

Abstract
Flaviviruses are the causative agents of severe diseases such as Dengue or Yellow fever. The replicative machinery used by the virus is based on few enzymes including a methyltransferase, located in the N-terminal domain of the NS5 protein. Flaviviral methyltransferases are involved in the last two steps of the mRNA capping process, transferring a methyl group from S-adenosyl-l-methionine onto the N7 position of the cap guanine (guanine-N7 methyltransferase) and the ribose 2′O position of the first nucleotide following the cap guanine (nucleoside-2′O methyltransferase). The RNA capping process is crucial for mRNA stability, protein synthesis and virus replication. Such an essential function makes methyltransferases attractive targets for the design of antiviral drugs. In this context, starting from the crystal structure of Wesselsbron flavivirus methyltransferase, we elaborated a mechanistic model describing protein/RNA interaction during N7 methyl transfer. Next we used an in silico docking procedure to identify commercially available compounds that would display high affinity for the methyltransferase active site. The best candidates selected were tested in vitro to assay their effective inhibition on 2′O and N7 methyltransferase activities on Wesselsbron and Dengue virus (Dv) methyltransferases. The results of such combined computational and experimental screening approach led to the identification of a high-potency inhibitor.
Abbreviations: RdRp, RNA dependent RNA polymerase; N7 MTase, guanine N7 methyltransferase; 2′O MTase, nucleoside-2′O methyltransferase; AdoMet, S-adenosyl-l-methionine; AdoHcy, S-adenosyl-l-homocysteine; Wv, Wesselsbron virus; NS, non-structural protein; GMP, guanosine monophosphate; GTP, guanosine triphosphate; Dv, Dengue virus; WNv, West Nile virus; LBS, putative low affinity RNA binding site; HBS, high affinity RNA binding site; ATA, aurintricarboxylic acid; PPNDS, pyridoxal-5′-phosphate-6-(2′-naphthylazo-6′-nitro-4′,8′-disulfonate) tetrasodium salt; PMSF, phenylmethylsulphonyl fluoride; TLC, thin-layer chromatography
Keywords: Viral methyltransferase, Flavivirus, Viral RNA capping, Methyltransferase inhibition, Virtual docking
1. Introduction
The genus Flavivirus comprises over 70 RNA viruses, many of which are important human pathogens. Dengue virus (Dv), as an example, is estimated to infect about 50 million people a year, causing 24,000 deaths (Guha-Sapir and Schimmer, 2005). Currently, no specific antiviral drugs are available against flaviviral infections, and their development has been an active field of research during recent years (Sampath and Padmanabhan, 2009).
The flaviviral genome consists of 11 kb; it is decorated with a ‘cap-1’ structure at the strictly conserved 5′ terminus N7MeGpppA2′OMeG-RNA. The genome encodes a 370-kDa polyprotein precursor, which is processed by viral and cellular proteases into three structural proteins and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) involved in virus replication (Fields et al., 2001). Among other proteins, the multi-functional protein NS5 is particularly important for viral replication. The C-terminal domain of NS5 is endowed with RNA-dependent RNA polymerase (RdRp) activity, while its N-terminal domain contains S-adenosyl-l-methionine (AdoMet)-dependent methyltransferase (MTase) activity. The MTase domain is involved in the last two steps of the capping process that starts with the conversion of the 5′-triphosphate end of the nascent RNA into a 5′-diphosphate (RNA triphosphatase activity), followed by the addition of a GMP unit via a 5′–5′ phosphodiester bond (guanylyltransferase activity). MTase, then, transfers a methyl group from AdoMet to the N7-atom of the cap guanine and, successively, to the ribose 2′O-atom of the first RNA nucleotide forming the ‘cap 0’ and ‘cap 1’ structures, respectively.
The crystal structures of the MTase domain from several viruses, in complex with AdoMet, S-adenosyl-l-homocysteine (the co-product of methyl transfer, AdoHcy), with GTP or with cap-analogues (Assenberg et al., 2007, Bollati et al., 2009, Egloff et al., 2002, Egloff et al., 2007) are taken here as starting data for an analysis of the main structural and mechanistic features related to MTase activity. Since flaviviral MTase displays both N7 and 2′O activities (Ray et al., 2006, Zhou et al., 2007), in order for capped RNA to be methylated at the two different sites the nucleic acid substrate must adopt two distinct binding modes relative to the enzyme active site. In particular, many structural results (Assenberg et al., 2007, Bollati et al., 2009, Egloff et al., 2002, Egloff et al., 2007) show that small capped RNA analogues bind to a MTase high affinity binding site (HBS), where they are supposed to be held during 2′O methylation. On the contrary, structural details on a distinct binding site occupied by capped RNA during N7 methylation are missing. Structural considerations, however, suggest the existence of a secondary, putative low affinity binding site (LBS) (Dong et al., 2008a, Mastrangelo et al., 2007) located in a positively charged region close to the AdoMet binding site. Here, we first present a model of a short capped RNA (GpppAGUp) bound to the LBS of Wesselsbron virus MTase (WvMTase), that is used to explore the details of protein/RNA interaction during N7 methyl transfer. Based on this analysis, we then discussed an in silico search that allowed to select three synthetic compounds, predicted to display low free energy of binding to WvMTase. The compounds selected were tested in vitro on WvMTase and DvMTase in 2′O and N7 methyltransferase activity assays. The results of the combined computational and experimental screening allowed us to identify aurintricarboxylic acid (ATA) as the most potent flaviviral MTase inhibitor known to date. The inhibitory effect of ATA is discussed here on the basis of existing models of the flaviviral MTase mechanism for N7 and 2′O cap methylation. Since flavivirus MTase has been shown by mutagenesis to be crucial for viral replication (Zhou et al., 2007), shedding light on MTase inhibition may prove a productive path towards the development of therapeutics against life threatening flaviviral diseases (Dong et al., 2008a, Dong et al., 2008b).
2. Materials and methods
2.1. Chemical database for virtual screening and reagents
The virtual Library of Pharmacologically Active Compounds (LOPAC) used for the docking analysis was provided by Sigma–Aldrich, and included 1280 commercially available compounds (www.sigmaaldrich.com). AdoMet was purchased from New England BioLabs. The compounds tested in vitro, ATA, nilutamide and pyridoxal-5′-phosphate-6-(2′-naphthylazo-6′-nitro-4′,8′-disulfonate) tetrasodium salt (PPNDS), were from Sigma–Aldrich. Compounds were dissolved at 20 mM in H2O, or at 8 mM in 0.1 mM NaOH (ATA), and stored at −20 °C.
2.2. Locating a short capped RNA in the LBS
In order to produce an MTase model suitable for the analysis of the N7 methylation process, the crystal structure of WvMTase in its complex with AdoMet and with an HBS-bound small capped RNA analog, N7MeGpppG, solved at 1.9 Å resolution (pdb 3ELW) (Bollati et al., 2009) was used. Hydrogen atoms and computed Gasteiger charges (Gasteiger and Marsili, 1978) were added after removing the HBS-bound RNA analog, maintaining the AdoMet cofactor bound to the protein. A squared grid (30 Å side) centered roughly in the LBS region between Ser56 and Arg84 (Autogrid4; step size 0.375 Å, 80 × 80 × 80 = 512,000 points) (Goodford, 1985) was subsequently built and used as the volume explored in the capped RNA docking searches. The short capped RNA GpppAGUp molecule was then built using the program Ghemical (http://www.bioinformatics.org/ghemical). Seventy genetic algorithm searches were performed using Autodock4, moving GpppAGUp within the described grid and using 32 active torsions in the ligand molecule (with 150 individuals in population and 27,000 generations; Morris et al., 1998). The search produced a list of 70 capped RNA positions within the explored LBS volume, ranked by means of the Estimated Free Energy of Binding (ΔG) that varied between −3.32 and −12.43 kcal/mol. Among the four best ΔG values (in the −11.74, −12.43 kcal/mol range) the model with the cap Guanine N7 atom closer to the AdoMet methyl group was chosen (−11.86 kcal/mol; AdoMet-CH3–N7-RNA distance 7.3 Å). Such model (WvMTase/GpppAGUp) was then used as starting structure for molecular dynamics (MD) simulations, performed using the program GROMACS (van der Spoel et al., 2005). Briefly, the protein with AdoMet and the docked capped RNA (GpppAGUp) were enclosed in a box of 61 Å × 68 Å × 91 Å filled with 11,688 water molecules and 6 Cl− ions for charge equilibration. Using the GROMACS force field (GROMOS-87 with corrections as in Mark et al., 1994), the energy was minimized with a steepest descent algorithm, and the system was then equilibrated at 10 K for 1 ps and at 100 K for 5 ps. The MD simulation was carried out with time step of 1 fs using a leap-frog algorithm and periodic boundary conditions; electrostatic interactions were treated with Fast Particle-Mesh Ewald algorithm, while van der Waals interactions were cut-off at 12 Å; the simulation was performed at fixed T (300 K) and P (1 atm) using Berendsen coupling (Berendsen et al., 1984). The structure produced after 9 ns was used as final model for the analysis of protein/RNA interaction.
2.3. In silico search for MTase inhibitors
The AutoDock4 software package (Morris et al., 1998) was used for a docking search using compounds from the LOPAC library, and Python Molecule Viewer 1.4.5 (MGL-tools package http://www.mgltools.scripps.edu/) to analyze the data. As docking model the atomic coordinates of WvMTase in complex with AdoMet and N7MeGpppG were chosen (pdb 3ELW) (Bollati et al., 2009), keeping both the protein and the AdoMet molecule and removing water and the cap-analogue. Hydrogen atoms and computed Gasteiger charges were added using the program Autodock4 (Gasteiger and Marsili, 1978). The protein model was then used to build a discrete grid within a box with dimensions 23 Å × 15 Å × 26 Å (Autogrid4; step size 0.375 Å, 60 × 40 × 70 = 168,000 points) (Goodford, 1985) for the compound docking search. The search (i.e. the grid center position) was centered in a wide crevice located between the AdoMet binding site and the RNA HBS, more precisely between residues Lys182 and Arg213. In this way the protein active site, where the methyl transfer from AdoMet occurs, is completely included in the search grid chosen. Twenty genetic algorithm searches were run using Autodock4 for each compound in the LOPAC library (with 150 individuals in the population and 27,000 generations) (Morris et al., 1998).
2.4. In vitro synthesis of capped RNA
The 7MeGpppAC5 RNA substrate, used for the 2′O MTase assay, was synthesized by incubating the DNA oligonucleotide CCCCGGGTCT25 with the bacteriophage T7 DNA primase in the presence of 7MeGpppA and CTP as described (Peyrane et al., 2007). The reaction products obtained after a 48 h incubation period were purified by reverse phase chromatography in HPLC. We collected the peak corresponding to 7MeGpppAC5 as described (Peyrane et al., 2007).
The RNA substrate used for N7 MTase assay corresponding to the authentic 5′-terminal 351 nucleotides of the Dv genome (Dv1–351 RNA) was obtained as follows. The 5′ UTR of Dv serotype 2, New Guinea C strain was amplified by PCR using the primers BamH1-Φ2.5Dv-5′(s) (CGGGATCCCAGTAATACGACTCACTATTAGTTGTTAGTCTACGTGGACC) and EcoR1-Dv-351(as) (GGAATTCGGTGGTGCAGATGAACTTCAG), and cloned in the pUC18 (Fermentas) plasmid using a standard BamH1/EcoR1 restriction-ligation procedure generating the pUC18-Φ2.5-Dv351 plasmid. The RNA transcription template was synthesized by PCR amplification performed on the pUC18-Φ2.5-Dv351 plasmid using the primers BamH1-Φ2.5Dv-5′(s) and Dv-351(as) (GGTGGTGCAGATGAACTTCAGGG). The PCR reaction product was purified on agarose gel using the QIAquick gel extraction kit (Qiagen). The Dv1–351 RNA substrate was generated by in vitro transcription using the MEGAshortscript T7 RNA polymerase (Ambion) that recognizes the T7 class II Φ2.5 promoter (underlined in primer) present in the PCR template and initiates RNA synthesis by pppAG (Coleman et al., 2004). After DNAse (Ambion) treatment, and purification by RNeasy mini kit (Qiagen), the Dv1–351 RNA was incubated for 1 h at 37 °C with vaccinia virus capping enzyme (Epicentre, Madison, WI) in presence of 10 μCi [α-32P]-GTP (PerkinElmer, Boston, MA) and 0.05 units of inorganic pyrophosphatase (Sigma–Aldrich). Radiolabeled capped Dv1–351 RNA was then purified with the RNeasy mini kit (Qiagen).
2.5. 2′O MTase inhibition assays
The activity of Wv and Dv MTases were tested by incubating purified proteins with the small capped RNA substrate 7MeGpppAC5 in the presence of [3H]AdoMet as described (Luzhkov et al., 2007). Briefly, the MTase activity assays were performed in 30 μl samples containing 40 mM Tris, pH 7.5, 5 mM DTT, 5 μM AdoMet (0.3–2 μCi [3H]AdoMet, Amersham Biosciences), 1 μM of Wv or Dv MTases and 2 μM of RNA substrate 7MeGpppAC5. Recombinant Wv and Dv MTases were produced as previously described (Bollati et al., 2009, Egloff et al., 2002, respectively). Enzymes were premixed with the inhibitor candidates and the reactions were started with a premix of AdoMet and RNA substrate. During a first test series two different final inhibitor concentrations (125 and 25 μM) were used. For the determination of IC50 values of the ATA compound a concentration range from 0.12 to 125 μM was tested. Reactions were incubated at 30 °C. Two 14-μl samples were spotted, immediately after mixing and after 3 h, into 96-well plates containing 100 μl of 20 μM ice cold AdoHcy to stop the reaction. The samples were then transferred to glass-fibre filtermats (DEAE filtermat, Wallac) by a Filtermat Harvester (Packard Instruments). Filtermats were washed twice with 0.01 M ammonium formate, pH 8.0, twice with water and once with ethanol, dried and transferred into sample bags. Liquid scintillation fluid was added and methylation of RNA substrates was measured in counts per minute (cpm) using a Wallac MicroBeta TriLux Liquid Scintillation Counter. The IC50 (inhibitor concentration at 50% activity) value of the inhibitor compounds was determined. Two independent experiments were done. Data were adjusted to a logistic dose–response function [% activity = 100/(1 + [I]/IC50)b, where b corresponds to the slope factor that determines the slope of the curve (DeLean et al., 1978)].
2.6. N7 MTase inhibition assays
N7 MTase inhibition assay was performed in 50 mM Tris, pH 7.0, 50 mM NaCl, 2 mM DTT, 80 μM AdoMet, 1.5 pmol Wv or Dv MTase and approximately 1 μg of radiolabeled capped Dv1–351 RNAs in a 10 μl reaction volume at 22 °C for 20 min and 1 h. As described in the 2′O MTase assay, enzymes were premixed with the inhibitor candidate and the reactions were started with a premix of AdoMet and RNA substrate. The reaction was stopped by heating at 70 °C for 5 min. Samples were treated overnight with proteinase K (0.1 μg/μl). Proteinase K was inactivated by heating at 70 °C (15 min) and addition of 5 mM PMSF. The methylation reaction mixtures were subsequently digested for 4 h with nuclease P1. The digestion products were separated on polyethyleneimine cellulose thin-layer chromatography (TLC) plates (Macherey Nagel) using 0.45 M (NH4)2SO4 as mobile phase. After drying the TLC plates, the non-labeled standards were visualized by UV shadowing and the cap structure release by nuclease P1 were visualized using phosphorimager (Fluorescent Image Analyzer FLA3000 (Fuji)). The quantification was performed using Image Gauge software.
3. Results
3.1. Model of WvMTase in complex with short capped RNA during N7 methylation: LBS
In order to analyze the MTase region proposed to act as a capped RNA binding site during N7 methylation (the LBS), we produced a docked model of WvMTase with a short-capped RNA (GpppAGUp; see Section 2), and subsequently relaxed such model through 9 ns MD simulation. During the simulation the starting energy of the system (−1.29 × 10−5 kcal/mol) reached its mean value (−1.05 × 10−5 kcal/mol) in about 10 ps, and remained constant thereafter [energy root mean square deviation (r.m.s.d.) 0.001 × 10−5 kcal/mol]. The WvMTase secondary structure elements were thoroughly conserved during the simulation, with the exclusion of part of the C-terminal α-helix (amino acids 275–283). Such C-terminal fluctuation is in agreement with observations on several flaviviral MTase crystal structures where the C-terminal helix was not located in the electron density, or displayed very high B-factor values (Bollati et al., 2009). In fact, the C-terminal helix present in WvMTase was shown to be part of the RdRp domain of West Nile virus (WNv) and Dv (Malet et al., 2007, Yap et al., 2007).
Grouping the structures produced during MD into clusters, according to their similarity, may help detecting the time evolution of the protein toward new overall conformations. In performing such analyses, we chose to discard few N- and C-terminal residues of the protein, focusing on amino acids 8–260, thus ignoring the motility of the protein terminals. We used a cut-off of 0.7 Å in Cα r.m.s.d. to cluster the different structures observed during MD time evolution. With such limitation we found that the structures generated during the simulation could be grouped into nine different clusters, and that the last cluster contained all the states produced after the first 70 ps, suggesting an early stabilization of the protein scaffold during the simulation. The same cluster analysis was carried out to investigate the structural evolution of capped RNA during the simulation. With a 0.8 Å cut-off, 32 capped RNA clusters were produced, the last one starting at 3.85 ns and lasting through the end of the simulation (9 ns). The 0–3.85 ns interval can be interpreted as the time needed for RNA to reach a stable conformation following interaction with the protein, starting from the docked model. R.m.s.d. values calculated for the simulated structures vs. the crystal structure, during MD, reached the mean value of 1.5 ± 0.1 Å r.m.s.d. after about 3 ns of equilibration. The protein regions with higher motility during the simulation (considering amino acids 8–260) are the HBS (N-terminal helix-loop-helix motif, amino acids 17–22), Gly58, and a loop (amino acids 106–109) shown to be highly mobile also in the crystal structures, and proposed to be involved in AdoMet binding/AdoHcy release after the methyl transfer to RNA (Bollati et al., 2009).
The structure produced after 9 ns of MD was used to analyze WvMTase/capped RNA interactions in the LBS region. In this model (Fig. 1A and B), the cap guanine was located close to AdoMet and to residues Glu149 and Arg213; other residues found to interact with capped RNA were: Arg37, Arg41, Leu44, Ser56, Arg57, Arg84, Glu149, Lys112, Ser150, Arg160, Ser215. It has been recently shown that point mutations of few amino acids dramatically inhibit N7 methyl transfer activity in WNvMTase (Dong et al., 2008a). In particular, seven amino acids (Arg37, Arg57, Arg84, Trp87, Glu149, Arg213, and Tyr220), whose mutation to Ala reduced the mentioned activity below 30%, have been identified. In this context, analyzing the role of interface residues indicated by our WvMTase/RNA simulated model, we propose to neglect Trp87 and Tyr220. In fact, these two residues are spatially close to each other, and partly buried in the protein core; it can be expected that their mutation to Ala might critically alter the correct MTase fold. Moreover, Tyr220 is located at the “floor” of the MTase catalytic tetrad region (K-D-K-E: Lys61-Asp146-Lys182-Glu218 in WvMTase), being hydrogen bonded to Lys61 (3.2 Å) and to Asp146 (2.9 Å); thus it may play a crucial role in the stabilization of the catalytic residues. As a result, our WvMTase/RNA interaction model focuses on the five remaining residues (Arg37, Arg57, Arg84, Glu149, and Arg213; Fig. 1B).
Fig. 1.
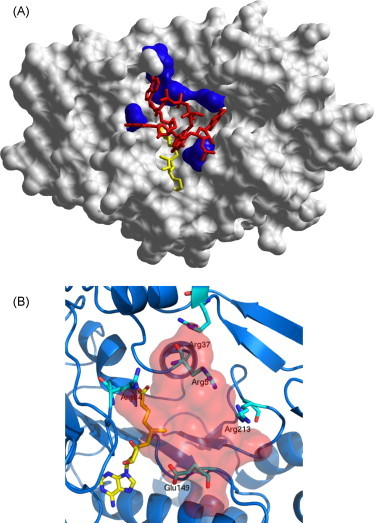
(A) Model of MTase-capped RNA interaction, at the LBS, after 9 ns of molecular dynamics. The protein is shown as white surface, AdoMet in yellow, capped RNA in red and five residues (Arg37, Arg59, Arg84, Glu149, and Arg213) shown by Dong et al. (2008a) to impair N7 methyltransferase activity, in blue. (B) A closer view into the LBS region showing key RNA recognition residues and AdoMet; the shaded area highlights the volume occupied by the capped RNA analog GpppAGUp. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
3.2. Docking compounds from the LOPAC library
Different criteria have been explored in the past for the discovery of effective MTase inhibitors (Dong et al., 2008a, Dong et al., 2008b). In particular, the search for compounds competing with AdoMet binding has been considered as one of the promising approaches (Luzhkov et al., 2007, Ray et al., 2006). AdoMet is the principal biological methyl donor in cells; under normal conditions in humans 6–8 g of AdoMet are generated per day, and most of these are used in transmethylation reactions whereby methyl groups are added to different compounds and AdoMet is converted to AdoHcy (Lu, 2000). We thus speculated that MTase inhibition based on AdoMet competition might be particularly difficult to achieve. Moreover, since many distinct enzymes require AdoMet as cofactor, an AdoMet mimetic compound (like sinefungin; IC50 of 14 μM on WNvMtase) (Dong et al., 2008b) may turn out to be generally toxic. Therefore, a different inhibitor search strategy could, in principle, be directed towards blockage of the MTase HBS; however, such class of inhibitory molecules would likely provide inhibition only for the 2′O MTase reaction, but not for the N7 MTase activity. Therefore, for the design of new inhibitors, we decided to target the active site of the enzyme with its bound AdoMet cofactor.
As for most enzymatic reactions, transfer of a methyl group from AdoMet to RNA, in each of the two MTase reactions, requires a precise location of the RNA substrate in the active site, such location being dictated by the position of the AdoMet transferrable methyl group. This consideration suggests that a small ligand, binding to a protein location close to (but not inside) the cofactor-binding site, would potentially impair both N7 and 2′O methylation reactions. Moreover, such a ligand could interact directly with AdoMet or AdoHcy, potentially slowing the release of the latter from the protein during enzyme turnover. As target for ligand docking, we thus selected the active site region, located between the AdoMet binding site, the HBS, and the LBS (Fig 2A), that had been explored by our simulated model described above. The selected region was explored using a library of small molecules (LOPAC library) as described in Section 2. The search produced a list of compounds with predicted free energy of binding (ΔG) ranging between +9.0 and −16.2 kcal/mol. The top three ranked compounds produced by the docking search (nilutamide, PPNDS and ATA, Fig. 2A), displaying the lowest binding energies (ΔG = −16.2, −13.4, and −12.4 kcal/mol, respectively), were used for in vitro testing of MTase inhibitory activity (Fig. 2B).
Fig. 2.
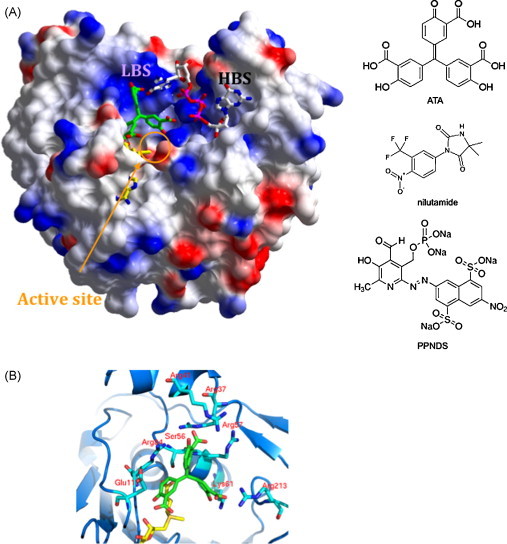
(A) Relative locations of HBS and LBS. The WvMTase surface representation is used to highlight the relative location of LBS and HBS (labeled). The HBS hosts 7MeGpppG observed in the crystal structure (pdb 3ELW; Bollati et al., 2009). The active site region includes AdoMet (partly hidden in its binding pocket) and the docked ATA molecule (green). Schematic formulas of ATA, nilutamide and PPNDS are shown on the right. (B) Details of the WvMTase/ATA interaction. A close up view of the active site region hosting the ATA inhibition as produced by the docking approach. ATA is green; residues held relevant for stabilization of the bond inhibitor are labeled. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
3.3. Inhibition of 2′O and N7 methylation activities in DvMTase and WvMTase
The inhibitory effect of the three best binders selected in silico was determined on DvMTase and WvMTase 2′O MTase activities. For this purpose, we first tested the inhibition of ATA, nilutamide and PPNDS at 125 and 25 μM by measuring the transfer of a [3H]-methyl group to the short 7MeGpppAC5 substrate yielding 7MeGpppA2′OMeC5. Fig. 3A shows that whereas nilutamide and PPDNS did not display efficient inhibition of the flaviviral MTases at the selected concentrations, ATA strongly inhibited both MTases even at 25 μM. We therefore determined the IC50 values for ATA in the 0.12–125 μM concentration range. Fig. 3B shows dose–response curves that yield IC50 values of 4.2 μM (±0.2 μM) for WvMTase and of 2.3 μM (±0.3 μM) for DvMTase. These values indicate that ATA can be considered the strongest inhibitor of flaviviral 2′O MTase activity known to date.
Fig. 3.
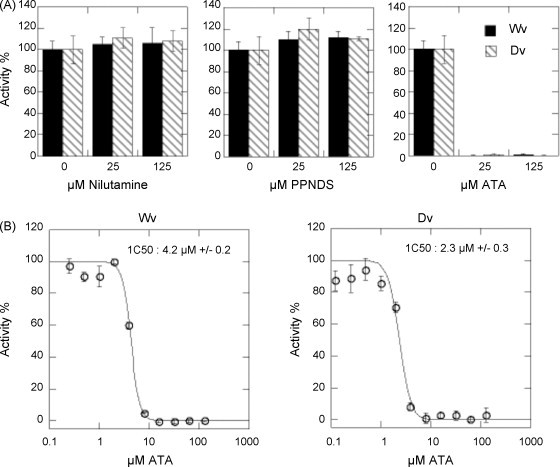
Inhibition of Wv and Dv 2′O MTase activity. Purified Wv and Dv MTases were incubated with 7MeGpppAC5 substrate in presence of [3H] AdoMet and inhibitor candidates. (A) Inhibition of nitulamine, PPNDS and ATA. Two concentrations of each inhibitor candidate were tested (125 and 25 μM) and a control reaction was carried out in absence of inhibitor candidate. The methyl transfer to the RNA substrate was stopped after 3 h and detected using a filter-binding assay that measured the amount of radioactivity, in cpm, that was transferred to the RNA substrates. MTase activity of 100% was arbitrarily attributed to the MTase activity in absence of inhibitor. The bar graph presents the results of three independent experiments. (B) Inhibition of Wv and Dv 2′O MTase activity using increasing concentrations of ATA (125 nM to 125 μM). The IC50 values were determined by fitting of the dose response curve as described in Section 2. Each reaction was carried out in triplicate and the standard deviation is plotted.
Flaviviral MTase also mediates guanine N7 methylation, when the enzymatic reaction is performed in the presence of an RNA substrate comprising more than 70 nucleotides including a stem loop structure conserved in the 5′ untranslated regions of flavivirus genomes (Ray et al., 2006, Dong et al., 2007). In order to determine whether ATA inhibited the flaviviral N7 MTase activity we synthesized an RNA corresponding to the first 351 nucleotides of the Dv genome (Dv1–351 RNA) using T7 polymerase, and capped the RNA by addition of [α-32P]-GTP using vaccinia virus guanylyltransferase (see Section 2). We expected that Dv1–351 RNA would be an appropriate substrate for WvMTase because the first three nucleotides of the capped RNA genomes of both viruses are GpppAGU and it had been shown that, besides the presence of the stem loop structure, the conservation of second and third nucleotide were especially important for specific N7 MTase activity (Dong et al., 2007). The capped Dv1–351 RNA was incubated with WvMTase in the presence of increasing ATA concentrations and the reaction products were analyzed after nuclease P1 digestion by TLC and autoradiography. Fig. 4A shows that in the absence of WvMTase (CTL-), radiolabeled GpppA released from the substrate RNA by nuclease P1 is detected at the expected position. In the presence of WvMTase (CTL+), almost 55% of GpppA was converted into a new product co-migrating with the N7MeGpppA cap analog, as standardly visualized by UV shadowing. At longer incubation times part of N7MeGpppA was further methylated into N7MeGpppA2′OMe (not shown). We conclude that, under these optimized reaction conditions, the RNA corresponding to the first 351 nucleotides of the Dv genome, is a good substrate to specifically follow the N7 MTase activity of WvMTase. We next determined the IC50 values of ATA inhibition on both MTases, by quantifying the GpppA conversion into N7MeGpppA in the presence of increasing ATA concentrations after 20 min, during the linear phase of the N7 MTase reaction. ATA inhibits the N7 WvMTase activity at higher concentrations than those observed in the 2′O MTase assay, the IC50 value being 38 μM (±2 μM) compared to 4.2 μM, respectively. As shown in Fig. 4B, ATA moderately inhibits the N7 DvMTase activity, with an IC50 value of 127 μM (±6 μM), compared to IC50 of 2.3 μM, for the inhibition of the 2′O DvMTase activity. Thus, our results indicate that ATA inhibits the N7 MTase activity of Dv and Wv to a minor extent compared to the 2′O MTase activity.
Fig. 4.
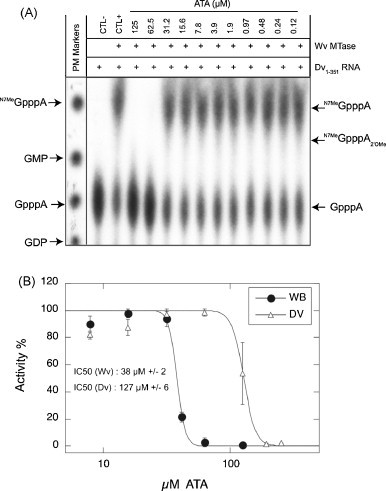
Inhibition of Wv and Dv N7 MTase activity by ATA. Purified Wv and Dv MTases were incubated with [α-32P]-G-capped Dv1–351 RNA in the presence of increasing concentrations of ATA (125 nM to 125 μM). The reaction, stopped after 60 min, was digested with nuclease P1 in order to release the radiolabeled cap structures. (A) TLC analysis of reaction products for N7 MTase activity performed with WvMTase. Two controls were carried out, one without enzyme and one without ATA and the molecular weight markers were visualized by UV shadowing. (B) Determination of the IC50 values for ATA on the N7 MTase activity of Wv and Dv MTases. The percentage of activity was determined after quantification of GpppA and 7MeGpppA. The IC50 value was determined by fitting of the dose–response curve as in Fig. 3.
3.4. Analysis of WvMTase/ATA complex docking model
Analysis of complex models obtained by docking ATA to WvMTase shows the inhibitor molecule facing the AdoMet binding site (Fig. 2B). In particular, ATA interacts with many positively charged amino acids conserved in flavivirus MTases (Arg37, Arg41 (mostly conserved), Arg57, Lys61, Arg84, and Arg213) and establishes a hydrogen bond with the 3′O atom of the AdoMet ribose, contributing to cofactor stabilization in its binding pocket. Interestingly, the docked model indicates a hydrogen bond between an ATA hydroxyl group and the side chain of Lys61, part of the conserved catalytic tetrad (K-D-K-E) that supports the 2′O MTase activity. Thus ATA, bound at the site predicted by docking and MD, may impair the correct location of RNA in the catalytic site through steric hindrance and through interaction with residues involved in RNA recognition.
4. Discussion
The results here reported show that basic hypotheses on the flaviviral MTase mechanism of action (more specifically, deepening our understanding of the role played by the LBS) could efficiently lead to the proposal of a novel binding site for inhibitors. Starting from our WvMTase crystal structure (Bollati et al., 2009), we proposed a model of the protein/RNA interaction during N7 methylation activity (no crystal structure available so far). We then used the proposed (LBS) RNA binding site to select three compounds out of 1280 that were screened by means of a docking analysis in silico. Among the three best ligands identified we showed by in vitro inhibitory assays that ATA is a potent inhibitor of flaviviral 2′O MTase activity for both Wv and Dv. ATA also inhibited the Wv and Dv N7 MTase activity on longer RNAs, but to a minor extent. The efficient inhibitory action of ATA against 2′O MTase activity is consistent with its specific interaction with residue Lys61, as indicated by docking. Indeed, this residue was previously shown to be important for 2′O MTase activity but not for N7 MTase activity (Zhou et al., 2007). Moreover, since N7 MTase and 2′O MTase assays were performed on RNA of different lengths, it is possible that the different IC50 values found reflect a difference in the affinity for the RNA substrates in the inhibition assays. Taken together, our results demonstrating the inhibitory capability of ATA (in the micromolar range) support the hypothesis that the LBS (adjacent to the MTase-bound AdoMet) which is targeted here as a new inhibitory site may be suitable for drug lead development.
Interestingly ATA has been reported to inhibit the replication of different kinds of viruses including human immunodeficiency virus (HIV) (Schols et al., 1989), influenza virus (Hung et al., 2009) and vaccinia virus (Myskiw et al., 2007). Moreover a recent patent (WO/2005/123965) indicates a possible inhibitory role of ATA against SARS corona virus. In the same patent, results indicating generic activity of ATA against flaviviruses are also reported. The authors of the patent, however, do not propose a model explaining the reported antiviral efficacy. The results here presented suggest a specific molecular target and a possible mechanism for the inhibitory activity of ATA against flaviviruses. As a cautionary note, it should be recalled that ATA has been generally reported in the literature as a non-specific inhibitor of several different enzymes (Bina-Stein and Tritton, 1976). In fact, ATA appears to be able to impair different kinds of protein/nucleotide interactions; therefore, it may also play yet unknown roles in the inhibition of other viral replication processes beyond RNA capping. Moreover, considering ATA's capability of inhibiting protein/nucleotide interactions, we cannot exclude different/further effects, even on MTase, such as the blockage of the HBS site (key RNA binding site for 2′O methylation), or effects resulting in a decreased protein affinity for AdoMet.
Acknowledgments
This work has been supported by the EU IP Project Vizier (CT 2004-511960, to BC and MB), and by the Italian Ministry for University and Scientific Research FIRB Project “Biologia Strutturale” (to MB). A research grant to BC from the Direction Générale de l’Armement is gratefully acknowledged.
References
- Assenberg R., Ren J., Verma A., Walter T.S., Alderton D., Hurrelbrink R.J., Fuller S.D., Bressanelli S., Owens R.J., Stuart D.I., Grimes J.M. Crystal structure of the Murray Valley encephalitis virus NS5 methyltransferase domain in complex with cap analogues. J. Gen. Virol. 2007;88:2228–2236. doi: 10.1099/vir.0.82757-0. [DOI] [PubMed] [Google Scholar]
- Berendsen H.J.C., Postma J.P.M., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- Bina-Stein M., Tritton T.R. Aurintricarboxylic acid is a nonspecific enzyme inhibitor. Mol. Pharmacol. 1976;12:191–193. [PubMed] [Google Scholar]
- Bollati M., Milani M., Mastrangelo E., Ricagno S., Tedeschi G., Nonnis S., Decroly E., Selisko B., de Lamballerie X., Coutard B., Canard B., Bolognesi M. Recognition of RNA cap in the Wesselsbron virus NS5 methyltransferase domain: implications for RNA capping mechanisms in Flavivirus. J. Mol. Biol. 2009;385:140–152. doi: 10.1016/j.jmb.2008.10.028. [DOI] [PubMed] [Google Scholar]
- Coleman T.M., Wang G., Huang F. Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 φ 2.5 promoter. Nucleic Acids Res. 2004;32:e14. doi: 10.1093/nar/gnh007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLean A., Munson P.J., Rodbard D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose–response curves. Am. J. Physiol. 1978;235:e97–e102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- Dong H., Ray D., Ren S., Zhang B., Puig-Basagoiti F., Takagi Y., Ho C.K., Li H., Shi P.-Y. Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. J. Virol. 2007;81:4412–4421. doi: 10.1128/JVI.02455-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H., Ren S., Zhang B., Zhou Y., Puig-Basagoiti F., Li H., Shi P.Y. West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substrate repositioning mechanism. J. Virol. 2008;82:4295–4307. doi: 10.1128/JVI.02202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H., Zhang B., Shi P.Y. Flavivirus methyltransferase: a novel antiviral target. Antiviral Res. 2008;80:1–10. doi: 10.1016/j.antiviral.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M.P., Benarroch D., Selisko B., Romette J.L., Canard B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff M.P., Decroly E., Malet H., Selisko B., Benarroch D., Ferron F., Canard B. Structural and functional analysis of methylation and 5′-RNA sequence requirements of short capped RNAs by the methyltransferase domain of Dengue virus NS5. J. Mol. Biol. 2007;372:723–736. doi: 10.1016/j.jmb.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Fields B.N., Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Straus S.E., Knipe D.M. 4th ed. Lippicott Williams & Wilkins; Philadelphia, PA: 2001. Fields Virology. [Google Scholar]
- Gasteiger J., Marsili M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978:3181–3184. [Google Scholar]
- Goodford P.J. A computational procedure for determining energetically favourable binding sites on biologically important macromolecules. J. Med. Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- Guha-Sapir D., Schimmer B. Dengue fever: new paradigms for a changing epidemiology. Emerg. Themes Epidemiol. 2005;2:1. doi: 10.1186/1742-7622-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung H.-C., Tseng C.-P., Yang J.-M., Ju Y.-W., Tseng S.-N., Chen Y.-F., Chao Y.-S., Hsiehd H.-P., Shih S.-R., Hsu J.T.-A. Aurintricarboxylic acid inhibits influenza virus neuraminidase. Antiviral Res. 2009;81:123–131. doi: 10.1016/j.antiviral.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.C. S-adenosylmethionine. Int. J. Biochem. Cell Biol. 2000;32:391–395. doi: 10.1016/s1357-2725(99)00139-9. [DOI] [PubMed] [Google Scholar]
- Luzhkov V.B., Selisko B., Nordqvist A., Peyrane F., Decroly E., Alvarez K., Karlen A., Canard B., Qvist J. Virtual screening and bioassay study of novel inhibitors for Dengue virus mRNA cap (nucleoside-2′O)-methyltransferase. Bioorg. Med. Chem. 2007;15:7795–7802. doi: 10.1016/j.bmc.2007.08.049. [DOI] [PubMed] [Google Scholar]
- Malet H., Egloff M.-P., Selisko B., Butcher R.E., Wright P.J., Roberts M., Gruez A., Sulzenbacher G., Vonrhein C., Bricogne G., Mackenzie J.M., Khromykh A.A., Davidson A.D., Canard B. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J. Biol. Chem. 2007;282:10678–10689. doi: 10.1074/jbc.M607273200. [DOI] [PubMed] [Google Scholar]
- Mark A.E., van Helden S.P., Smith P.E., Janssen L.H.M., van Gunsteren W.F. Convergence properties of free energy calculations: α-cyclodextrin complexes as a case study. J. Am. Chem. Soc. 1994;116:6293–6302. [Google Scholar]
- Mastrangelo E., Bollati M., Milani M., Selisko B., Peyrane F., Canard B., Grard G., de Lamballerie X., Bolognesi M. Structural bases for substrate recognition and activity in Meaban virus nucleoside-2′-O-methyltransferase. Protein Sci. 2007;16:1133–1145. doi: 10.1110/ps.072758107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- Myskiw C., Deschambault Y., Jefferies K., He R., Cao J. Aurintricarboxylic acid inhibits the early stage of vaccinia virus replication by targeting both cellular and viral factors. J. Virol. 2007;81:3027–3032. doi: 10.1128/JVI.02531-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrane F., Selisko B., Decroly E., Vasseur J.J., Benarroch D., Canard B., Alvarez K. High-yield production of short GrppA- and 7MeGpppA-capped RNAs and HPLC-monitoring of methyltransfer reactions at the guanine-N7 and adenosine-2′ O positions. Nucleic Acids Res. 2007;35:e26. doi: 10.1093/nar/gkl1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D., Shah A., Tilgner M., Guo Y., Zhao Y., Dong H., Deas T.S., Zhou Y., Li H., Shi P.Y. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J. Virol. 2006;80:8362–8370. doi: 10.1128/JVI.00814-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath A., Padmanabhan R. Molecular targets for flavivirus drug discovery. Antiviral Res. 2009;81:6–15. doi: 10.1016/j.antiviral.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schols D., Baba M., Pauwels R., Desmyter J., De Clercq E. Specific interaction of aurintricarboxylic acid with the human immunodeficiency virus/CD4 cell receptor. Proc. Natl. Acad. Sci. U.S.A. 1989;86:3322–3326. doi: 10.1073/pnas.86.9.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J.C. GROMACS: fast, flexible and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Yap T.L., Xu T., Chen Y.L., Malet H., Egloff M.P., Canard B., Vasudevan S.G., Lescar J. Crystal structure of the Dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 2007;81:4753–4765. doi: 10.1128/JVI.02283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Ray D., Zhao Y., Dong H., Ren S., Li Z., Guo Y., Bernard K.A., Shi P.Y., Li H. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007;81:3891–3903. doi: 10.1128/JVI.02704-06. [DOI] [PMC free article] [PubMed] [Google Scholar]