

Abstract
Some viruses have the ability to modulate the development of autoimmune diseases. Virus infections have long been associated with the exacerbation of autoimmune disease, however, there is also evidence that viruses can actually protect against autoimmune disease. Several experimental models have been developed to investigate how some virus infections can prime for and trigger autoimmunity whereas others ameliorate the pathway leading to clinical disease. It is possible that the type I interferons, via interleukin 12, provide the link between viruses and autoimmunity.
Keywords: virus, virus infection, autoimmune disease, autoimmunity
Autoimmune disease occurs when the immune system is fooled into attacking ‘self’. Almost any organ or tissue in the body represents ‘self’ and can therefore be a target for autoimmune destruction. In the USA, ∼1 in 30, or >8.5 million, individuals have one or more autoimmune diseases 1. Thus, autoimmunity is a prevalent problem that impacts on many individuals worldwide.
Although autoimmune diseases are multifactorial, genetics plays an important role. Twin studies have demonstrated that if one monozygotic twin develops an autoimmune disease, the other twin has roughly 25–60% concordance, depending on the autoimmune disease in question 2, 3, 4, 5, 6. Genetics therefore contributes to disease susceptibility. More than 12 genes contribute to autoimmunity but the contribution of each gene varies. Interestingly, when human leukocyte antigen (HLA) genes are studied, in most cases class II HLA molecules appear to be the restricting element, and these molecules appear to be associated with disease susceptibility 3, 7, 8. The one exception is HLA-B27 and its association with ankylosing spondylitis (AS) 9. AS is associated with this class I HLA molecule, but the mechanism by which HLA-B27 relates to the pathogenesis of this particular autoimmune disease is still unclear.
The key role of genetics in susceptibility to autoimmune disease presumably involves the presentation of self peptides by particular class II HLA molecules. However, other factors such as contributions from the environment (e.g. infections) have an essential role in the development of autoimmunity. Here, we will consider multiple sclerosis (MS), the human central nervous system (CNS) autoimmune demyelinating disease. Epidemiological studies indicate a paucity of MS around the equatorial regions of the earth 10, and the prevalence of MS is higher as you move further north or south from the equator. This phenomenon is connected not only with the genetics of the populations who inhabit different parts of the world but also with environmental agents present in these areas. Migration studies are cited as evidence for environmental factors and their timing being important in MS. An individual moving from a high-risk area to a lower-risk area after the age of 15 keeps the high-risk phenotype 11, 12. If that individual moves before age 15, they acquire the risk of the area to which he/she has moved. However, other studies have shown the reverse: that an individual moving from a low-risk area to a high-risk area after age 15 keeps the low-risk phenotype, but if they move before age 15 they acquire the high-risk rate of that region. In addition, MS is often diagnosed in individuals between the ages of 20 and 40. These data have been interpreted to indicate that environmental factors such as infections and the timing of these infections can influence susceptibility to MS (Refs 13, 14).
Along these lines, various investigators have made an association between virus and/or virus infections and the initiation and/or exacerbation of autoimmune disease. Such reports can be divided into two categories. The first involves reports in which investigators have isolated viruses from individuals with autoimmune diseases. Although in MS >16 viral agents have been isolated from individuals with the disease, no virus has been confirmed as the causative agent. By contrast, in diabetes, another autoimmune disease, a Coxsackievirus has been isolated from the pancreas of a patient who died of acute diabetes. When mice were infected with this virus, they developed diabetes. Clearly, in this case, the virus was the causative agent of disease 15. Similarly, congenital rubella virus infection is also associated with the occurrence of diabetes 16, 17, 18, 19; in one study >12% of children with congenital rubella virus infection acquired diabetes 20. Fortunately, the incidence of diabetes owing to rubella in young children has fallen dramatically because of vaccination against rubella. Thus, in certain cases, there is supporting evidence that virus infections can induce autoimmunity. However, Coxsackievirus and rubella virus persistence cannot explain the vast majority of type I diabetes.
One potential explanation for the development of virus-induced autoimmunity is that viruses with a particular tropism for the pancreas or CNS could initiate inflammation via the local induction of proinflammatory cytokines. The presence of the virus would drive presentation of self antigens by activated antigen-presenting cells, leading to autoimmune reactivity. This has been documented in two experimental models of virus-induced CNS autoimmune disease. Rats infected with a rodent-adapted strain of measles virus develop T cells reactive to myelin basic protein (MBP) 21. More recently, infection of mice with Theiler's murine encephalomyelitis virus was shown to induce autoreactive T cells against myelin proteolipid protein (PLP) late in the demyelinating phase (reviewed in Ref. 22). In both examples, virus persisted in the CNS and was not cleared. Alternatively, because virus could be isolated from the target organ, the disease could have arisen from an antiviral immune response in the target organ and, if this is the case, should not be considered a strict autoimmune disease. This possibility has been discussed in a previous review article 23.
The second group of studies involving viruses and autoimmunity 24, 25, 26, 27 also show an association between virus infection and exacerbation of disease, but the virus is no longer present or isolatable. For example, in approximately 1 in 1000 cases of acute measles virus infections of humans, post-infectious encephalomyelitis develops where T cells reactive to MBP are present. These patients clear the virus, and it is no longer isolatable, yet autoreactive cells are retained 28. Interestingly, in exacerbations in 20 MS patients, Beck et al. 29 found that before the exacerbation, increased interferon (IFN)-γ and tumor necrosis factor (TNF) production preceded disease relapses, suggesting an infectious association. Furthermore, Panitch 25 concluded that infectious illnesses, largely of non-specific viral origin, were responsible for the majority of exacerbations in a population of 30 relapsing–remitting MS patients. This evidence is indirect, and a problem with such associations is that a cause and effect conclusion is often difficult to substantiate 30. However, several experimental animal models have been developed to address the issue of how virus infections can protect from or potentiate autoimmune diseases.
1. Beneficial viral infections
Two experimental models in which virus infections protect against autoimmune disease will be discussed. The first model takes advantage of the non-obese diabetic (NOD) mouse strain 31 and the BB rat 32, 33, both of which spontaneously develop type I diabetes. Infection of either animal with lymphocytic choriomeningitis virus (LCMV) reduces the incidence of diabetes compared with sham-infected animals. In this work 31, 32, 33, it was shown that both insulitis and the normally elevated levels of insulin in the urine were markedly reduced. This was one of the first models to demonstrate that virus infections can protect against autoimmune disease. More recently, another group injected NOD mice with CpG-containing oligonucleotides or plasmid DNA containing CpG motifs and were also able to modulate diabetes 34. The mechanism of inhibition and actual viral infection is probably similar, and will be discussed later.
The second experimental model is an animal model of MS known as experimental allergic/autoimmune encephalomyelitis (EAE). In EAE, animals are sensitized with myelin, myelin proteins such as MBP or PLP, or encephalitogenic peptides (derived from the myelin proteins) in complete Freund's adjuvant (CFA), which potentiates immune responses. Animals can develop a relapsing–remitting clinical disease with CNS lesions similar to the acute inflammatory lesions seen in MS patients 35. Infection with a recombinant vaccina virus can protect mice from EAE. This model takes advantage of molecular mimicry 36, 37 as the recombinant virus shares an immunological determinant with a host CNS protein (Fig. 1 ). Molecular mimicry occurs when a microorganism and its host share an immunological epitope; the epitope can be a similar amino acid sequence or a comparable conformational determinant. An immune response induced by infection with a virus having a cross-reactive determinant also recognizes the similar host epitope. Another example of this is the cross-reaction between the M protein of Streptococcus and heart myosin – infection with certain strains of Streptococcus can lead to myocarditis owing to a cross-reacting immune response (reviewed in Refs 38, 39).
Fig. 1.
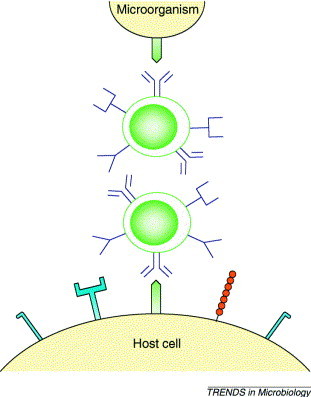
Molecular mimicry. Molecular mimicry occurs when a microorganism and its host share an immunological epitope. Infection with a virus having molecular mimicry (a cross-reacting determinant) with a self epitope could lead to an autoimmune response. Cross-reacting antibodies are known to react with conformational as well as linear epitopes and, more recently, it has been described that cross-reacting T cells recognize not only linear epitopes but also epitopes having similar conformations presented by major histocompatibility complex (MHC) molecules.
Mice were vaccinated with a recombinant vaccinia virus encoding an encephalitogenic epitope of MBP (Ref. 40). This infection alone did not cause autoimmune CNS disease. When these vaccinated mice were sensitized with the same encephalitogenic peptide or the entire MBP, they did not develop the symptoms or pathological changes of EAE (Ref. 40), whereas non-vaccinated mice and mice vaccinated with a control virus did develop EAE. Vaccinated mice had decreased lymphoproliferative and delayed-type hypersensitivity (DTH) responses to MBP. CD4+ T cells from these animals could not adoptively transfer EAE to naive mice, whereas T cells from mice infected with the control virus (vaccinia virus encoding β-galactosidase, VVSC11) could transfer disease. It is likely that recombinant vaccina virus can protect from EAE because acetylation of the first amino acid is important for the ability of this peptide to induce EAE (Ref. 41), and in the virus-encoded MBP peptide this residue is not acetylated. The recombinant virus presents an altered peptide ligand to the mouse, resulting in protection from autoimmune disease 42, 43, 44. It should be noted that another possible explanation is that T cells specific for the cross-reacting epitope were inappropriately stimulated during infection leading to anergy or unresponsiveness owing to activation-induced cell death 45. However, when taken together, these data indicate that viral infections can protect individuals against autoimmune disease; the types of infections described above might explain the paucity of MS observed in the equatorial regions of the world.
2. Detrimental viral infections
The other side of the coin is that viral infections can induce or initiate autoimmune disease. Two laboratories have developed an interesting model involving transgenic mice encoding the LCMV glycoprotein (GP) or nucleoprotein (NP) 46, 47. The viral gene is under the control of the rat insulin promoter; the viral protein is therefore expressed in the pancreas and, because it is present in the mouse from inception, it is considered to be a ‘self’ protein. When mice are infected with LCMV, they develop diabetes and this LCMV infection also generates an immune response to the ‘self’ LCMV GP or NP present in the pancreas. Thus, the murine immune system is fooled into attacking a self protein present in the pancreas. The effector cell in this model is the CD8+ T cell 48. A class I major histocompatibility complex (MHC) blocking peptide can inhibit the CD8+ T cells from mediating diabetes 49, 50.
This cross-reactive immune response leads to inflammation, insulitis and diabetes. If the ‘self’ (LCMV) protein is expressed in the thymus as well as the pancreas, CD4+ T cells are required to expand and activate the CD8 population sufficiently to induce overt disease 51. The hypothesis here is that an individual becomes infected very early in life with a virus having a specific tropism for an organ such as the pancreas or brain and this virus then persists in that organ. A later infection, with the same virus or a related virus having a similar epitope, leads to immune-mediated attack of virus-infected cells in that organ. Periodic infections with the same or related viruses could lead to increased damage and frank clinical disease. A similar scenario could occur if animals were infected with a virus having molecular mimicry with a pancreatic epitope. The immune-mediated damage could in itself result in sufficient tissue destruction to cause functional disease (i.e. diabetes) or immune-mediated attack of the target cells could release other self antigens. The release of other self antigens in this inflammatory milieu would lead to the presentation of additional self peptides to the immune system, initiating an autoimmune response different from that of the original inciting self determinant. This is often referred to as determinant or epitope spreading 52.
Another model of a virus potentiating an autoimmune disease is a variant of the EAE model. Here, mice infected with a recombinant vaccinia virus encoding myelin PLP did not develop CNS disease with infection by this virus alone. However, at a later time, mice were challenged with an encephalitogenic peptide, PLP139–151, to induce EAE and developed exacerbated, acute disease 53. This suggests that infection with virus capable of molecular mimicry with a self protein could prime for autoimmune disease.
Viral proteins are synthesized within infected cells. Both viral proteins and cellular proteins are ubiquitinated and transported to proteasomes. In the proteasome, proteins are degraded into peptides and can be presented on the cell surface by MHC molecules to T cells, which recognize viral peptides presented in association with MHC molecules. Ubiquitination of PLP targets it to the proteasome 54. Three-week-old female SJL/J mice were injected with a cDNA construct encoding ubiquitinated PLP (pUPLP) 55. Injection of young mice with pUPLP leads to the synthesis of PLP within cells, with PLP degradation via proteasomes (similar to an infection with a virus sharing cross-reacting epitopes with PLP) occurring early in life. The mice were rested and then challenged with a non-specific immunological stimulus (the adjuvant CFA). Interestingly, 20% of the mice developed EAE. One interpretation of these data is that viruses having molecular mimicry with self CNS proteins could prime mice for disease early in life and a non-specific immunological challenge such as an infection could initiate or exacerbate disease at a later time. To further test this hypothesis, mice were inoculated with pUPLP and rested; they were then infected with a recombinant vaccinia virus encoding β-galactosidase, an antigen that does not cross-react with any CNS protein, instead of being given CFA as a non-specific challenge. Again, 20% of these mice developed CNS inflammatory lesions. Therefore, a non-specific virus infection could replace CFA as an immunological stimulus. As a corollary to these experiments, mice were infected with recombinant vaccinia virus encoding PLP (VVPLP), rested and then given CFA as an immunological stimulus. The majority of mice developed inflammatory lesions within the CNS. Under these conditions, viruses encoding cross-reacting epitopes could silently prime mice for autoimmune disease and, at a later time, a viral infection could exacerbate the disease 55. This could explain why no virus has been identified as the causative agent in many autoimmune diseases such as MS and why infections are often seen in temporal association with attacks.
3. An hypothesis
I speculate that certain kinds of virus infections such as LCMV infection can inhibit or reduce spontaneous autoimmune disease in NOD mice or BB rats by inducing the production of large amounts of IFN-α/β, the type 1 IFNs. The production of type 1 IFNs downregulates the production of interleukin (IL)-12 by monocytes and dendritic cells 56. In many experimental models of autoimmune disease, IL-12 exacerbates disease. For example, IL-12 can exacerbate diabetes when given to NOD mice 57. Similarly, IL-12 can initiate an attack of EAE (Ref. 58). Therefore, IL-12 plays an important role in the expansion and activation of autoreactive CD4+ T cells. Thus, in the NOD–LCMV model, where viruses have a beneficial effect, we propose that the induction of IFN-α/β by infection results in the downregulation of IL-12, leading to a decrease in spontaneous disease.
In the experimental model where viruses with molecular mimicry to CNS proteins can prime mice for autoimmune disease, and a non-specific immunological challenge can exacerbate disease, we predict that certain kinds of viral infections – those that produce very little type 1 IFN and enhance IL-12 production – could exacerbate autoimmune disease (Fig. 2 ). Interestingly, viruses that stimulate the production of IL-12, including herpes simplex virus, human herpesvirus 6, influenza virus and coronavirus 59, 60, 61, 62, 63, 64, 65, 66, 67, have been isolated from or been associated with exacerbation of MS (Refs 25, 68). Virus-stimulated IL-12 production could lead to early NK cell activation. These NK cells produce large amounts of IFN-γ (Refs 69, 70). IFN-γ and IL-12 have been shown to be two prerequisites for the activation of CD4+ T cells 71, 72. If autoreactive CD4+ T cells have been previously expanded by virus infections having molecular mimicry with self CNS proteins, then infections with one of the viruses that induce high levels of IL-12 could now lead to further expansion of the autoreactive cells. Having these cells above a certain threshold could manifest as autoimmune disease.
Fig. 2.
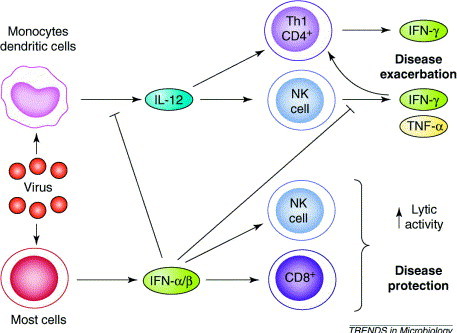
Proposed model showing why some virus infections can protect against autoimmune disease and others exacerbate disease. Certain viruses induce the production of large amounts of type I interferon (IFN-α/β). Type I IFNs downregulate interleukin (IL)-12 production leading to downregulation of disease. By contrast, other viruses induce the production of IL-12, which in turn can activate natural killer (NK) cells. IFN-γ produced by NK cells, together with IL-12, could stimulate and further expand autoreactive CD4+ T cells, which then exacerbate disease. This could explain why some infections are associated with exacerbations of autoimmune disease.
Acknowledgements
I wish to thank Jane E. Libbey for technical assistance, J. Lindsay Whitton for thoughtful discussions and Kathleen Borick for preparation of this manuscript. This research is supported by NIH AI42525 and NS34497.
References
- 1.Jacobson D.L. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol. 1997;84:223–243. doi: 10.1006/clin.1997.4412. [DOI] [PubMed] [Google Scholar]
- 2.Ebers G.C. A population-based study of multiple sclerosis in twins. New Engl. J. Med. 1986;315:1638–1642. doi: 10.1056/NEJM198612253152603. [DOI] [PubMed] [Google Scholar]
- 3.McFarland H.F. Twin studies and multiple sclerosis. Ann. Neurol. 1992;32:722–723. doi: 10.1002/ana.410320603. [DOI] [PubMed] [Google Scholar]
- 4.Kyvik K.O. Concordance rates of insulin dependent diabetes mellitus: a population based study of young Danish twins. Br. Med. J. 1995;311:913–917. doi: 10.1136/bmj.311.7010.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar D. North-American twins with IDDM. Genetic, etiological, and clinical significance of disease concordance according to age, zygosity, and the interval after diagnosis in first twin. Diabetes. 1993;42:1351–1363. doi: 10.2337/diab.42.9.1351. [DOI] [PubMed] [Google Scholar]
- 6.Olmos P. The significance of the concordance rate for type 1 (insulin-dependent) diabetes in identical twins. Diabetologia. 1988;31:747–750. doi: 10.1007/BF00274777. [DOI] [PubMed] [Google Scholar]
- 7.Zamani M., Cassiman J.J. Reevaluation of the importance of polymorphic HLA class II alleles and amino acids in the susceptibility of individuals of different populations to type I diabetes. Am. J. Med. Genet. 1998;76:183–194. doi: 10.1002/(sici)1096-8628(19980305)76:2<183::aid-ajmg12>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 8.Martin R. Genetics of multiple sclerosis – how could disease-associated HLA-types contribute to pathogenesis? J. Neural Transm. 1997;49(Suppl.):177–194. doi: 10.1007/978-3-7091-6844-8_19. [DOI] [PubMed] [Google Scholar]
- 9.Ebringer A., Wilson C. HLA molecules, bacteria and autoimmunity. J. Med. Microbiol. 2000;49:305–311. doi: 10.1099/0022-1317-49-4-305. [DOI] [PubMed] [Google Scholar]
- 10.Kurtzke J.F. Geography in multiple sclerosis. J. Neurol. 1977;215:1–26. doi: 10.1007/BF00312546. [DOI] [PubMed] [Google Scholar]
- 11.Hammond S.R. The age-range of risk of developing multiple sclerosis: evidence from a migrant population in Australia. Brain. 2000;123:968–974. doi: 10.1093/brain/123.5.968. [DOI] [PubMed] [Google Scholar]
- 12.Kurtzke J.F. Multiple sclerosis in North African migrants to France. Acta Neurol. Scand. 1998;98:302–309. doi: 10.1111/j.1600-0404.1998.tb01738.x. [DOI] [PubMed] [Google Scholar]
- 13.Delasnerie-Laupretre N., Alperovitch A. Childhood infections in multiple sclerosis: a study of North African-born patients who migrated to France. The French Collaborative Group on Multiple Sclerosis. Neuroepidemiology. 1990;9:118–123. doi: 10.1159/000110760. [DOI] [PubMed] [Google Scholar]
- 14.Alter M. Multiple sclerosis and childhood infections. Neurology. 1986;36:1386–1389. doi: 10.1212/wnl.36.10.1386. [DOI] [PubMed] [Google Scholar]
- 15.Yoon J.-W. Virus-induced diabetes mellitus. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. New Engl. J. Med. 1979;300:1173–1179. doi: 10.1056/NEJM197905243002102. [DOI] [PubMed] [Google Scholar]
- 16.Forrest J.M. High frequency of diabetes mellitus in young adults with congenital rubella. Lancet. 1971;2:332–334. doi: 10.1016/s0140-6736(71)90057-2. [DOI] [PubMed] [Google Scholar]
- 17.Johnson G.M., Tudor R.B. Diabetes mellitus and congenital rubella infection. Am. J. Dis. Child. 1970;120:453–455. doi: 10.1001/archpedi.1970.02100100117014. [DOI] [PubMed] [Google Scholar]
- 18.Plotkin S.A. Diabetes mellitus and congenital rubella. Pediatrics. 1970;46:650–651. [PubMed] [Google Scholar]
- 19.Forrest J.M. Diabetes mellitus and congenital rubella. Pediatrics. 1969;44:445–447. [PubMed] [Google Scholar]
- 20.Ginsberg-Fellner F. Diabetes mellitus and autoimmunity in patients with the congenital rubella syndrome. Rev. Infect. Dis. 1985;7(Suppl. 1):S170–S176. doi: 10.1093/clinids/7.supplement_1.s170. [DOI] [PubMed] [Google Scholar]
- 21.Liebert U.G. Induction of autoimmune reactions to myelin basic protein in measles virus encephalitis in Lewis rats. J. Neuroimmunol. 1988;17:103–118. doi: 10.1016/0165-5728(88)90018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller S.D. Evolution of the T-cell repertoire during the course of experimental immune-mediated demyelinating diseases. Immunol. Rev. 1995;144:225–244. doi: 10.1111/j.1600-065x.1995.tb00071.x. [DOI] [PubMed] [Google Scholar]
- 23.Whitton J.L., Fujinami R.S. Viruses as triggers of autoimmunity: facts and fantasies. Curr. Opin. Microbiol. 1999;2:392–397. doi: 10.1016/s1369-5274(99)80069-1. [DOI] [PubMed] [Google Scholar]
- 24.Sibley W.A. Clinical viral infections and multiple sclerosis. Lancet. 1985;1:1313–1315. doi: 10.1016/S0140-6736(85)92801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panitch H.S. Influence of infection on exacerbations of multiple sclerosis. Ann. Neurol. 1994;36(Suppl.):S25–S28. doi: 10.1002/ana.410360709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Keyser J. Effects of influenza vaccination and influenza illness on exacerbations in multiple sclerosis. J. Neurol. Sci. 1998;159:51–53. doi: 10.1016/s0022-510x(98)00139-7. [DOI] [PubMed] [Google Scholar]
- 27.Andersen O. Viral infections trigger multiple sclerosis relapses: a prospective seroepidemiological study. J. Neurol. 1993;240:417–422. doi: 10.1007/BF00867354. [DOI] [PubMed] [Google Scholar]
- 28.Johnson R.T. Measles encephalomyelitis – clinical and immunologic studies. New Engl. J. Med. 1984;310:137–141. doi: 10.1056/NEJM198401193100301. [DOI] [PubMed] [Google Scholar]
- 29.Beck J. Increased production of interferon γ and tumor necrosis factor precedes clinical manifestation in multiple sclerosis: do cytokines trigger off exacerbations? Acta Neurol. Scand. 1988;78:318–323. doi: 10.1111/j.1600-0404.1988.tb03663.x. [DOI] [PubMed] [Google Scholar]
- 30.von Herrath M.G. Obstacles to identifying viruses that cause autoimmune disease. J. Neuroimmunol. 2000;107:154–160. doi: 10.1016/s0165-5728(00)00227-7. [DOI] [PubMed] [Google Scholar]
- 31.Oldstone M.B.A. Prevention of type I diabetes in nonobese diabetic mice by virus infection. Science. 1988;239:500–502. doi: 10.1126/science.3277269. [DOI] [PubMed] [Google Scholar]
- 32.Dyrberg T. The incidence of diabetes in BB rats is decreased following acute LCMV infection. Adv. Exp. Med. Biol. 1988;246:397–402. doi: 10.1007/978-1-4684-5616-5_48. [DOI] [PubMed] [Google Scholar]
- 33.Dyrberg T. Inhibition of diabetes in BB rats by virus infection. J. Clin. Invest. 1988;81:928–931. doi: 10.1172/JCI113405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quintana F.J. Vaccination with empty plasmid DNA or CpG oligonucleotide inhibits diabetes in nonobese diabetic mice: modulation of spontaneous 60-kDa heat shock protein autoimmunity. J. Immunol. 2000;165:6148–6155. doi: 10.4049/jimmunol.165.11.6148. [DOI] [PubMed] [Google Scholar]
- 35.Tsunoda I., Fujinami R.S. Two models for multiple sclerosis: experimental allergic encephalomyelitis and Theiler's murine encephalomyelitis virus. J. Neuropathol. Exp. Neurol. 1996;55:673–686. doi: 10.1097/00005072-199606000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Fujinami R.S. Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc. Natl. Acad. Sci. U. S. A. 1983;80:2346–2350. doi: 10.1073/pnas.80.8.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujinami R.S., Oldstone M.B.A. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- 38.Cunningham M.W. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunningham M.W. Molecular mimicry between streptococcal M protein and cardiac myosin and the immunopathogenesis of rheumatic fever. In: Cunningham M.W., Fujinami R.S., editors. Molecular Mimicry, Microbes and Autoimmunity. ASM Press; 2000. pp. 39–56. [Google Scholar]
- 40.Barnett L.A. Virus encoding an encephalitogenic peptide protects mice from experimental allergic encephalomyelitis. J. Neuroimmunol. 1996;64:163–173. doi: 10.1016/0165-5728(95)00165-4. [DOI] [PubMed] [Google Scholar]
- 41.Zamvil S.S. T cell specificity for class II (I-A) and the encephalitogenic N-terminal epitope of the autoantigen myelin basic protein. J. Immunol. 1987;139:1075–1079. [PubMed] [Google Scholar]
- 42.Nicholson L.B. An altered peptide ligand mediates immune deviation and prevents autoimmune encephalomyelitis. Immunity. 1995;3:397–405. doi: 10.1016/1074-7613(95)90169-8. [DOI] [PubMed] [Google Scholar]
- 43.Santambrogio L. Altered peptide ligand modulation of experimental allergic encephalomyelitis: immune responses within the CNS. J. Neuroimmunol. 1998;81:1–13. doi: 10.1016/s0165-5728(97)00138-0. [DOI] [PubMed] [Google Scholar]
- 44.Ruiz P.J. Microbial epitopes act as altered peptide ligands to prevent experimental autoimmune encephalomyelitis. J. Exp. Med. 1999;189:1275–1284. doi: 10.1084/jem.189.8.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Parijs L., Abbas A.K. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 1998;280:243–248. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- 46.Oldstone M.B.A. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65:319–331. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 47.Ohashi P.S. Ablation of ‘tolerance’ and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 48.Laufer T.M. Autoimmune diabetes can be induced in transgenic major histocompatibility complex class II-deficient mice. J. Exp. Med. 1993;178:589–596. doi: 10.1084/jem.178.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.von Herrath M.G. In vivo treatment with a MHC class I-restricted blocking peptide can prevent virus-induced autoimmune diabetes. J. Immunol. 1998;161:5087–5096. [PubMed] [Google Scholar]
- 50.Aichele P. Peptide-induced T-cell tolerance to prevent autoimmune diabetes in a transgenic mouse model. Proc. Natl. Acad. Sci. U. S. A. 1994;91:444–448. doi: 10.1073/pnas.91.2.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.von Herrath M.G. How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in a transgenic model. Immunity. 1994;1:231–242. doi: 10.1016/1074-7613(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 52.Sercarz E.E. Dominance and crypticity of T cell antigenic determinants. Annu. Rev. Immunol. 1993;11:729–766. doi: 10.1146/annurev.iy.11.040193.003501. [DOI] [PubMed] [Google Scholar]
- 53.Barnett L.A. Enhancement of autoimmune disease using recombinant vaccinia virus encoding myelin proteolipid protein. J. Neuroimmunol. 1993;44:15–25. doi: 10.1016/0165-5728(93)90263-x. [DOI] [PubMed] [Google Scholar]
- 54.Rodriguez F., Whitton J.L. Enhancing DNA immunization. Virology. 2000;268:233–238. doi: 10.1006/viro.2000.0209. [DOI] [PubMed] [Google Scholar]
- 55.Theil, D.J. et al. Viruses can silently prime for and trigger central nervous system autoimmune disease. J. Neurovirology (in press) [DOI] [PubMed]
- 56.Biron C.A. Initial and innate responses to viral infections – pattern setting in immunity or disease. Curr. Opin. Microbiol. 1999;2:374–381. doi: 10.1016/s1369-5274(99)80066-6. [DOI] [PubMed] [Google Scholar]
- 57.Trembleau S. Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J. Exp. Med. 1995;181:817–821. doi: 10.1084/jem.181.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith T. Interleukin-12 induces relapse in experimental allergic encephalomyelitis in the Lewis rat. Am. J. Pathol. 1997;150:1909–1917. [PMC free article] [PubMed] [Google Scholar]
- 59.Kanangat S. Herpes simplex virus type 1-mediated up-regulation of IL-12 (p40) mRNA expression. Implications in immunopathogenesis and protection. J. Immunol. 1996;156:1110–1116. [PubMed] [Google Scholar]
- 60.Li C. Interferon-γ (IFN-γ) regulates production of IL-10 and IL-12 in human herpesvirus-6 (HHV-6)-infected monocyte/macrophage lineage. Clin. Exp. Immunol. 1997;109:421–425. doi: 10.1046/j.1365-2249.1997.4661362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arena A. Altered cytokine production after human herpes virus type 6 infection. New Microbiol. 1999;22:293–300. [PubMed] [Google Scholar]
- 62.Coutelier J.P. Interleukin-12 gene expression after viral infection in the mouse. J. Virol. 1995;69:1955–1958. doi: 10.1128/jvi.69.3.1955-1958.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Malmgaard L. Herpes simplex virus type 2 induces secretion of IL-12 by macrophages through a mechanism involving NF-κB. J. Gen. Virol. 2000;81:3011–3020. doi: 10.1099/0022-1317-81-12-3011. [DOI] [PubMed] [Google Scholar]
- 64.Cousens L.P. Interferon-α/β inhibition of interleukin 12 and interferon-γ production in vitro and endogenously during viral infection. Proc. Natl. Acad. Sci. U. S. A. 1997;94:634–639. doi: 10.1073/pnas.94.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Germann T., Rude E. Interleukin-12. Int. Arch. Allergy Immunol. 1995;108:103–112. doi: 10.1159/000237126. [DOI] [PubMed] [Google Scholar]
- 66.Monteiro J.M. Role of interleukin-12 in primary influenza virus infection. J. Virol. 1998;72:4825–4831. doi: 10.1128/jvi.72.6.4825-4831.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsutsumi H. Respiratory syncytial virus-induced cytokine production by neonatal macrophages. Clin. Exp. Immunol. 1996;106:442–446. doi: 10.1046/j.1365-2249.1996.d01-874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson R.T. Viral Infections of the Nervous System. Lippincott–Raven; 1998. [Google Scholar]
- 69.Biron C.A. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 70.Hafner M. Interleukin-12 activates NK cells for IFN-γ-dependent and NKT cells for IFN-γ-independent antimetastatic activity. Eur. Cytokine Netw. 1999;10:541–548. [PubMed] [Google Scholar]
- 71.Macatonia S.E. Dendritic cells and macrophages are required for Th1 development of CD4+ T cells from αβ TCR transgenic mice: IL-12 substitution for macrophages to stimulate IFN-γ production is IFN-γ-dependent. Int. Immunol. 1993;5:1119–1128. doi: 10.1093/intimm/5.9.1119. [DOI] [PubMed] [Google Scholar]
- 72.Wenner C.A. Roles of IFN-γ and IFN-α in IL-12-induced T helper cell-1 development. J. Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]