

Abstract
Microbial infections are dependent on the panoply of interactions between pathogen and host and identifying the molecular basis of such interactions is necessary to understand and control infection. Phage display is a simple functional genomic methodology for screening and identifying protein–ligand interactions and is widely used in epitope mapping, antibody engineering and screening for receptor agonists or antagonists. Phage display is also used widely in various forms, including the use of fragment libraries of whole microbial genomes, to identify peptide–ligand and protein–ligand interactions that are of importance in infection. In particular, this technique has proved successful in identifying microbial adhesins that are vital for colonization.
Phage display technology
Display on filamentous phage
One of the first genetic techniques developed to study protein–ligand interactions was phage display, described by Smith in 1985 [1]. This method displays recombinant peptides or proteins on the surface of phage particles, which can then be selected for (‘panned’ for – Figure 1 ) by enabling the phage to interact with selected immobilized ligands. The power of phage display lies in its ability to: (i) maintain a physical link between the displayed protein and the DNA sequence encoding it; and (ii) screen libraries containing billions of unique peptides and proteins. Escherichia coli filamentous phage of the Ff class including strains M13, fd and f1 have been extensively used to develop and exploit this technology. These phage are composed of a circular single-stranded DNA genome that is encased in a long tube composed of thousands of copies of a single major coat protein, with four additional minor capsid proteins at the tips (Figure 2 ).
Figure 1.
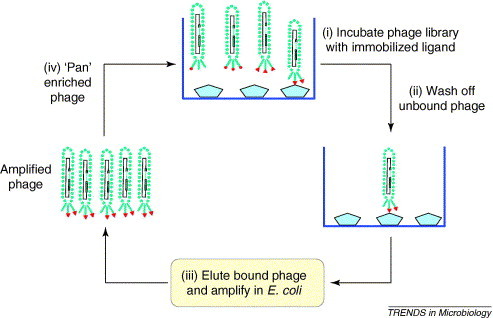
A cycle of affinity selection of filamentous phage display libraries to identify phage that display fusion proteins (pIII fusions) that bind to the chosen ligand or ligands.
Figure 2.
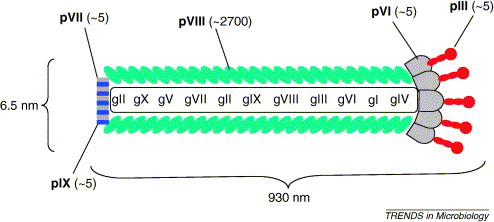
Dimensions and architecture of filamentous bacteriophage fd. The copy number of each protein is shown in brackets.
Phage display involves the fusion of foreign DNA sequences to the phage genome such that the resulting foreign proteins are expressed in fusion with one of the coat proteins. Although all five coat proteins have been used to display proteins or peptides, gene VIII protein (pVIII) and gene-III-encoded adsorption protein (pIII) are by far the most commonly used [2]. A viable wild-type phage expresses ∼2700 copies of pVIII and 3–5 copies of pIII (Figure 2) [3], although this does depend on the size of the phage genome.
Phage display libraries can be constructed using vectors based on the natural Ff phage sequence (i.e. phage vectors) or by using ‘phagemids’, which are hybrids of phage and plasmid vectors 2, 3. Such phagemids are designed with the origin of replication (ori) from the Ff phage, a plasmid origin of replication from E. coli, gene III and/or gene VIII for fusion formation, a multiple cloning site and an antibiotic resistance gene [2]. However, they lack all other phage genes that encode the structural and non-structural proteins that are required to produce a complete phage. Phagemids can be grown as plasmids in E. coli and packaged as recombinant Ff phage DNA with the aid of helper phage, which provide all of the necessary components for phage assembly.
Filamentous phage versus alternative systems for phage display
The key feature of filamentous phage (as applied to phage display) is that, in contrast to the lytic bacteriophages, filamentous phage are assembled in the cytoplasmic membrane and secreted from infected bacteria without cell lysis [2] (Figure 3 ). However, the characteristics of the filamentous phage life cycle has limitations for the display of proteins, the properties of which prevent the correct transfer of the hybrid capsid protein across the lipid bilayer of the inner membrane of E. coli [4]. Alternative bacteriophage display systems have been developed using bacteriophage such as T4, T7, λ [4] and P4 [5], which have lytic life cycles so that the proteins of the phage capsids are assembled and folded in the cytoplasm rather than being secreted through the membrane [4].
Figure 3.
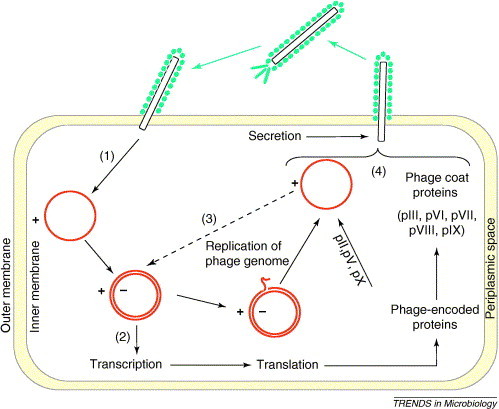
Life cycle of filamentous bacteriophage in Escherichia coli. The drawing is schematic and not to scale. (1) Phage bind to the E. coli cell through the pIII coat protein. The single-stranded viral genome (+ strand, single circle) is injected into the cell and a complementary strand (− strand) is synthesized to form a double-stranded phage genome (double circle). (2) Subsequently, all ten phage-encoded proteins are produced by host-mediated protein synthesis, including coat proteins (pIII, pVI, pVII, pVIII and pIX), proteins for replication (pII, pV and pX) and proteins involved in assembly and export (pI and pIV). (3) The phage genome is replicated using the (+)-strand as a primer and the (−)-strand as a template. (4) Virions are assembled and exported across the bacterial membranes.
Display of proteins encoded by cDNA fragments on phage
Because the most common approaches to phage display (described earlier) involve N-terminal fusion to the gene III or gene VIII products of filamentous phage, they are unsuitable for surface expression of proteins coded by intact cDNA inserts that have stop codons 6, 7. Hence, most phage libraries of cDNA fragments are constructed in alternative display systems. However, a modified filamentous phage display system based on the high-affinity interactions between the Jun and Fos leucine zipper proteins was developed by Crameri and Suter [6]. In this system, the Jun leucine zipper protein is fused to the N terminus of the pIII coat protein so that the Jun protein is displayed on the surface of the phage, and the cDNA libraries are cloned as a C-terminal fusion to the Fos leucine zipper protein [8]. The pIII-anchored Jun protein is bound by the soluble Fos fusion in the periplasm, thus, the cDNA-encoded protein is bound indirectly to pIII.
Natural peptide libraries versus random peptide libraries
There are two types of phage display library: random peptide libraries (RPLs) and natural peptide libraries (NPLs). The repertoire of peptides displayed in RPLs is encoded by synthetic random degenerate oligonucleotide inserts 9, 10, 11 and these libraries have been extensively used to identify linear antigenic epitopes (see later). The advantage of this type of library is their universal nature, which enables many applications of each library. The main disadvantage of RPLs is that, because of the way in which they are constructed, peptide sequences that are not found within the antigen or intact pathogen can be displayed [12]. By contrast, NPLs are constructed from randomly fragmented DNA from the genomes of selected organisms such as pathogenic microbes. Thus, the phage particles in these libraries display fragments of natural proteins. Although the majority of clones in NPLs are nonfunctional (only one in 18 clones will be correctly in frame with the vector sequences: one clone in three will start correctly, one clone in three will end correctly and one clone in two will be in the correct orientation), peptides or proteins selected from NPLs are more successful in eliciting an antibody response that crossreacts with the native intact pathogen than those selected from RPLs [13]. Therefore, NPLs provide important alternatives to RPLs for applications such as the identification of vaccine components and, recently, have been used effectively in the identification of bacterial adhesins (see later).
Phage display and infectious diseases
Phage display technology has been used in a wide variety of applications including: the identification of peptide agonists and antagonists for receptors [14], the identification of targets for the inhibition of tumour-specific angiogenesis [15], the identification of peptide drug candidates [16], vaccine development [17] and the isolation and engineering of recombinant antibodies [18]. However, in recent years, numerous studies have used phage display to address specific aspects of infectious disease. The purpose of this review is to focus on the growing number of ways in which phage display technology can be applied to the study of infectious diseases and to evaluate the impact of the use of this technology.
Investigation of host–pathogen interactions
Infectious diseases are absolutely dependent upon host–pathogen interactions, the extent of which determines the infectious process both for the pathogen and the host. As discussed later, epitope mapping of infectious agents has been used by numerous research groups, particularly to focus on the bacterial adhesins that enable host colonization.
Phage display technology has been used to good effect in malaria research to investigate host–pathogen interactions in both hosts of the malaria parasite: Homo sapiens and the mosquito Anopheles. Panning of a Plasmodium falciparum (the causative agent of malaria) cDNA phage display library against immobilized human erythrocyte membrane proteins identified seven parasite proteins that are potentially involved in the entry into or exit from human erythrocytes by P. falciparum [19]. Such proteins could become vaccine targets.
The search for the mechanisms used by Plasmodium species to cause infection has also prompted novel uses for phage display libraries. The Plasmodium parasite completes its life cycle in the mosquito and it is this interaction, rather than that of the human host, that has been investigated by Ghosh et al. [20]. Because the development of the Plasmodium parasite in the mosquito involves the crossing of both midgut and salivary gland epithelia, this study investigated the hypothesis that such trafficking requires specific host–pathogen interaction. A phage display library of random dodecapeptides fused to the N terminus of phage coat protein VIII was injected into Anopheles, followed by dissection of the organs of interest and elution of bound phage. A 12-residue peptide from the eluted phage was identified and designated ‘salivary gland and midgut peptide 1 (SM1)’ [20]. This peptide strongly inhibited Plasmodium invasion of salivary-gland and midgut epithelia, thereby hindering the development of the parasite in Anopheles. This study illustrates another variation of phage display technology – the use of phage display libraries to investigate specific phage-binding targets within the whole organism. In vivo panning was first described by Pasqualini and Ruoslahti [21], who isolated phage-displayed peptides that bound to vascular beds in vivo. To date, there are limited reports of such in vivo panning but this could have an important future role for the identification of specific tissue receptors for microbes or their products.
A variation on this theme is the panning of phage display libraries against ex vivo biomaterials that have been used in a range of clinical applications including prosthetic heart valves and intravenous catheters. Bacteria such as Staphylococcus aureus and Staphylococcus epidermidis are frequently isolated from biomaterial infections [22]. A phage display library constructed from the genomic DNA of S. aureus was panned against a central intravenous catheter that had been removed from a patient [23]. Numerous clones from the S. aureus library were recovered through panning and, after the second and third rounds, the enriched phage encoded fragments of bacterial proteins known to bind to fibrinogen and β2-glycoprotein I, which, unsurprisingly, were the most abundant host proteins deposited on the catheter.
Laminin-binding motifs in the surface virulence factor [plasminogen activator (PLA)] of Yersinia pestis were mapped by Benedek et al. [24] using an RPL. This approach involved identification of phage-displayed amino acid sequences that bound to laminin, then comparison of these sequences to the sequence of PLA to identify the specific binding site of PLA to laminin. This study demonstrates how phage display can be used to map specific binding sites within a protein of interest.
Epitope mapping and identification of potential vaccine candidate antigens
The term epitope can be used to describe the contacting points of any molecular interaction but it is more often used to describe the region on an antigen that elicits an immune response. The identification of epitopes from microbial pathogens has obvious importance in the study of infectious disease, particularly for the development of novel vaccines. Phage display technology is especially suited to epitope mapping and has been widely used in infectious-disease research (for review, see Ref. [17]).
Much of the work on epitope mapping and identification of mimotopes of infectious agents has used RPLs. Mimotopes are peptide sequences that are capable of inducing immune responses by mimicking the structural features of a non-linear protein epitope or of a non-protein (e.g. carbohydrate) epitope structure. These libraries have been panned against immune sera to identify disease-specific epitopes. Selected examples of these applications are listed in Table 1 . In addition, phage display has successfully identified peptide mimics of polysaccharides. This approach has important implications for the development of novel vaccines against encapsulated bacteria because the polysaccharide capsules of these bacteria are often antigenic. Peptide mimics of the capsular polysaccharide of three of the Neisseria meningitidis serotypes A [25], B [26] and C [27] have been identified using RPLs. The results of these studies are encouraging in that the mimotopes identified in this way did elicit immune responses in murine models and could be useful in the development of novel vaccines against this bacterium.
Table 1.
Surface display of antigenic epitopes and mimotopes on filamentous phage
| Library | Screened against | Results | Refs |
|---|---|---|---|
| 12mer RPL displayed on pIII | Polyclonal sera specific for Burkholderia pseudomallei protease | Identification of linear and discontinuous protease epitopes of B. pseudomallei | [49] |
| 7mer RPLs displayed on pIII | Purified anti-Mycoplasma hyopneumoniae IgG | Identification of M. hyopneumoniae antigens. Phage clones identified by screening induced immune responses in murine model | [50] |
| 12mer RPL displayed on pIII | Monoclonal IgA1 specific for the capsule of Streptococcus pneumoniae | Identification of mimotopes of the capsular polysaccharide of type 8 S. pneumoniae. When conjugated to tetanus toxoid, mimotopes induced a type 8 capsular-polysaccharide-specific antibody response in mice | [51] |
| 7mer RPL displayed on pIII | Sera from swine infected with Nipah virus | Identification of several putative epitopes within the nucleocapsid protein of Nipah virus | [52] |
| 12mer RPL displayed on pIII | Polyclonal IgG specific for neutral polysaccharides of Mycoplasma tuberculosis | Isolation of phage clones that were antigenic mimotopes of B-cell epitopes of M. tuberculosis sugars. One clone invoked immune responses in rabbits | [53] |
The pathogenesis of S. aureus has been investigated by panning an RPL against the RNAIII-activating protein (RAP), which autoinduces toxin production in this bacterium 28, 29. Initial work demonstrated that selected peptides could attenuate infection by S. aureus in murine models [28]. More recent studies have identified a peptide mimic for the RAP protein that, when displayed on E. coli and used to vaccinate mice, prevented mortality caused by S. aureus infection [29].
Infection with Pseudomonas aeruginosa is a major problem in patients with cystic fibrosis (CF). The development of vaccines against this organism must take into account that antibiotic treatment of chronic P. aeruginosa infections rarely results in clearance of the bacterium. This is in contrast to treatment of early P. aeruginosa infections in which the bacteria can be eradicated [30]. Antigens expressed early in P. aeruginosa infections of CF patients were investigated by panning an RPL against sera from non-infected and infected patients [31]. In conjunction with gene-array data and bioinformatic analysis, this study identified several genes that encode secreted proteins and outer-membrane proteins that are potential targets for vaccine development against P. aeruginosa infection [31]. Alterations in the panning technique used, such as subjecting the library to initial negative selection, further increases the potential uses of these libraries. Gnanasekar et al. [32] constructed a T7 phage display library of the cDNA of the parasite Brugia malayi (a causative agent of lymphatic filariasis). To ensure that the clones of interest (i.e. those binding to infection-specific antibodies) were enriched, the phage library was first subjected to three negative-selection steps using non-infected human sera followed by a positive-selection step using sera from patients infected with B. malayi. Further experiments revealed that one of the five antigens identified using this strategy, B. malayi NIP3-like protein, conferred protection against challenge infections in animal models [32].
Single chain variable fragment (scFv) libraries have been used in several ways to study infectious diseases. Identification of specific scFvs that bind to microbial antigens forms the basis for the development of novel vaccines and diagnostic reagents [3]. Panning of a scFv library against purified severe acute respiratory syndrome-associated coronavirus (SARS-CoV) identified two antibody fragments that bind to SARS-CoV with high specificity [33]. These could potentially be used in the development of detection assays for the virus or to elucidate potential vaccine candidate antigens. Phage-display scFv libraries have also been used successfully to develop immunodiagnostic and detection methods for microbial products or spores. One of the most impressive aspects of these applications is the high specificity of selected clones for the microbe. For example, panning of a scFv library against spores of Bacillus subtilis resulted in the selection of clones that bound to only one of 11 spore strains [34]. Similarly, clones selected from a phage display scFv library screened against Clostridium difficile toxin B were highly specific and showed no cross-reactivity with strains of C. difficile that are toxin-B negative. The single-chain antibody could also be used to detect as little as 10 ng of toxin B when used in ELISAs [35].
Identification of bacterial adhesins
Microbial infections are initiated by molecular interactions between the pathogen (through cell surface adhesins) and receptor molecules on host cells and the extracellular matrix (ECM), resulting in microbial adhesion, which can be followed by internalization 36, 37. It is well established that the adhesion of enteric, oral and respiratory bacteria is required for colonization [37]. Furthermore, when bacteria adhere to surfaces, they are substantially more resistant to host antimicrobial defences [38]. Adherence to structures such as the ECM is, therefore, a key step in the development of disease. The ECM consists of a complex mixture of macromolecules including collagens, fibronectin, fibrinogen, vitronectin, laminin and heparin sulfate [38], all of which function as ligands for bacterial adhesion.
With the continued rise in antibiotic resistance in bacteria, there is an urgent need to find alternative ways to combat microbial and, specifically, bacterial infection. Inhibition of bacterial adhesion is one potential therapeutic approach but usually requires an intimate knowledge of the adhesins involved in infection.
Phage display lends itself perfectly to the investigation of possible binding partners for a particular ligand and this application of phage display was first exploited by Jacobsson and Frykberg 39, 40 to identify genes encoding bacterial proteins that interact with host proteins. By constructing libraries from random fragments of bacterial genomic DNA (i.e. shotgun phage display) and subsequent panning against components of the host, such as proteins of the ECM, host serum or plasma, it is possible to identify bacterial genes that encode cell surface adhesins. Microbial proteins (including cell-bound and soluble proteins) that bind to mammalian target proteins were termed ‘receptins’ by Kronvall and Jonsson [41]. Following the initial description of gene-III-based phage display to identify genes coding for ligand-binding domains of bacterial receptins by Jacobsson and Frykberg [39], many genes involved in host–pathogen interactions (from a variety of bacterial species) have been discovered using phage-display systems (Table 2 ).
Table 2.
Identification of bacterial receptins by phage display screening
| Organism | Vector | Ligand | Gene product identified | Refs |
|---|---|---|---|---|
| Helicobacter pylori | pG8SAET | Plasminogen | PgbB | [54] |
| Staphylococcus aureus | pHen1 | Human IgG | Sbi | [46] |
| pG8SAET | von Willebrand factor | vWBp | [55] | |
| pG8SAET | Platelets | FnBPA, FnBPB | [56] | |
| pG8SAET | Fibronectin | FnBPA | [57] | |
| Staphylococcus epidermidis | pG8SAET | Fibronectin | Embp | [58] |
| pG8SAET | Fibronectin and osteoblast cell line MC3T3-E1 | Second fibronectin-binding domain within FnBPA and FnBPB | [59] | |
| pG8H6 | Fibrinogen | Fbe | [60] | |
| Streptococcus agalactiae | pG3H6 | Fibronectin | ScpB | [61] |
| pG8SAET | Fibrinogen | FgagV1, FgagV2, FgagV3 | [62] | |
| Streptococcus dysgalactiae | pG8H6 | Fibronectin | DemA | [63] |
| Streptococcus equi | pG8SAET | Fibronectin | FnBP | [64] |
| Staphylococcus lugdunensis | pG8SAET | von Willebrand factor | VWB1 | [65] |
| pG8SAET | Fibrinogen | Fb1 | [66] |
The majority of these studies have been carried out using gene-VIII-based vectors such as the pG8H6 or pG8SAET phagemid vectors developed by Jacobsson and Frykberg 40, 42. Panning of pIII-based libraries tends to select for fusion proteins with the strongest interaction with the ligand of choice but only a small fraction of pIII libraries contain functional inserts [43]. Use of pVIII generates phage with multiple copies of recombinant proteins but the selection process tends to select for lower affinity interactions.
Shotgun phage display has also been used in the mapping of binding domains within proteins coded by genes of interest. Instead of random fragmentation of genomic DNA from a particular species, a previously identified gene of interest is fragmented by sonication and used to generate a single-gene phage display library. This approach has been used to map fibronectin-binding activity to two C-terminal domains of the fibronectin-binding protein FNZ of Streptococcus equi [44], the IgG and β 2-glycoprotein-I-binding domains of the Sbi protein of S. aureus 42, 45 and the IgG and albumin-binding domains in two cell-surface receptors, MAG and ZAG, from group C streptococci [46]. The application of shotgun phage display to the identification of bacterial virulence factors has been further extended by Frykberg and colleagues, with the development of phagemid vectors that only enable the display of bacterial exported proteins 47, 48. Many exported microbial proteins are adhesins, enzymes or toxins, all of which might have a role in pathogenicity.
The libraries constructed by the shotgun method might not achieve full coverage of bacterial genomes for two reasons: (i) the low transformation efficiency of E. coli limits the number of primary clones (i.e. those containing unique inserts); and (ii) only one in 18 clones displays fusion proteins (see earlier). However, despite these limitations, the success of this method is obvious considering the numbers of shotgun phage display libraries that have been used to identify novel genes that encode putative bacterial receptins and protein binding domains (Table 2). Use of this technique identifies multiple microbial genes encoding proteins that potentially interact with the host in a single panning experiment and such results provide a starting point to determine the importance of various bacterial genes in pathogenesis. It is likely that phage display NPLs will become even more widely used in the study of infectious diseases, particularly because the growing number of available bacterial genomes means that the DNA fragments that encode phage-displayed peptides and proteins can be easily mapped to a particular bacterial gene.
Concluding remarks
The study of infectious diseases has benefited greatly from phage display technology. This technique has many advantages: the link between phenotype and genotype, the enormous diversity of variant proteins displayed within a single library and the flexibility of selection that can be performed in vitro or in vivo. Earlier limitations on this technique (such as the unsuitability of pIII- or pVIII-based filamentous phage display systems for the display of cDNA coding for peptides or problems with protein conformation) have been circumvented by continual development of the technology. The versatility of phage display means that it can be adapted easily to many different areas of research, as highlighted by the variety of biological questions to which phage display has been applied. It has yielded interesting and important results in the study of infectious diseases, not least in epitope mapping, the identification of potential vaccine candidates and bacterial adhesins. However, the vast potential of phage display is such that there are many more ways of applying the technology to these areas of research. There is certainly scope for developing new strategies to identify host receptors for microbial products by panning in vivo in animal models or against ex vivo tissues or organs. There are numerous examples of synergistic relationships between opportunistic bacteria that could be examined by panning phage display libraries against each other to investigate interactions between microbes. Perhaps the most exciting potential use of phage display in the study of infectious diseases is in comparative genomics. The number of sequenced microbial genomes has increased greatly in the past ten years and continues to do so as more genomes are sequenced. This will enable detailed bioinformatic analysis of genes identified by panning of NPLs and a comparison of the genes implicated in pathogenicity between groups of closely related bacteria.
References
- 1.Smith G.P. Filamentous fusion phage – novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 2.Russel M. Introduction to phage display and phage biology. In: Clackson T., Lowman H., editors. Phage Display: A Practical Approach. Oxford University Press; 2004. pp. 1–26. [Google Scholar]
- 3.Azzazy H.M.E., Highsmith J.W.E. Phage display technology: clinical applications and recent innovations. Clin. Biochem. 2002;35:425–445. doi: 10.1016/s0009-9120(02)00343-0. [DOI] [PubMed] [Google Scholar]
- 4.Castagnoli L. Alternative bacteriophage display systems. Comb. Chem. High Throughput Screen. 2001;4:121–133. doi: 10.2174/1386207013331174. [DOI] [PubMed] [Google Scholar]
- 5.Lindqvist B.H., Naderi S. Peptide presentation by bacteriophage P4. FEMS Microbiol. Rev. 1995;17:33–39. doi: 10.1111/j.1574-6976.1995.tb00185.x. [DOI] [PubMed] [Google Scholar]
- 6.Crameri R., Suter M. Display of biologically-active proteins on the surface of filamentous phages – a cDNA cloning system for selection of functional gene-products linked to the genetic information responsible for their production. Gene. 1993;137:69–75. doi: 10.1016/0378-1119(93)90253-y. [DOI] [PubMed] [Google Scholar]
- 7.Crameri, R. (1997) pJUFO: a phage surface display system for cloning genes based on protein–ligand interaction. In Gene Cloning and Analysis: Current Innovations (Schaefer, B., ed), Horizon Scientific Press
- 8.Crameri R. Display of expression products of cDNA libraries on phage surfaces – a versatile screening system for selective isolation of genes by specific gene-product ligand interaction. Eur. J. Biochem. 1994;226:53–58. doi: 10.1111/j.1432-1033.1994.tb20025.x. [DOI] [PubMed] [Google Scholar]
- 9.Cwirla S.E. Peptides on phage – a vast library of peptides for identifying ligands. Proc. Natl. Acad. Sci. U. S. A. 1990;87:6378–6382. doi: 10.1073/pnas.87.16.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott J.K., Smith G.P. Searching for peptide ligands with an epitope library. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 11.Devlin J.J. Random peptide libraries – a source of specific protein-binding molecules. Science. 1990;249:404–406. doi: 10.1126/science.2143033. [DOI] [PubMed] [Google Scholar]
- 12.Lundin K. Peptides isolated from random peptide libraries on phage elicit a neutralizing anti-HIV-1 response: analysis of immunological mimicry. Immunology. 1996;89:579–586. doi: 10.1046/j.1365-2567.1996.d01-772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthews L.J. Immunogenically fit subunit vaccine components via epitope discovery from natural peptide ligands. J. Immunol. 2002;169:837–846. doi: 10.4049/jimmunol.169.2.837. [DOI] [PubMed] [Google Scholar]
- 14.Pillutla R.C. Peptides identify the critical hotspots involved in the biological activation of the insulin receptor. J. Biol. Chem. 2002;277:22590–22594. doi: 10.1074/jbc.M202119200. [DOI] [PubMed] [Google Scholar]
- 15.Trepel M. In vivo phage display and vascular heterogeneity: implications for targeted medicine. Curr. Opin. Chem. Biol. 2002;6:399–404. doi: 10.1016/s1367-5931(02)00336-8. [DOI] [PubMed] [Google Scholar]
- 16.Ladner R.C. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov. Today. 2004;9:525–529. doi: 10.1016/S1359-6446(04)03104-6. [DOI] [PubMed] [Google Scholar]
- 17.Wang L.F., Yu M. Epitope identification and discovery using phage display libraries: applications in vaccine development and diagnostics. Curr. Drug Targets. 2004;5:1–15. doi: 10.2174/1389450043490668. [DOI] [PubMed] [Google Scholar]
- 18.Benhar I. Biotechnological applications of phage and cell display. Biotechnol. Adv. 2001;19:1–33. doi: 10.1016/s0734-9750(00)00054-9. [DOI] [PubMed] [Google Scholar]
- 19.Lauterbach S.B. Construction and use of Plasmodium falciparum phage display libraries to identify host parasite interactions. Malar. J. 2003;2:47. doi: 10.1186/1475-2875-2-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh A.K. Targeting Plasmodium ligands on mosquito salivary glands and midgut with a phage display peptide library. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13278–13281. doi: 10.1073/pnas.241491198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasqualini R., Ruoslahti E. Organ targeting in vivo using phage display libraries. Nature. 1996;380:364–366. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- 22.von Eiff C. Pathogenesis of infections due to coagulase-negative staphylococci. Lancet Infect. Dis. 2002;2:677–685. doi: 10.1016/s1473-3099(02)00438-3. [DOI] [PubMed] [Google Scholar]
- 23.Bjerketorp J. Sorting a Staphylococcus aureus phage display library against ex vivo biomaterial. J. Med. Microbiol. 2004;53:945–951. doi: 10.1099/jmm.0.45638-0. [DOI] [PubMed] [Google Scholar]
- 24.Benedek O. Identification of laminin-binding motifs of Yersinia pestis plasminogen activator by phage display. Int. J. Med. Microbiol. 2005;295:87–98. doi: 10.1016/j.ijmm.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Grothaus M.C. Selection of an immunogenic peptide mimic of the capsular polysaccharide of Neisseria meningitidis serogroup A using a peptide display library. Vaccine. 2000;18:1253–1263. doi: 10.1016/s0264-410x(99)00390-4. [DOI] [PubMed] [Google Scholar]
- 26.Park I. Peptide mimotopes of Neisseria meningitidis group B capsular polysaccharide. Yonsei Med. J. 2004;45:755–758. doi: 10.3349/ymj.2004.45.4.755. [DOI] [PubMed] [Google Scholar]
- 27.Prinz D.M. Two different methods result in the selection of peptides that induce a protective antibody response to Neisseria meningitidis serogroup C. J. Immunol. Methods. 2004;285:1–14. doi: 10.1016/j.jim.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Yang G. Inhibition of Staphylococcus aureus pathogenesis in vitro and in vivo by RAP-binding peptides. Peptides. 2003;24:1823–1828. doi: 10.1016/j.peptides.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 29.Yang G. A novel peptide screened by phage display can mimic TRAP antigen epitope against Staphylococcus aureus infections. J. Biol. Chem. 2005;280:27431–27435. doi: 10.1074/jbc.M501127200. [DOI] [PubMed] [Google Scholar]
- 30.Gibson R.L. Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003;167:841–849. doi: 10.1164/rccm.200208-855OC. [DOI] [PubMed] [Google Scholar]
- 31.Beckmann C. Use of phage display to identify potential Pseudomonas aeruginosa gene products relevant to early cystic fibrosis airway infections. Infect. Immun. 2005;73:444–452. doi: 10.1128/IAI.73.1.444-452.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gnanasekar M. Novel phage display-based subtractive screening to identify vaccine candidates of Brugia malayi. Infect. Immun. 2004;72:4707–4715. doi: 10.1128/IAI.72.8.4707-4715.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z-X. Identification of single-chain antibody fragments specific against SARS-associated coronavirus from phage-displayed antibody library. Biochem. Biophys. Res. Commun. 2005;329:437–444. doi: 10.1016/j.bbrc.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knurr J. Peptide ligands that bind selectively to spores of Bacillus subtilis and closely related species. Appl. Environ. Microbiol. 2003;69:6841–6847. doi: 10.1128/AEM.69.11.6841-6847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng X.K. Recombinant single-chain variable fragment antibodies directed against Clostridium difficile toxin B produced by use of an optimized phage display system. Clin. Diagn. Lab. Immunol. 2003;10:587–595. doi: 10.1128/CDLI.10.4.587-595.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soto G.E., Hultgren S.J. Bacterial adhesins: common themes and variations in architecture and assembly. J. Bacteriol. 1999;181:1059–1071. doi: 10.1128/jb.181.4.1059-1071.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ofek I. American Society for Microbiology Press; 2003. Bacterial Adhesion to Animal Cells and Tissues. [Google Scholar]
- 38.Patti J.M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 1994;48:585–617. doi: 10.1146/annurev.mi.48.100194.003101. [DOI] [PubMed] [Google Scholar]
- 39.Jacobsson K., Frykberg L. Cloning of ligand-binding domains of bacterial receptors by phage display. Biotechniques. 1995;18:878–885. [PubMed] [Google Scholar]
- 40.Jacobsson K., Frykberg L. Phage display shot-gun cloning of ligand-binding domains of prokaryotic receptors approaches 100% correct clones. Biotechniques. 1996;20:1070–1081. doi: 10.2144/96206rr04. [DOI] [PubMed] [Google Scholar]
- 41.Kronvall G., Jonsson K. Receptins: a novel term for an expanding spectrum of natural and engineered microbial proteins with binding properties for mammalian proteins. J. Mol. Recognit. 1999;12:38–44. doi: 10.1002/(SICI)1099-1352(199901/02)12:1<38::AID-JMR378>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L. A second IgG-binding protein in Staphylococcus aureus. Microbiology. 1998;144:985–991. doi: 10.1099/00221287-144-4-985. [DOI] [PubMed] [Google Scholar]
- 43.Jacobsson K. Shotgun phage display – selection for bacterial receptins or other exported proteins. Biol. Proced. Online. 2003;5:123–135. doi: 10.1251/bpo54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lindmark H. Fibronectin-binding protein of Streptococcus equi subsp zooepidemicus. Infect. Immun. 1996;64:3993–3999. doi: 10.1128/iai.64.10.3993-3999.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang L. Staphylococcus aureus expresses a cell surface protein that binds both IgG and β2-glycoprotein I. Microbiology. 1999;145:177–183. doi: 10.1099/13500872-145-1-177. [DOI] [PubMed] [Google Scholar]
- 46.Jacobsson K. Shot-gun phage display mapping of two streptococcal cell-surface proteins. Microbiol. Res. 1997;152:121–128. doi: 10.1016/S0944-5013(97)80002-X. [DOI] [PubMed] [Google Scholar]
- 47.Rosander A. Phage display as a novel screening method to identify extracellular proteins. J. Microbiol. Methods. 2002;51:43–55. doi: 10.1016/s0167-7012(02)00052-0. [DOI] [PubMed] [Google Scholar]
- 48.Karlstrom A. Identification of a novel collagen-like protein, SclC, in Streptococcus equi using signal sequence phage display. Vet. Microbiol. 2004;104:179–188. doi: 10.1016/j.vetmic.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 49.Chan S.W., Nathan S. Epitope mapping of Burkholderia pseudomallei serine metalloprotease: identification of serine protease epitope mimics. FEMS Immunol. Med. Microbiol. 2005;43:37–44. doi: 10.1016/j.femsim.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 50.Yang W-J. Epitope mapping of Mycoplasma hyopneumoniae using phage displayed peptide libraries and the immune responses of the selected phagotopes. J. Immunol. Methods. 2005;304:15–29. doi: 10.1016/j.jim.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 51.Buchwald U.K. A peptide mimotope of type 8 pneumococcal capsular polysaccharide induces a protective immune response in mice. Infect. Immun. 2005;73:325–333. doi: 10.1128/IAI.73.1.325-333.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eshaghi M. Identification of epitopes in the nucleocapsid protein of Nipah virus using a linear phage-displayed random peptide library. J. Med. Virol. 2005;75:147–152. doi: 10.1002/jmv.20249. [DOI] [PubMed] [Google Scholar]
- 53.Gevorkian G. Peptide mimotopes of Mycobacterium tuberculosis carbohydrate immunodeterminants. Biochem. J. 2005;387:411–417. doi: 10.1042/BJ20041139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jonsson K. Molecular cloning and characterization of two Helicobacter pylori genes coding for plasminogen-binding proteins. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1852–1857. doi: 10.1073/pnas.0307329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bjerketorp J. A novel von Willebrand factor binding protein expressed by Staphylococcus aureus. Microbiology. 2002;148:2037–2044. doi: 10.1099/00221287-148-7-2037. [DOI] [PubMed] [Google Scholar]
- 56.Heilmann C. Staphylococcus aureus fibronectin-binding protein (FnBP)-mediated adherence to platelets, and aggregation of platelets induced by FnBPA but not by FnBPB. J. Infect. Dis. 2004;190:321–329. doi: 10.1086/421914. [DOI] [PubMed] [Google Scholar]
- 57.Ingham K.C. Interaction of Staphylococcus aureus fibronectin-binding protein with fibronectin – affinity, stoichiometry, and modular requirements. J. Biol. Chem. 2004;279:42945–42953. doi: 10.1074/jbc.M406984200. [DOI] [PubMed] [Google Scholar]
- 58.Williams R.J. Identification of a fibronectin-binding protein from Staphylococcus epidermidis. Infect. Immun. 2002;70:6805–6810. doi: 10.1128/IAI.70.12.6805-6810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williams R.J. Staphylococcus aureus fibronectin binding proteins A and B possess a second fibronectin binding region that may have biological relevance to bone tissues. Calcif. Tissue Int. 2002;70:416–421. doi: 10.1007/s00223-001-2073-z. [DOI] [PubMed] [Google Scholar]
- 60.Nilsson M. A fibrinogen-binding protein of Staphylococcus epidermidis. Infect. Immun. 1998;66:2666–2673. doi: 10.1128/iai.66.6.2666-2673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beckmann C. Identification of novel adhesins from group B streptococci by use of phage display reveals that C5a peptidase mediates fibronectin binding. Infect. Immun. 2002;70:2869–2876. doi: 10.1128/IAI.70.6.2869-2876.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobsson K. A novel family of fibrinogen-binding proteins in Streptococcus agalactiae. Vet. Microbiol. 2003;96:103–113. doi: 10.1016/s0378-1135(03)00206-2. [DOI] [PubMed] [Google Scholar]
- 63.Vasi J. M-like proteins of Streptococcus dysgalactiae. Infect. Immun. 2000;68:294–302. doi: 10.1128/iai.68.1.294-302.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lindmark H., Guss B. SFS, a novel fibronectin-binding protein from Streptococcus equi, inhibits the binding between fibronectin and collagen. Infect. Immun. 1999;67:2383–2388. doi: 10.1128/iai.67.5.2383-2388.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nilsson M. A von Willebrand factor-binding protein from Staphylococcus lugdunensis. FEMS Microbiol. Lett. 2004;234:155–161. doi: 10.1016/j.femsle.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 66.Nilsson M. A fibrinogen-binding protein of Staphylococcus lugdunensis. FEMS Microbiol. Lett. 2004;241:87–93. doi: 10.1016/j.femsle.2004.10.008. [DOI] [PubMed] [Google Scholar]