHz, CDCl3) δ 9.27 (d, J = 8.88 Hz, 1H, H-1), 8.29 (d, J = 8.88 Hz, 1H, H-6), 7.74 (d, J = 8.59 Hz, 1H, H-7), 7.56 (dd, J = 8.59, 7.16 Hz, 1H, H-2), 7.38 (d, J = 6.87 Hz, 1H, H-3), 4.95 (t, J = 9.16 Hz, 1H, H-16b), 4.43 (dd, J = 8.88, 6.01 Hz, 1H, H-16a), 3.63 (d, J = 4.87 Hz, 1H, H-15, 2.68 (s, 3H, H-18), 1.38 (d, J = 9.16 Hz, 3H, H-17). 13C NMR (125 MHz, CDCl3) δ 184.6, 176.0, 170.9, 135.2, 135.0, 132.3, 132.2, 130.7, 129.1, 128.4, 126.3, 125.2, 120.5, 81.9, 34.9, 30.0, 20.1, 19.1.
4.2.7. Rosmariquinone (7)
Red powder; mp 98–105 °C; ESI-MS m/z = 333 [M+Na]+, [calcd C19H22O2, 332]; 1H NMR (600 MHz, CDCl3) δ 7.56 (d, J = 8.25 Hz, 1H, H-6), 7.10 (d, J = 7.56 Hz, 1H, H-7), 7.10 (s, H, H-14), 3.15 (t, J = 6.19 Hz, 1H, H-1), 3.00 (m, 1H, H-15), 1.77 (m, 2H, H-3), 1.62 (m, 2H, H-2), 1.27 (s, 3H, H-18, 19), 1.13 (d, J = 6.19 Hz, 3H, H-16, 17). 13C NMR (150 MHz, CDCl3) δ 182.6, 181.7, 149.9, 145.2, 144.7, 140.1, 134.6, 134.0, 128.4, 128.1, 38.0, 34.7, 32.0, 30.1, 27.1, 21.7, 19.3.
4.3. Preparations of SARS-CoV 3CLpro and PLpro
The two genes encoding the 3CL protease and PL protease of SARS-CoV were synthesized based on the sequence reported by Sun et al23 and Han et al.13 The synthesized genes were inserted into the cloning sites of pET-23d (+) (Novagen, Madison, WI, USA) containing the 6x histidine-tag at the C-terminus. E. coli BL21 (DE3) CodonPlus-RIL cells (Stratagene, La Jolla, CA, USA), containing the constructed plasmid were grown in Luria-Bertani broth medium containing 100 μg/mL of ampicilline at 37 °C to an absorbance of 0.5 at 600 nm and then induced with 0.5 mM isopropyl β-d-thiogalactopyranoside for 12 h at 16 °C. After collection by centrifugation at 6000×g, the cells were suspended in buffer A (20 mM sodium phosphate, 300 mM NaCl, 20 mM imidazole, 0.2% Triton X-100, pH 7.5). The suspended cells were sonicated and centrifuged at 15,000×g for 30 min. The supernatant was loaded on a Ni-Sepharose column equilibrated with buffer A. The column was washed with buffer A, and the sample protein was eluted with 200 mM imidazole. The elution buffer was then changed to 20 mM Tris–HCl buffer (pH 7.5) with 1 mM dithiothreitol, and the protein was concentrated using an Amicon Ultra filter with a 10,000 molecular weight cut-off (Millipore, Billerica, MA, USA). The enzyme concentration was determined from its absorbance at 280 nm. The purified protein was stored at −80 °C before use in any of the assays.
4.4. SARS-CoV 3CLpro inhibition assay
The inhibitory effect of prepared samples on SARS-CoV 3CLpro was measured using a FRET method developed and described previously by our group.24 In this assay, the 12-mer fluorogenic peptide Dabcyl-KNSTLQSGLRKE-Edans (Anygen Co., Republic of Korea) was used as a substrate, and the enhanced fluorescence due to cleavage of this substrate catalyzed by the protease was measured at 590/40 nm with excitation 360 nm using a fluorescence plate reader (BioTeck Instrument Inc., USA). The IC50 values of isolated compounds were measured in a reaction mixture containing 10 μg/mL of the 3CLpro (final concentration, 2.5 μg), the test compounds (from 0 to 200 μM), and 10 μM of fluorogenic 12-mer peptide substrate in 20 mM Bis–Tris buffer (pH 7.5). The reactions were run for 60 min at 37 °C while continuously monitoring the fluorescence. The initial velocities of the inhibited reactions were plotted against the different concentrations of inhibitor to obtain the IC50 values by properly fitting the data according to the analysis method previously reported.11
4.5. SARS-CoV PLpro inhibition assay
The inhibition assay was first optimized The inhibition assay was first optimized in a 96-well plate format to establish suitable assay conditions and incubation times. The fluorogenic peptide substrate, Arg-Leu-Arg-Gly-Gly-AMC (RLRGG-AMC), was purchased from Bachem Bioscience. The enhanced fluorescence emission upon substrate cleavage was monitored at excitation and emission wavelengths of 360 and 460 nm, respectively, in a SpectraMax M2e Multimode Reader (Molecular Devices Co., USA).16 The reaction mixture contained 54 nM PLpro in 20 mM Tris–HCl buffer (pH 6.8) in a total volume of 200 μL. After the addition of 30 μM substrate to the reaction mixture, the increase in fluorescence at 460 nm was continuously monitored at 37 °C. For the inhibition studies, 54 nM PLpro and 0-200 μM of the individual compounds were mixed with the substrate (30 μM) at 37 °C, and the fluorescence intensity was monitored.
4.6. Protease inhibition mechanism studies
To study the kinetics of SARS-3CLpro activation, various concentrations of fluorogenic peptide as substrate were added to 3CLpro in a reaction buffer. The kinetin study of SARS-PLpro used RLRGG-AMC as the substrate. The data acquisition was initiated within 20 min, and the reaction was monitored continuously every 2 min for up to 2 h. To study the kinetics of 3CLpro and PLpro inhibition by compounds 1–7, various concentrations of compounds 1–7 were added to 3CLpro or PLpro in assay buffer containing the predetermined substrate. Like the order kinetic trials, the data acquisition was initiated within 20 min, and the reaction was monitored continuously every 2 min for up to 2 h. All reactions were run in at least triplicate, and the results are presented as means ± S.D.
4.7. Analysis of SARS-CoV 3CLpro and PLpro inhibition progress curves
The Time-dependent assays and progress curves were carried out using predetermined recombinant proteases and substrates in optimal buffers. The enzyme activities were measured continuously for 120 min with a spectrometer. To determine the kinetic parameters associated with the time dependent inhibition of the proteases, progress curves were obtained at several inhibitor concentrations using fixed substrate concentrations. The data were analyzed using a nonlinear regression program, Simga Plot (SPCC Inc., Chicago, IL), to find the individual parameters for each curve; v i (initial velocity), v s (steady-state velocity), k obs (apparent first-order rate constant for the transition from v i to v s), and K i app (apparent K i) according to the following equations:21
| (1) |
| (2) |
| (3) |
| (4) |
4.8. SARS-CoV PLpro deubiquitination assay
For the deubiquitination assay, purified PLpro (54 nM) was mixed with fixed compound concentration of 0–200 μM before the substrate, Ubiquitin-AMC (Enzo Life sciences Inc., USA), was added in 20 mM Tris–HCl buffer (pH 6.8). All assays were performed at 37 °C in 96-well plate format. The enzyme activities were determined by monitoring the enhanced fluorescence emission upon substrate cleavage at excitation and emission wavelengths of 360 and 460 nm, respectively, in a SpectraMax M2e Multimode Reader (Molecular Devices Co., USA). The substrate concentration was 100 nM, and release of AMC was measured in the same manner as for the IC50 measurements described above.14, 16
4.9. Other proteases assays
Chymotrypsin activity was evaluated according to a method described by Francisco et al.,25 using BTEE as the substrate, where 20 μL of enzyme solution was mixed with 980 μL of 1 mM BTEE (N-benzoyl-l-tyrosine ethyl ester) in 80 mM Tris–HCl buffer at pH 7.8 with 10 mM CaCl2 at 25 °C. For the inhibition studies, chymotrypsin and 0–200 μM of the individual compounds were mixed with BTEE at 25 °C. The production of benzoyl-tyrosine was measured by monitoring the absorbance at 260 nm every 10 s for 6 min. Papain in a solution of 50 mM sodium phosphate (pH 6.2) was incubated with various concentrations of compounds for 5 min at 25 °C. Then, 20 mM BAPNA (N-α-benzoyl-dl-arginine-p-nitroanilide) was added, and the increase in absorbance at 410 nm with time was monitored. The HIV protease assay kits and HIV-1 protease were purchased from AnasPec Inc., USA. HIV-1 protease in assay buffer was pre-incubated with each compound for 10 min at 37 °C. The solution was then mixed with the FRET substrate, and the fluorescence intensity was measured with time at Ex/Em = 490 nm/520 nm.
Acknowledgments
This research was supported by National Research Foundation Grant founded by Korea government (MEST) (No. 2012-0001110) and KRIBB Research Initiative Program, Republic of Korea.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2012.07.038.
Contributor Information
Woo Song Lee, Email: wslee@kribb.re.kr.
Young Bae Ryu, Email: ybryu@kribb.re.kr.
Supplementary data
Supplementary data
References and notes
- 1.Donnelly A.C., Ghani A.C., Leung G.M., Hedley A.J., Fraser C., Riley S., Abu-Radda L.J., Ho L.-M., Thach T.-Q., Chau P., Chan K.-P., Lam T.-H., Tse L.-Y., Tsang T., Liu S.-H., Kong J.H.B., Lau E.M.C., Ferguson N.M., Anderson R.M. Lancet. 2003;361:1761. doi: 10.1016/S0140-6736(03)13410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stadler K., Vega M., Eickmann M., Becker S., Abrignami S., Klenk S.H.-D., Rappuoli R. Nat. Rev. Microbiol. 2003;1:209. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zieburh J. Curr. Opin. Microbiol. 2004;7:412. doi: 10.1016/j.mib.2004.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Grobalenya A.E., Ziebuhr J.J. Gen. Virol. 2003;84:2305. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 5.(a) Chou K., Wei D., Zhong W. Biochem. Biophys. Res. Commun. 2003;308:48. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh A.K., Xi K., Johnson M.E., Baker S.C., Mesecar A.D. Ann. Rep. Med. Chem. 2006;41:184. doi: 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. USA. 2003;1000:13190. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu C.-Y., Jan J.-T., Ma H.-H., Kuo C.-J., Juan H.-F., Cheng Y.-S.E., Hsu H.-H., Huang H.-C., Wu D., Brik A., Liang F.-S., Liu R.-S., Fang J.-M., Chen S.-T., Liang P.-H., Wong C.-H. Proc. Natl. Acad. Sci. USA. 2004;101:10012. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kao R.Y., Tsui W.H.W., Lee T.S.W., Tanner J.A., Watt R.M., Huang J.-D., Hu L., Chen G., Chen Z., Zhang L., He T., Chan K.-H., Tse H., To A.P.C., Ng L.W.Y., Wong B.C.W., Tsoi H.-W., Yang D., Ho D.D., Yuen K.-Y. Chem. Biol. 2004;11:1293. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shie J.-J., Fang J.-M., Kuo C.-J., Kuo T.-H., Liang P.-H., Huang H.-J., Yang W.-B., Lin C.-H., Chen J.-L., Wu Y.-T., Wong C.-H. J. Med. Chem. 2004;48:4469. doi: 10.1021/jm050184y. [DOI] [PubMed] [Google Scholar]
- 10.Ryu Y.B., Park S.-J., Kim Y.M., Lee J.-Y., Seo W.D., Chang J.S., Park K.H., Rho M.-C., Lee W.S. Bioorg. Med. Chem. Lett. 1873;2010:20. doi: 10.1016/j.bmcl.2010.01.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryu Y.B., Jeong H.J., Kim J.H., Park J.-Y., Kim D., Naguyen T.T.H., Park S.-J., Chang J.S., Park K.H., Rho M.-C., Lee W.S. Bioorg. Med. Chem. 2010;18:7940. doi: 10.1016/j.bmc.2010.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Clercq E. J. Clin. Virol. 2004;30:115. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]; (a) Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.T., Baker S.C., Li K.J. Biol. Chem. 2007;282:32208. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lindner H.A., Fotouhi-Aradakani N., Lytvyn V., Lachance P., Sulea T., Menard R.J. Virol. 2005;79:15199. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Proc. Natl. Acad. Sci. USA. 2006;103:5717. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Barretto N., Jukneliene D., Ratia K., Chen Z., Mesear A.D., Baker S.C.J. Virol. 2005;79:15189. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han Y.S., Chang G.G., Juo C.G., Lee H.J., Yeh S.H., Hsu J.T., Chen X. Biochemistry. 2005;44:10349. doi: 10.1021/bi0504761. [DOI] [PubMed] [Google Scholar]
- 14.Chou C.-Y., Chien C.-H., Han Y.-S., Prebanda M.T., Hsieh H.-P., Turk B., Chang G.-G., Chen X. Biochem. Pharmacol. 2008;75:1601. doi: 10.1016/j.bcp.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghoshi A.K., Takayama J., Aubin Y., Ratia K., Chaudhuri R., Baez Y., Sleeman K., Coughlin M., Nichols D.B., Mulhearn D.C., Pradhakar B.S., Baker S.C., Johnson M.E., Mesecar A.D. J. Med. Chem. 2009;52:5228. doi: 10.1021/jm900611t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E., Baker S.C., Ghosh A.K., Mesecar A.D. Proc. Natl. Acad. Sci. USA. 2008;105:16119. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou L., Zuo Z., Chow M.S.S. J. Clin. Pharmacol. 2005;45:1345. doi: 10.1177/0091270005282630. [DOI] [PubMed] [Google Scholar]
- 18.Wang B.-Q.J. Med. Plant. Res. 2010;4:2813. [Google Scholar]
- 19.Abd-Elazem I.S., Chen H.S., Bates R.B., Huang R.C.C. Antiviral Res. 2002;55:91. doi: 10.1016/s0166-3542(02)00011-6. [DOI] [PubMed] [Google Scholar]
- 20.Ma Z., Zhang M., Song Z. Rapid Commun. Mass Spectrom. 2009;23:2857. doi: 10.1002/rcm.4195. [DOI] [PubMed] [Google Scholar]
- 21.Morrison J.F., Walsh C.T. Adv. Enzymol. 1988;61:201. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 22.Li S.-W., Lai C.-C., Ping J.-F., Tsai F.-J., Wan L., Lin Y.-J., Kung S.-H., Lin C.-W.J. Gen. Virol. 2011;92:1127. doi: 10.1099/vir.0.028936-0. [DOI] [PubMed] [Google Scholar]
- 23.Sun H., Luo H., Yu C., Sun T., Chen J., Peng S., Qin J., Shen J., Yang Y., Xie Y., Chen K., Wang Y., Shen X., Jiang H. Protein Expr. Purif. 2003;32:302. doi: 10.1016/j.pep.2003.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Virus Res. 2008;133:63. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castillo-Yáñez F.J., Pacheco-Aguilar R., García-Carreño F.L., Toro M., Delos A.N.-D., López M.F. Food Chem. 2006;99:252. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data