

Abstract
Severe acute respiratory syndrome (SARS) has been globally reported. A novel coronavirus (CoV), SARS‐CoV, was identified as the etiological agent of the disease. SARS‐CoV 3C‐like protease (3CLpro) mediates the proteolytic processing of replicase polypeptides 1a and 1ab into functional proteins, playing an important role in viral replication. In this study, we demonstrated the expression of the SARS‐CoV 3CLpro in Escherichia coli and Vero cells, and then characterized the in vitro trans‐cleavage and the cell‐based cis‐cleavage by the 3CLpro. Mutational analysis of the 3CLpro demonstrated the importance of His41, Cys145, and Glu166 in the substrate‐binding subsite S1 for keeping the proteolytic activity. In addition, alanine substitution of the cleavage substrates indicated that Gln‐P1 in the substrates mainly determined the cleavage efficiency. Therefore, this study not only established the quantifiable and reliable assay for the in vitro and cell‐based measurement of the 3CLpro activity, but also characterized the molecular interaction of the SARS‐CoV 3CLpro with the substrates. The results will be useful for the rational development of the anti‐SARS drugs.
Keywords: SARS, severe acute respiratory syndrome; CoV, coronavirus; 3CLpro, 3C-like protease; S-I (II), substrate-I (II); Luc, luciferase; SARS-coronavirus; 3C-like protease; trans- and cis-cleavage; Substrate specificity
1. Introduction
Severe acute respiratory syndrome (SARS) with high fever, malaise, headache, dry cough. and a progress of generalized, interstitial infiltrates in the lung has recently been reported over 32 countries around the world, including Taiwan, China, Hong Kong, Vietnam, and Canada [1, 2, 3, 4]. SARS was rapidly transmitted through aerosols, causing 8447 reported cases with 811 deaths worldwide in a short period from February to June, 2003 [5, 6, 7]. For successful control of the SARS outbreak, developing effective therapies and vaccines becomes medically important efforts.
A novel coronavirus (CoV), SARS‐coronavirus (SARS‐CoV), was identified as the etiological agent of the disease [1, 2, 3, 4]. SARS‐CoV particles contain a single positive‐stranded RNA genome that is approximately 30 kb in length and has a 5′ cap structure and 3′ poly(A) tract [8, 9, 10]. The SARS‐CoV genome encodes for replicase, spike, envelope, membrane, and nucleocapsid. The replicase gene encodes two large overlapping polypeptides (replicase 1a and 1ab, ∼450 and ∼750 kDa, respectively), including 3C‐like protease (3CLpro), RNA‐dependent RNA polymerase, and RNA helicase for viral replication and transcription [11]. The SARS‐CoV 3CLpro mediates the proteolytic processing of replicase polypeptides 1a and 1ab into functional proteins, playing an important role in viral replication. Eleven cleavage sites of the 3CLpro on the viral polyprotein have been mapped using the computer prediction based on the substrate conservation among CoV main proteases [12], being confirmed by the in vitro trans‐cleavage of 11 substrate peptides [13]. Therefore, the SARS‐CoV 3CLpro becomes an attractive target for developing effective drugs against SARS.
In this study, we characterized the in vitro trans‐cleavage and the cell‐based cis‐cleavage with the SARS‐CoV 3CLpro (Fig. 1A and B ). For the trans‐cleavage assay, the functional 3CLpro and three mutants at the substrate‐binding sites were used to test their proteolytic activity with the cleavage substrate‐I (S‐I, TVRLQAGNAT) fused at the N‐terminus of the SARS‐CoV non‐structure protein 7 (nsp7). For the cis‐cleavage assay, the in‐frame construction of the 3CLpro, the substrate‐II (S‐II, SAVLQSGFRK), and the luciferase (Luc) was transfected into the Vero cells. In addition, the mutations at the substrate conserved residues Leu and Gln were performed for the examination of the substrate specificity. In this study, the in vitro trans‐cleavage and cell‐based cis‐cleavage activities of the SARS‐CoV 3CLpro had been determined using the quantitative methods of an Enzyme‐Linked Immunosorbent Assay (ELISA) and a Luc assay, which will be useful for large‐scale screening of inhibitors against SARS.
Figure 1.
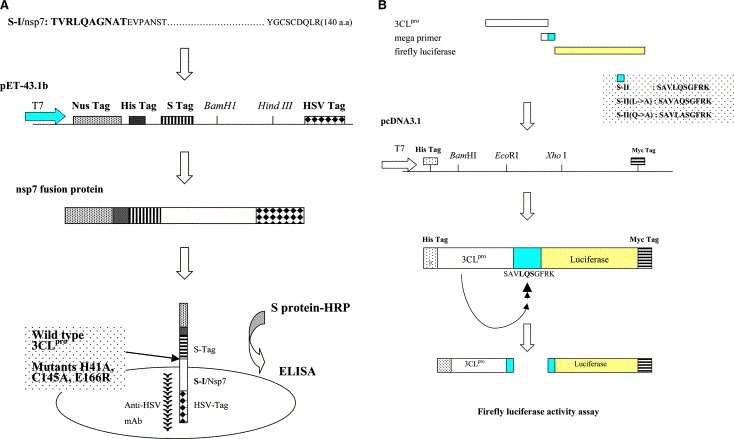
Flowcharts for the in vitro trans‐cleavage ELISA assay (A) and the cell‐based cis‐cleavage Luc assay (B). For trans‐cleavage assay (A), the substrate‐I/nsp7 (S‐I/nsp7) gene was cloned into the pET43.1b expression vector for generation of the in‐frame S Tag‐nsp7‐HSV Tag fusion proteins in E. coli. Subsequently, the trans‐cleavage product of the fusion protein S‐I/nsp7 by the 3CLpro and the mutants (H41A, C145A, and E166R) was captured onto the anti‐HSV mAb coated microwells. The non‐cleavage fusion protein was detected using the S protein‐HRP conjugate and ABTS/H2O2 substrates. For cis‐cleavage assay (B), the in‐frame construction of the 3CLpro, the substrate‐II, and Luc plus the pEGFP‐N1 was transfected into the Vero cells. The Luc activity was referred to monitor the cis‐cleavage of the fusion protein, the 3CLpro‐S‐II‐Luc.
2. Materials and methods
2.1. Construction, expression, and purification of SARS‐CoV 3CLpro
The 3CLpro gene located within the nucleotides 9985–10 902 of the SARS‐CoV TW1 strain genome (GenBank Accession No. genbank:AY291451) [14] was amplified using the reverse‐transcriptase polymerase chain reaction (RT‐PCR) with specific primers 5′‐CCCGGATCCAGTGGTTTTAGGAAAATGGCATTC‐3′ and 5′‐GGTGCTCGAGTTGGAAGGTAACACCAGAGCATTG‐3′. The forward primer mentioned above contained an EcoRI restriction site and the reverse primers included an XhoI restriction site. Each RT‐PCR product was digested with EcoRI and XhoI, and then ligated into the EcoRI/XhoI cleavage sites of the pET24a vector (Novagen) and expressed as a histidine tag fusion protein. The Escherichia coli strain BL21(DE3) was transformed with the resulting plasmid, pET24a‐3CLpro, then cultured in LB medium in the presence of 100 × g of kanamycin per ml at 37 °C. Once the cultures reached an absorbance of 0.5–0.6 at 600 nm, they were induced by the addition of 4 mM isopropyl‐β‐d‐thiogalactopyranoside for 4 h at 22 °C. Finally, the bacteria were harvested by centrifugation at 10 000 rpm for 15 min at 4 °C and resuspended in the 10 mM imidazole Bind buffer for sonication. The supernatant was purified with the HisTrap Kit and buffer kit (Amersham) after centrifugation (10 000 × g for 20 min). The concentration of the purified protein was determined using the Bio‐Rad protein assay reagent.
2.2. SDS–PAGE and Western blotting
The samples from fractions of each purification step were then dissolved in 2× SDS–PAGE sample buffer without 2‐mercaptoethanol and boiled for 10 min. Proteins were resolved on 12% SDS–PAGE gels and stained with Coomassie Brilliant Blue (Sigma). Moreover, the electrophoretically separated proteins were transferred to nitrocellulose paper. The resultant blots were blocked with 5% skimmed milk and then reacted with the appropriately diluted Anti‐His Tag monoclonal antibody (mAb) (Serotec) for a 3‐h incubation. The blots were then washed with 1% TBS containing 0.05% Tween 20 (TBST) three times and overlaid with a 1/5000 dilution of goat anti‐mouse IgG antibodies conjugated with alkaline phosphatase (Perkin–Elmer Life Sciences, Inc.). Following a 1‐h incubation at room temperature, the blots were developed with TNBT/BCIP (Gibco).
2.3. Azocasein digestion of SARS‐CoV 3CLpro
The protease activity of the SARS‐CoV 3CLpro was determined spectrophotometrically following the digestion of azocasein (Sigma) as the substrate [15]. 150 μl of samples was added to 150 μl of chromogen reagent containing 2% azocasein in 50 mM Tris–HCl, pH 8.5. After 2‐h incubation at 37 °C, non‐digested azocasein was precipitated by adding 350 μl of 10% trichloroacetic acid (TCA) (Merck). For determining the proteolytic activity, 350 μl of the resulting supernatants centrifuged at 10 000 × g for 10 min, was mixed with 300 μl of 1 N NaOH, and then the absorbance of the above mixture at 440 nm was measured. The blank was obtained by precipitating the substrate plus the sample in TCA without incubation.
2.4. Site‐directed mutagenesis of SARS‐CoV 3CLpro
His41, Cys145, and Glu166 within the catalytic sites of the SARS‐CoV 3CLpro protein were mutated by the PCR method similar to our previous report [16]. Site‐directed mutagenesis was conducted by using paired complementary oligonucleotides for the desired point mutations to generate specific mutations into the SARS‐CoV 3CLpro protein. The pairs of primers used were 5′‐TGTCCAAGAGCCGTCATTTGC‐3′ and 5′‐GCAAATGACGGCTCTTGGACA‐3′ for the substitution of His41 with alanine, 5′‐AATGGATCAGCCGGTAGTGTT‐3′ and 5′‐AACACTACCGGCTGATCCATT‐3′ for the replacement of Cys145 by alanine, and 5′‐CATCATATGCGGCTTCCAACA‐3′ and 5′‐TGTTGGAAGCCGCATATGATG‐3′ for the mutation of Glu166 with arginine. The correct sequence of each mutant of the SARS‐CoV 3CLpro protein constructs was confirmed using a DNA sequence analysis.
2.5. Construction and expression of SARS‐CoV nsp7 fusion protein
The nsp7 gene located in the nucleotides 12 937–13 356 of the TW1 strain genome. The cleavage S‐I (TVRLQAGNATE) for the 3CLpro protein located within the junction of nsp6 and nsp7, being fused at the N‐terminus of the nsp7 protein (Fig. 1A). The S‐I/nsp7 gene was amplified using PCR with specific paired primers 5′‐CGTGGATCCGGCTACAGTACGTCTTCAGGCT‐3′ and 5′‐CGCaagcttGCGGAGTTGGTCACAACTACA‐3′. The forward primer mentioned above contained a BamHI restriction site and the reverse primers included a HindIII restriction site. Each RT‐PCR product was digested with BamHI and HindIII, and then ligated into the BamHI/HindIII cleavage sites of the pET43.1b vector (Novagen). The resultant plasmids were transformed into the E. coli strain BL21(DE3). The S‐I/nsp7 fusion protein expressed in E. coli was purified using the HisTrap Kit (Amersham).
2.6. In vitro trans‐cleavage activity of the 3CLpro determined by ELISA
For determining the trans‐acting proteolytic assay, the SARS‐CoV 3CLpro reacted with the S‐I/nsp7 fusion protein captured onto the microtiter plates (Fig. 1A). The wells of a 96‐well plate were coated with 100 μl of diluted anti‐HSV mAb (Novagen) and incubated overnight at 4 °C. Following each incubation and subsequent layer of the ELISA, the wells were washed three times with TBST. After blocking by incubation with 5% skimmed milk in TBST for 2 h at room temperature (200 μl/well), 100 μl of the mixture containing the S‐I/nsp7 fusion protein (10 μg/ml) and the 3CLpro (300 μg/ml) was added into anti‐HSV mAb‐coated wells for the 3‐h incubation. The intact form of the S‐I/nsp7 fusion protein was detected using the S protein conjugated to peroxidase (Novagen) for 1 h at room temperature. The ELISA products were developed with a chromogen solution containing 2,2′‐azino‐di‐(3‐ethylbenzthiazoline‐6‐sulfonate) (ABTS) and hydrogen peroxide. The relative trans‐cleavage activity was calculated as 1 −
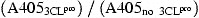 .
.
2.7. The cell‐based cis‐cleavage activity of the 3CLpro
For examining the cis‐acting proteolytic assay, the 3CLpro was fused in‐frame with a cleavage site and a Luc at the C‐terminus (Fig. 1B). The 3CLpro gene was amplified using PCR with the paired primers 5′‐CCCGGATCCAGTGGTTTTAGGAAAATGGCATTC‐3′ and 5′‐TGCAGAATTCTTTCCTAAAACCACTCTGCAGAACAGCAGATTGGAAGGTAACACCAGAGCATTG‐3′. The forward primer mentioned above contained a BamHI restriction site and the reverse primer included an EcoRI restriction site and an in‐frame gene encoding for the C‐terminus of the 3CLpro and the S‐II (SAVLQSGFRK). The PCR product was digested with BamHI and EcoRI, and then cloned into the pcDNA3.1 vector (Amersham). The resulting plasmid was named as Pro/S‐II. Meanwhile, the substitutions of Leu and Gln in the S‐II by alanine with the plasmid Pro/S‐II were also constructed using the reverse primers 5′‐TGCAGAATTCTTTCCTAAAACCACTCTGCGCAACAGCAGA‐3′ and 5′‐TGCAGAATTCTTTCCTAAAACCACTCGCCAGAACAGCAGA‐3′, which plasmids were designed as Pro/S‐II(L → A) and Pro/S‐II(Q → A), respectively. Subsequently, the firefly Luc gene in the pGL3(R2.1)‐Basic vector (Promega) was amplified by PCR with the primers 5′‐GGTGGAATTCATGGAAGACGCCAAAAACATAAAG‐3′ and 5′‐TAGACTCGAGTTACACGGCGATCTTTCCGCCCTT‐3′. The Luc gene was cloned into the EcoRI/XhoI restriction enzyme sites of the plasmids Pro/S‐II, Pro/S‐II(L → A) and Pro/S‐II(Q → A). These recombinant plasmids were designed as Pro/S‐II/Luc, Pro/S‐II(L → A)/Luc and Pro/S‐II(Q → A)/Luc for detecting the cis‐acting proteolytic activity of the 3CLpro in Vero cells. Vero cells at 60–90% confluency in 6‐well plates were transfected with 5 μg of total plasmids (0.5 μg of the indicated expression vector pEGFP‐N1 plus 4.5 μg of pcDNA3.1), Pro/S‐II, Pro/S‐II/Luc, Pro/S‐II(L → A)/Luc or Pro/S‐II(Q → A)/Luc, using the GenePorter reagent. According to the manufacturer's direction (Gene Therapy Systems, San Diego, CA), the transfected cells were maintained in 2 ml of Dulbeccos modified Eagle's medium (DMEM) containing 20% bovine serum (FBS) after 5‐h incubation with the mixture of the plasmid DNA and GenePorter reagent. Transfected cells were selected using the DMEM containing 10% FBS and 800 μg/ml of G418 for one month, and then G418‐resistant cell clones were confirmed by the expression of green fluorescent protein (EGFP). Firefly Luc activity in the transfected cells was measured using the dual Luciferase Reporter Assay System (Promega) and the Luminometer TROPIX TR‐717 (Applied Biosystems).
3. Results
3.1. Expression and purification of the recombinant SARS‐CoV 3CLpro in E. coli
To examine the expression of the 3CLpro protein in E. coli, the C‐terminal His tagged 3CLpro protein was detected using Western blotting with anti‐His Tag monoclonal antibody. Western blot revealed that a 34‐kDa protein in the supernatant and the pellet fractions was found in the recombinant E. coli (data not shown). The 34‐kDa recombinant protein was in agreement with the theoretical molecular weight (33.8 kDa) of the recombinant 3CLpro fusion protein using the Compute pI/Mw tool (http://tw.expasy.org).
Subsequently, the soluble 3CLpro protein expressed in the E. coli was harvested from the supernatant of the sonicated cells and then purified using the immobilized‐metal affinity chromatography (IMAC). The Coomassie blue‐stained gel revealed that one major band of the recombinant 3CLpro protein was eluted with imidazole ranging from 100 to 500 mM (Fig. 2A , lanes 5–9). The high purity of the 3CLpro was observed in the last eluted fraction with 500 mM imidazole (Fig. 2A, lane 9). Interestingly, Western blotting demonstrated that an about 68‐kDa immuno‐reactive band, except a 34‐kDa band, was found at more than 200 μg/ml concentration of our purified 3CLpro, which was in the eluted fraction with imidazole ranging from 300 to 500 mM (Fig. 2B, lanes 5–7). However, no 68‐kDa immuno‐reactive band was observed at lower than 200 μg/ml concentration, being in the eluted fraction with 200 mM imidazole (Fig. 2B, lane 4). The result was in agreement with a previous report in which the purified SARS‐CoV 3CLpro existed at more than 200 μg/ml protein concentration as a mixture of the inactive monomer (major) and the active dimer (minor) [13].
Figure 2.
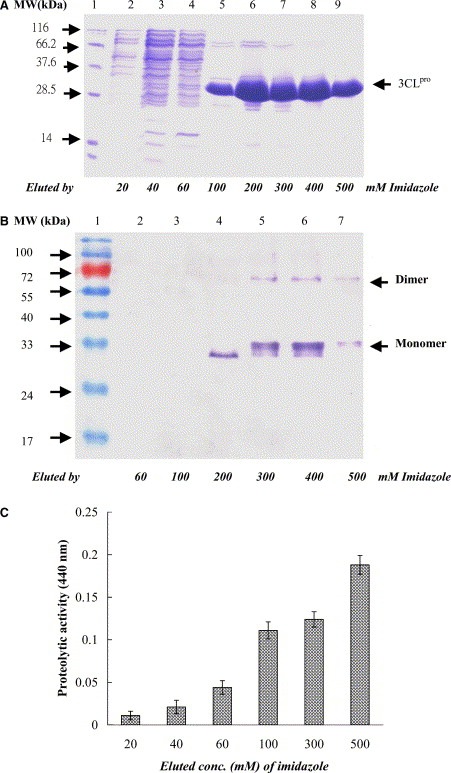
SDS–PAGE (A), Western blotting (B), and enzyme activity (C) of the recombinant SARS‐CoV 3CLpro in each purified step. (A) The supernatant of the sonicated cells transformed with the pET24a‐3CLpro plasmid was purified by IMAC. 20 μl of each eluted fraction was analyzed by 12% SDS–PAGE with Coomassie blue staining. Lanes 2–9 indicated that the samples from the fractions eluted with 20, 40, 60, 100, 200, 300, 400, and 500 mM imidazole, respectively. Lane 1 was the molecular marker. kDa, kilodaltons. (B) 5 μl of each eluted fraction was analyzed by 12% SDS–PAGE, then electrophorectically transferred onto nitrocellulose paper. The blot was probed with mouse anti‐His tag antibodies, and developed with an alkaline phosphatase‐conjugated secondary antibody and NBT/BCIP substrates. Lanes 2–7 indicated that the samples from the fractions eluted with 60, 100, 200, 300, 400, and 500 mM imidazole, respectively. (C) The digestion of azocasein by the 3CLpro protein was measured at 440 nm. The blank was obtained by precipitating the substrate plus the sample in TCA without incubation.
3.2. Azocasein hydrolysis of the SARS‐CoV 3CLpro
To test the protease activity of the recombinant 3CLpro protein, the azocasein hydrolysis of the 3CLpro protein in each eluted fraction was further performed (Fig. 2C). The azocasein proteolytic profile revealed that the 3CLpro protein with a high purity eluted at 500 mM imidazole has highest proteolytic activity compared to those eluted fractions at 20, 40, 60, 100, and 300 mM imidazole (Fig. 2C). Moreover, the azocasein proteolytic assay showed a dose‐dependent ability of the 3CLpro protein using the serial 2‐fold dilution ranging from 200 to 800 μg/ml (data not shown). The results showed the enzyme activity of the purified 3CLpro protein.
3.3. In vitro trans‐cleavage activity of the 3CLpro
The proteolytic specificity of the SARS‐CoV 3CLpro was examined using the in vitro trans‐cleavage of the S‐I (TVRLQAGNATE mapped at the junction of nsp6 and nsp7). The S‐I was fused in‐frame with a Nus‐Tag and an S‐Tag at the N‐terminus and the nsp7 and an HSV‐Tag at the C‐terminus (Fig. 1A). According to the structure knowledge [17], mutations of His‐41 by Ala (H41A), Cys‐145 by Ala (C145A), and Glu166 by Arg (E166R) within the substrate‐binding site SI were also performed and tested the effects on the enzyme activity. The trans‐cleavage of the S‐I/nsp7 fusion protein (Nus Tag/S‐Tag/S‐I/nsp7/HSV‐Tag) by the 3CLpro and the mutants was analyzed using the Western blotting with the S‐protein conjugated to peroxidase. An immuno‐band for the cleavage product, the Nus Tag/S‐Tag protein, was detected in the trans‐cleavage by 3CLpro, but not in the reactions by the 3CLpro mutants H41A, C145A, and E166R (data not shown). For quantification of the in vitro trans‐cleavage, the mixture of the 3CLpro and the S‐I/nsp7 fusion protein was incubated in the anti‐HSV mAb‐coated wells. Subsequently, the non‐cleavage form of the S‐I/nsp7 fusion protein was captured and then detected using the ELISA with the S‐protein conjugated to peroxidase (Fig. 3 ). The relative trans‐cleavage ability revealed that the 3CLpro mutants H41A, C145A, and E166R lose more than 50% activity compared to the wild type 3CLpro. Furthermore, the enzyme activity of the 3CLpro was significantly inhibited by the serine protease inhibitor 4‐(2‐aminoethyl)benzenesulfonyl fluoride hydrochloride (ABF) (Fig. 3). These results demonstrated the trans‐cleavage specificity of the 3CLpro and the importantly functional role of the residues His41, Cys145, and Glu166 within the substrate‐binding site S1.
Figure 3.
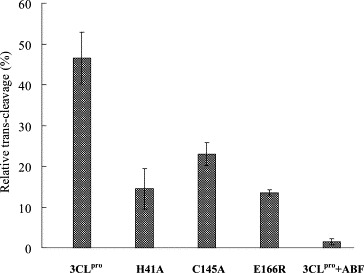
ELISA for the trans‐cleavage of the S‐I/nsp7 fusion protein by the 3CLpro and the mutants H41A, C145A, and E166E. After a 3‐h incubation, the mixture of the fusion protein Nus‐Tag/S‐Tag/S‐I/nsp7/HSV‐Tag and the 3CLpro was added into the anti‐HSV mAb‐coated wells. The inhibitor ABF was also added into the trans‐cleavage assay. The non‐cleavage S‐I/nsp7 fusion protein captured onto 96‐well plates with anti‐HSV mAb was detected using the S protein‐HRP conjugate and ABTS/H2O2 substrates. The ELISA product was measured at A405 nm. The relative trans‐cleavage activity was calculated as 1 −
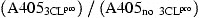 .
.
3.4. cis‐cleavage activity of the 3CLpro in the cell‐based assays
For the cell‐based cis‐cleavage assay, the in‐frame construction (Pro/S‐II/Luc) of the 3CLpro, the S‐II (SAVLQSGFRK), and the Luc plus pEGFP‐N1 was transfected into Vero cells (Fig. 4D ). Since the fusion of the firefly Luc with a more than 30 kDa protein fused as the N‐terminus resulted in a dramatic decrease of Luc activity [18], the detection of Luc activity could be referred to the measurement of the cis‐cleavage by the SARS‐CoV 3CLpro. For examining the substrate specificity, alanine substitution of the substrate conserved residues Leu‐P2 and Gln‐P1, designed as Pro/S‐II(L → A)/Luc and Pro/S‐II(Q → A)/Luc, were also carried out (Fig. 4E and F). Western blotting of the cell lysates with the anti‐His Tag mAb demonstrated that three immuno‐bands, a 94‐kDa band for the fusion protein 3CLpro/S‐II/Luc, a 68‐kDa band for the 3CLpro dimer and a 34‐kDa band for the 3CLpro monomer, were clearly observed in the Vero cells transfected with Pro/S‐II/Luc, Pro/S‐II(L → A)/Luc, and Pro/S‐II(Q → A)/Luc (data not shown). However, the relative Luc activity in the transfected cells showed that the Vero cells transfected with the plasmid Pro/S‐II/Luc (100 689 ± 408 light units) showed 4‐ and 25‐fold higher Luc activity compared to the cells carrying Pro/S‐II(L → A)/Luc (23 972 ± 582 light units) and Pro/S‐II(Q → A)/Luc (3780 ± 282 light units), respectively (Fig. 4G). The results demonstrated the cis‐cleavage of the fusion protein 3CLpro/substrate‐II/Luc. Moreover, mutational analysis indicated that the conserved residue Gln at the P1 position mainly determined the substrate cleavage efficiency of the 3CLpro.
Figure 4.
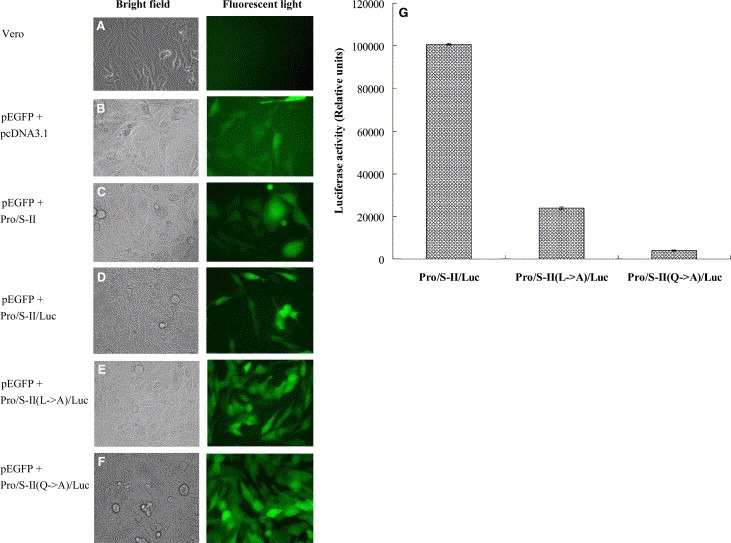
Expression of the in‐frame gene 3CLpro/S‐II/Luc for the cis‐cleavage analysis. (A–F) Microscopical analysis of the Vero cells with bright field (left) and fluorescent light (right). Vero cells (B–F) transfected with pEGFP‐N1 plus pcDNA3.1, Pro/S‐II, Pro/S‐II/Luc, Pro/S‐II(L → A)/Luc or Pro/S‐II(Q → A)/Luc were compared to the un‐transfected cells (A). (G) Equal amounts (100 × g) of cell lysates were used to determine the Luc activity using the dual Luciferase Reporter Assay System. The reference cells transfected with pcDNA3.1 or Pro/S‐II were used to subtract background noise. Relative Luc activity in the test cells transfected with Pro/S‐II/Luc, Pro/S‐II(L → A) or Pro/S‐II(Q → A)/Luc was determined as the value of light unitstest‐reference.
4. Discussion
In this study, we demonstrated the expression and functional activity of the SARS‐CoV 3CLpro in E. coli and Vero cells, and also characterized the trans‐ and cis‐cleavage of the substrates TVRLQAGNAT and SAVLQSGFRK in the fusion proteins by the 3CLpro. In addition, we examined the active site SI and substrate specificity of the 3CLpro using site‐directed mutagenesis, providing the insight into molecular recognition of the SARS‐CoV 3CLpro with the substrates for the rational design of anti‐SARS drugs.
Our results indicated that the recombinant SARS‐CoV 3CLpro exists as a mixture of monomers (major) and dimers (minor) in the solutions (Fig. 2B), being in agreement with other studies on the recombinant protease of human CoV (HCoV) and the related porcine transmissible gastroenteritis (corona)virus (TGEV) [19, 20]. Furthermore, alanine substitution at the Cys145–His41 catalytic dyad resulted in a significant loss of the 3CLpro enzyme activity (Fig. 3), revealing the importance of the Cys145–His41 catalytic dyad. According to the crystallographic data [17], the Glu166 in the substrate‐binding subsite, SI, of the SARS‐CoV 3CLpro has a salt bridge with His172 and hydrogen bonds with the NH group of the other monomer Ser1, being important for the substrate binding and the 3CLpro dimerization. Mutational analysis of Glu166 conformed the importance of Glu166 in the enzymatic function of the SARS‐CoV 3CLpro (Fig. 3). These results showed the important role of the substrate‐binding subsite S1 in the anti‐SARS drug design.
The identified cleavage site of the SARS‐CoV 3CLpro contains a LQA(S,N) motif recognized by most other CoV proteases [12, 21], which leads us to suggest that the proteolytic processing of the SARS‐CoV replicase polyproteins could be similar to those of other CoVs, such as HCoV and TGEV. Based on the conserved LQA(S,N) motif, Leu‐P2 and Gln‐P1 were selected for mutational analysis of the substrate specificity by the 3CLpro. Alanine substitution at Leu‐P2 and Gln‐P1 revealed that Gln‐P1 dominantly determined the cleavage efficiency of the substrates by the SARS‐CoV 3CLpro (Fig. 4G). We will further characterize the substrate specificity by the systematically mutational analysis for the molecular‐based design on the anti‐SARS drugs.
In this study, we establish the in vitro trans‐cleavage assay and the cell‐based cis‐cleavage assay with the recombinant 3CLpro protein. The azocasein and the substrate fusion protein Nus‐Tag/S‐Tag/S‐I/nsp7/HSV‐Tag were used as the substrates in the in vitro trans‐cleavage assay, providing the rapid and quantifiable assay for large‐scale screening of SARS‐CoV 3CLpro inhibitors. Furthermore, the Luc activity referred to the cell‐based cis‐cleavage activity will be useful for the examination of the inhibitory effects on the SARS‐CoV 3CLpro in the Vero cells. Therefore, this study not only characterizes the molecular interaction of the SARS‐CoV 3CLpro with the substrates, but also provides reliable assays for screening the anti‐SARS drugs.
Acknowledgements
We thank the National Science Council (Taiwan) and China Medical University for financial supports (NSC 92‐2314‐B‐039‐030, NSC 92‐2751‐B‐039‐009‐Y, and CMU92‐MT‐03).
Lin Cheng-Wen,Tsai Chang-Hai,Tsai Fuu-Jen,Chen Pei-Jer,Lai Chien-Chen,Wan Lei,Chiu Hua-Hao and Lin Kuan-Hsun(2004), Characterization of trans- and cis-cleavage activity of the SARS coronavirus 3CLpro protease: basis for the in vitro screening of anti-SARS drugs, FEBS Letters, 574, doi: 10.1016/j.febslet.2004.08.017
References
- 1. Poutanen S.M., Low D.E., Henry B., Finkelstein S., Rose D., Green K., Tellier R., Draker R., Adachi D., Ayers M., Chan A.K., Skowronski D.M., Salit I., Simor A.E., Slutsky A.S., Doyle P.W., Krajden M., Petric M., Brunham R.C., McGeer A.J., N. Engl. J. Med., 348, (2003), 1995– 2005. [DOI] [PubMed] [Google Scholar]
- 2. Lee N., Hui D., Wu A., Chan P., Cameron P., Joynt G.M., Ahuja A., Yung M.Y., Leung C.B., To K.F., Lui S.F., Szeto C.C., Chung S., Sung J.J., N. Engl. J. Med., 348, (2003), 1986– 1994. [DOI] [PubMed] [Google Scholar]
- 3. Tsang K.W., Ho P.L., Ooi G.C., Yee W.K., Wang T., Chan-Yeung M., Lam W.K., Seto W.H., Yam L.Y., Cheung T.M., Wong P.C., Lam B., Ip M.S., Chan J., Yuen K.Y., Lai K.N., N. Engl. J. Med., 348, (2003), 1977– 1985. [DOI] [PubMed] [Google Scholar]
- 4. Hsueh P.R., Chen P.J., Hsiao C.H., Yeh S.H., Cheng W.C., Wang J.L., Chiang B.L., Chang S.C., Chang F.Y., Wong W.W., Kao C.L., Yang P.C., Emerg. Infect. Dis., 10, (2004), 489– 493. [DOI] [PubMed] [Google Scholar]
- 5. Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J., N. Engl. J. Med., 348, (2003), 1953– 1966. [DOI] [PubMed] [Google Scholar]
- 6. Peiris J.S., Chu C.M., Cheng V.C., Chan K.S., Hung I.F., Poon L.L., Law K.I., Tang B.S., Hon T.Y., Chan C.S., Chan K.H., Ng J.S., Zheng B.J., Ng W.L., Lai R.W., Guan Y., Yuen K.Y., Lancet, 361, (2003), 1767– 1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W., N. Engl. J. Med., 348, (2003), 1967– 1976. [DOI] [PubMed] [Google Scholar]
- 8. Lai M.M.C., Holmes K.V., Knipe D.M. Howley P.M. Fields Virology (2001), Lippincott Williams and Wilkins ; New York: [Google Scholar]
- 9. Enjuanes L., Brian D., Cavanagh D., Holmes K., Lai M.M.C., Laude H., Masters P., Rottier P., Siddell S.G., Spaan W.G.M., Taguchi F., Talbot P., van Regenmortal M.H.V. Fauqet C.M. Bishop D.H.L. Carstens E.B. Estes M.K. Lemon S.M. Mayo M.A. McGeoch D.J. Pringle C.R. Wickner R.B. Virus Taxonomy (2000), Academic Press ; New York: [Google Scholar]
- 10. Holmes K.V., Knipe D.M. Howley P.M. Fields Virology (2001), Lippincott Williams and Wilkins ; New York: [Google Scholar]
- 11. Ziebuhr J., Snijder E.J., Gorbalenya A.E., J. Gen. Virol., 81, (2000), 853– 879. [DOI] [PubMed] [Google Scholar]
- 12. Gao F., Ou H.Y., Chen L.L., Zheng W.X., Zhang C.T., FEBS Lett., 553, (2003), 451– 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L., J. Bio. Chem., 279, (2004), 1637– 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsueh P.R., Hsiao C.H., Yeh S.H., Wang W.K., Chen P.J., Wang J.T., Chang S.C., Kao C.L., Yang P.C., Emerg. Infect. Dis., 9, (2003), 1163– 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tomarelli R.M., Charney J., Harding M.L., J. Lab. Clin. Med., 34, (1949), 428– 433. [PubMed] [Google Scholar]
- 16. Lin C.W., Wu S.C., J. Virol., 77, (2003), 2600– 2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z., Proc. Natl. Acad. Sci. USA, 100, (2003), 13190– 13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Joubert P., Pautigny C., Madelaine M.F., Rasschaert D., J. Gen. Virol., 81, (2000), 481– 488. [DOI] [PubMed] [Google Scholar]
- 19. Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R., EMBO J., 21, (2002), 3213– 3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R., Science, 300, (2003), 1763– 1767. [DOI] [PubMed] [Google Scholar]
- 21. Hegyi A., Ziebuhr J., J. Gen. Virol., 83, (2002), 595– 599. [DOI] [PubMed] [Google Scholar]