

Graphical abstract
Phosphoramidate ProTides of (fluoro) deoxyribose are reported for the first time. Their biology and stability is reported.

Keywords: Sugars, Deoxyribose, Phosphorylation, Prodrugs, Fluorination
Abstract
We report in this Letter the synthesis of prodrugs of 2-fluoro-2-deoxyarabinose-1-phosphate and 2,2-difluoro-2-deoxyribose-1-phosphate. We demonstrate the difficulty of realising a phosphorylation step on the anomeric position of 2-deoxyribose, and we discover that introduction of fluorine atoms on the 2 position of 2-deoxyribose enables the phosphorylation step: in fact, the stability of the prodrugs increases with the degree of 2-fluorination. Stability studies of produgs of 2-fluoro-2-deoxyribose-1-phosphate and 2,2-difluoro-2-deoxyribose-1-phosphate in acidic and neutral conditions were conducted to confirm our observation. Biological evaluation of prodrugs of 2,2-difluoro-2-deoxyribose-1-phosphate for antiviral and cytotoxic activity is reported.
Glycosyl-1-phosphates are important compounds in many life processes. They are essential constituents of larger biomolecules and are key intermediates in the metabolism of sugars and their transformation into nucleosides. Among them, 2-deoxy-α-d-ribose-1-phosphate is a catabolic product of thymidine phosphorylase (EC 2.4.2.4), an enzyme involved in the metabolism of pyrimidine nucleosides and which may promote angiogenesis.1 Thymidine phosphorylase is also involved in the degradation of antiviral agents such as BVDU.2 It is recognized that the therapeutic potential of drugs bearing a phosphate, phosphinate or phosphonate moiety is considerably reduced due to their low membrane permeation because each of these groups carry negative charges at physiological pH.3 Several prodrug technologies have been investigated to overcome this problem4 and, among them, the phosphoramidate ProTide technology has been successfully applied to various nucleoside analogues.5 Furthermore the phosphoramidate technology has been recently applied to sugars (mainly glucosamine)6 in order to improve their therapeutic potential. The synthesis of phosphoroamidate prodrugs of 2-deoxy-α-d-ribose-l-phosphate is of great interest, as they could act as inhibitors of thymidine phosphorylase or regulators in associated metabolic processes.7
Many papers have reviewed the introduction of a phosphate group at the anomeric position of a glycosyl unit.8 Three main strategies are observed: (a) the sugar, activated at the anomeric position, acts as an electrophilic compound and a nucleophilic displacement reaction is performed thanks to a phosphate anion. In this case, the electrophilic compounds are generally glycosyl halides,9 glycosyl-trichloroacetimidates,10 glycosyl nitrates,11 or glycosyl thioimidoyl derivatives;12 (b) the anomeric hydroxyl group of the sugar acts as the nucleophilic component and attacks an activating phosphate or phosphite in the presence of a base; (c) another alternative strategy consists in synthesising P(III) intermediates: the most famous approaches are the phosphoramidite methodology followed by oxidation of the intermediate phosphite;13 or the H-phosphonate approach which entails phosphitylation of the hydroxylic sugar followed by oxydation of the resulting H-phosphonate.14 However, these strategies are mainly applied to glycosyls in their pyranose form. In the case of the furanosyl family, the phosphorylation step has been successfully applied in the case of the ribo or arabinofuranosyl analogues,15 but it is hardly described in the case of 2-deoxyribose derivatives: due to the absence of a 2-hydroxyl group, the anomeric phosphate is more labile, so its synthesis is quite difficult. In fact, to the best of our knowledge, only one strategy concerning the phosphorylation step at the anomeric position of 2-deoxyribose was described:16 under acidic conditions and with an excess of phosphoric acid, 2-deoxyribose-1-chloride 1 was phosphorylated in good yield to give compound 2 (Scheme 1 ).
Scheme 1.

Phosphorylation of 1-choro-3,5-di-para-chlorobenzoyl-2-deoxyribose.
However it is well-known that the presence of a fluorine substituent adjacent to the anomeric centre stabilizes glycosyl phosphates or nucleosides15 and excellent reviews on fluorinated sugars describing their high potential and their broad field of applications have been published.17 In this paper we describe our attempts to phosphorylate the anomeric position of 3,5-dibenzyl-2-deoxyribose and how the gem-difluorination of the sugar significantly stabilises the resulting phosphate. We were particularly interested to prepare C1-phosphoramidates as prodrugs of the free phosphate, following procedures we have widely reported for nucleosides.5, 18
The strategy used to synthesise prodrugs of 2-deoxyribose-1-phosphate is shown in Scheme 2 : the anomeric position of 2-deoxyribose 3 was first quantitatively protected as a methyl glycoside in standard conditions. Protection of the diol was accomplished in 74% yield and removal of the methyl ether was realised under acidic conditions. The next step was the coupling of compound 4 with different suitable phosphorochloridates in order to obtain the desired phosphoramidates. We tried this reaction under different conditions (−78 °C or rt) and with different bases (DMAP, NMI, tBuMgCl) but unfortunately this coupling reaction did not work.
Scheme 2.

Synthesis of prodrugs of 2-deoxyribose-1-phosphate. (a) CH3COCl, MeOH, rt, 1 h, 100%, (b) BnCl, KOH, THF, reflux, 31 h, 74%, (c) AcOH/H2O 8:2, 36 h, 50 °C, 75% (d) appropriate phosphorochloridate, tBuMgCl or NMI, THF.
We knew from our previous experience16b and from the literature16a that a phosphorylation reaction was possible on the 1-chloro-3,5-di-para-chlorobenzoyl-2-deoxyribose. Encouraged by this preliminary result, we decided to study the phosphorylation of 3,5-di-benzyl-2-deoxyribose. Results are summarized in Table 1 in the Supplementary data. The phosphorylation step was first tried in classical conditions using POCl3, phenyldichlorophosphate19 or chlorodiphenyl phosphate20 in the presence of different bases and activating agents such as PyBOP.21 We also investigated the P(III) strategy: we tried to use the phosphoramidite,22 phosphoradiamidite23 and H-phosphonate24 methodologies. We decided to try the phosphorylation with charged phosphorylating agents such as methyl pyridinium dichlorophosphate25 or of the dibarium salt of 2-cyanoethylphosphate26 in order to stabilize the resulting phosphate. To finish our study, we decided to invert our strategy by placing the leaving group on the anomeric position of the sugar(a), (b), 27 and to use the phosphate reagent as the attacking agent. But all these attempts did not lead to the desired products, which lead to the conclusion that final compounds are not stable under the reaction conditions.
Table 1.
Stability study of compound 14b and 25c
| 2-Fluoro-2-deoxyarabinose family compound 14b | 2,2-Fluoro-2-deoxyribose family compound 25c | |
|---|---|---|
| Stability study at pH 2 | t1/2 = 64 min | — |
| Stability study at pH 5 | — | No degradation observed over 2 days |
| Stability study at pH 7 | t1/2 = 120 min | No degradation observed over 2 days |
| Stability study at −20 °C | Purity after 1 h: 68% Purity after 13 h: 30% Purity after 20 h: 8% |
No degradation observed after 4 months |
At this stage, we concluded that the problem in all these phosphorylation reactions came from the 2-deoxyribose sugar: the lack of a group at the 2 position destabilizes the resulting phosphate. This hypothesis is confirmed by the fact that the phosphorylation reaction was described several times on ribose and 2-fluoro-2-deoxyribose derivatives in the literature.15 Considering this information and the fact that the fluorine atom is a good isostere of the hydrogen atom, we decided to synthesize prodrugs of 2-fluoro-2-deoxyarabinose-1-phosphate. The synthesis started from commercially 1,3,5-tri-benzoyl-2-fluoro-2-deoxyribose 6 (Scheme 3 ): bromination of the anomeric position28 followed by the alcohol formation in the presence of triethylamine, water and dimethylformamide23 was realized in quantitative yield. Protection of the resulting alcohol as a silyl ether29 yielded compound 9 in a α/β 1:0.9 ratio. We then needed to change the benzoyl protecting groups of the sugar since the basic conditions used to remove them are not compatible with the presence of the future phosphoramidate group. In a recent paper, Schinazi and co-workers30 explained that a benzyloxycarbonyl protecting group was well-adapted with synthesis of nucleoside aryloxy phosphoramidate prodrugs: this protecting group is easy to introduce, and the neutral conditions used to remove it are compatible with the presence of the phosphoramidate group. So we decided to apply this group to our strategy of synthesis. Compound 9 was deprotected under basic conditions and the resulting diol was protected in the presence of benzyl chloroformate to yield quantitatively compound 11 in a α/β ratio 1:1. Deprotection of the anomeric position was tried in several conditions (TBAF, NH4F, HCl, TFA) but the yields were low (0–20%) in all these conditions. Thus, we finally decided to perform the deprotection in the presence of HF-pyridine,29 and in this case the yield was quantitative and the α/β ratio changed to 1:0.2. Thanks to the literature,31 it was easy to discriminate the α from the β diastereoisomers: the α anomers had J 1,2 = 0 Hz, confirming that H1 and H2 were trans to each other, so the NMR signal of H2 was a doublet with J 2-F = 49-50 Hz. In the case of the β anomer, J 1-F = 0 Hz, which confirmed that H1 and 2F were trans to each other, and the signal of H2 was in this case a ddd (with a general range of J 1,2 = 4 Hz, J 2-F = 50 Hz, J 2,3 = 6 Hz). Finally, coupling of compound 12 with the appropriate aryloxy phosphorochloridates followed by deprotection of the benzyloxy carbonyl protecting groups25 yielded the desired 2-fluoro-2-deoxyarabinose-1-phosphate prodrugs 14a–h. Their successful isolation highlights the change in stability upon 2-fluorination.
Scheme 3.

Synthesis of 2-f luoro-2-deoxy-arabinose-1-phosphate prodrugs. (a) HBr, DCM, rt, 13 h, (b) Et3N, H2O, DMF, rt, 1.5 h, 100% over two-steps, (c) TIPS-OTf, 2,6-lutidine, CH2Cl2, 0 °C, 2.5 h, 100%, (d) Et3N, H2O, MeOH, rt, 3 days, 86%, (e) CbzCl, DMAP, CH2Cl2, rt, 12 h, 95%, (f) HF-pyr, pyridine, rt, 44 h, 100%, (g) appropriated phosphorochloridate, tBuMgCl, THF, rt, 15 h, (h) H2, Pd/C, EtOH, rt, 2 h.
Surprisingly, only the α prodrugs were isolated. However, during the purification of the final prodrugs, we experienced considerable difficulty in isolating pure compound and we discovered that a second purification, either by preparative TLC, column chromatography or preparative HPLC, led to a degradation of the prodrugs. Thus, we conducted a preliminary stability study of compound 14b. One hour after purification by column chromatography the purity of compound 14b was 68%, after one night at −20 °C, the purity of the compound decreased dramatically to 30%, and after 20 h it was only 8%. At pH 2, the half-life of compound 14b was only 64 min, while at pH 7 it was 120 min (Fig. 1 ).
Figure 1.
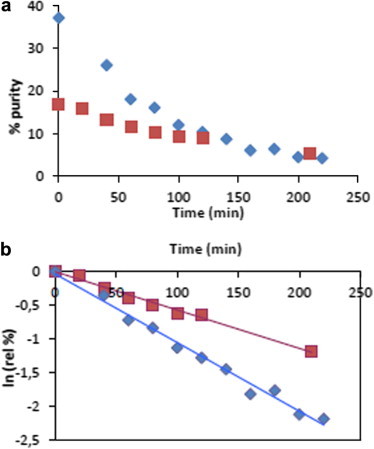
Stability studies of compound 14b. Blue experiment was done at pH 2. Red experiment was done at pH 7. (a) Percentages of purity of compound 14b according to the time at different pH values. (b) Calculation of t1/2 of compound 14b at different pH values.
Since all our 2-fluoro-2-deoxyarabinose-1-phosphate prodrugs were not stable, they could not be biologically evaluated but we were encouraged by these results: indeed by adding one fluorine atom on the 2 position of 2-deoxyribose, the phosphorylation was possible on the anomeric position, even if the resulting prodrugs were not stable enough to be tested. So we decided to explore the 2,2-difluoro-2-deoxyribose family. We hypothesized that the addition of a second fluorine atom at the 2 position would further increase the stability of the resulting prodrugs.
The strategy used to synthesize prodrugs of 2,2-difluoro-2-deoxyribose-1-phosphate was the same as the one previously used in the case of the 2-fluoro-2-deoxyribose family (Scheme 4 ). Reduction of the lactone 15 was achieved in the presence of LiAl(OtBu)3H and the resulting alcohol was protected as a silyl ether with 46% yield. We were also able to isolate a mixture of the monobenzoylated sugars 18 and 19 with 31% yield but this partial deprotection was acceptable as the next step consisted in removing all the benzoyl esters. Protection of the resulting 3 and 5 alcohols was achieved in the presence of benzyl chloroformate and deprotection of the silyl ether at the anomeric position was done with HF-Pyridine and yielded compound 22 in a 1:0.6 α/β ratio. The coupling reaction of the compound 22 with the appropriate phosphorochloridates was done in the presence of tBuMgCl, and the final step consisted in the hydrogenolysis of the benzyloxycarbonyl groups which led to the desired prodrugs of 2,2-difluoro-2-deoxyribose-1-phosphate (with yields of between 24% and 68% over the two last steps). For each compound, α anomers 25 and β anomers 24 were isolated as a mixture of PS and PR diastereoisomers. In order to characterize the two anomers we performed different experiments. We first examined the NOE effects between the H1 and H4 protons, without success. So we based our study on the literature:32 in the β anomer the signal of the fluorines are close to each other (−125 and −127 ppm in 19F NMR), whereas in the α anomer, the two signals are much more separated (−110 and −126 ppm in 19F NMR).
Scheme 4.

Synthesis of 2,2-difluoro-2-deoxyribose-1-phosphate prodrugs. (a) LiAl(OtBu)3H, THF/H2O 4:1, −78 °C, 2 h, (b) TIPS-OTf, 2,6-lutidine, CH2Cl2, 0 °C, 2 h, 67% over 2 steps, (c) Et3N, H2O, MeOH, rt, 4 days, 70%, (d) CbzCl, DMAP, CH2Cl2, rt, 12 h, 100%, (e) HF-pyr, pyridine, rt, 100%, (f) appropriate phosphorochloridate, tBuMgCl, THF, rt, 15 h, (g) H2, Pd/C, EtOH, rt, 2 h.
As expected, the 2,2-difluoro-2-deoxyribose-1-phosphate prodrugs are much more stable than the 2-fluoro-2-deoxyribose-1-phosphate prodrugs. Compound 25c showed no degradation at all after 4 months at −20 °C whereas the corresponding mono-fluoro prodrug was almost completely degradated after only 24 h (Table 1). Stability studies of compound 25c showed no degradation at all after 2 days in acidic (pH 5) and neutral (pH 7) conditions. Thus, difluorination of the sugar affords striking stabilization of the anomeric phosphate prodrugs.
2,2-Difluoro-2-deoxyribose-1-phosphate prodrugs were subjected to biological evaluation because it could be expected that such ribose phosphate analogues can interfere with several enzymes of the purine and pyrimidine nucleoside metabolism. Unfortunately, these compounds did not show biological activity. They had no antiviral activity and were not cytotoxic in HEL cell cultures (herpes simplex virus-1 (KOS), herpes simplex virus-2 (G), vaccinia virus, vesicular stomatitis virus, herpes simplex virus-1 TK− KOS ACV, varicella-zoster virus (OKA, 07-1), cytomegalovirus (AD-169, Davis), EC50 >20–100 μM, MCC ⩾100 μM), and they had no activity against feline corona virus (FIPV) and feline herpes virus in CRFK cell cultures (EC50 >20–100 μM, CC50 >100 μM), nor anti HIV-1 (IIIB) and HIV-2 (ROD) activity in human T-lymphocyte CEM cell cultures (EC50 >50 μM for the naphthyl series, EC50 >250 μM for the phenyl series). They were not significant inhibitors of the proliferation of murine leukemia cells (L1210) and human T-lymphocyte cells (CEM) (IC50 from 30 to >250 μM).
As a conclusion, we have demonstrated that introducing a phosphate group on the anomeric position of 2-deoxyribose is difficult due to the lack of a stabilizing group at the 2 position. However, by introducing one or two fluorine atoms on the 2 position, we increased the stability of the resulting prodrug: the higher the degree of fluorination on the 2 position, the more stable is the prodrug. We have confirmed this tendency by performing a pH stability study of the 2,2-difluoro-2-deoxyribose-1-phosphate prodrug and the 2-fluoro-2-deoxyarabinose-1-phosphate prodrug: the difluoro prodrugs are stable for 2 days under acidic conditions whereas the mono-fluorinated derivative had a half-life of 64 min. Moreover, the 2,2-difluoro prodrugs are stable at −20 °C for more than 4 months whereas the mono-fluorinated analogs were almost fully degradated within 24 h. Unfortunately, biological evaluation of the pro-drugs of the 2,2-difluoro-2-deoxyribose-1-phosphate showed that they had no inhibitory activity against a variety of virus infections and cancer cell proliferation.
Acknowledgments
We thank Helen Murphy for secretarial assistance and Leentje Persoons, Frieda De Meyer, Lizette van Berckelaer and Leen Ingels for excellent technical assistance. The biological evaluations were supported by the KU Leuven (GOA no. 10/014).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2013.02.117.
Supplementary data
Data on the phosphorylation of the anomeric position of 2,3-di-benzyl-2-deoxyribose-1-phosphate; experimental procedures, 1H, 13C, 19F and 31P NMR spectra of compounds 14a–h, 24a–h and 25a–h; HPLC analysis of compounds 24a–h and 25a–h are given in the supplementary data.
References and notes
- 1.Griffiths L., Stratford I.J. Br. J. Cancer. 1997;76:689. doi: 10.1038/bjc.1997.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Clercq E. Biochem. Pharmacol. 2004;68:2301. doi: 10.1016/j.bcp.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 3.He G.-X., Krise J.P. Biotechnol. Pharm. Aspects. 2007;5:923. [Google Scholar]
- 4.Carston R., Carston R.Wagner, Vidhya V. Iyer, McIntee E.J. Med. Res. Rev. 2000;20:417. doi: 10.1002/1098-1128(200011)20:6<417::aid-med1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 5.Mehellou Y., Balzarini J., McGuigan C. ChemMedChem. 2009;4:1779. doi: 10.1002/cmdc.200900289. [DOI] [PubMed] [Google Scholar]
- 6.Serpi M., Bibbo R., Rat S., Roberts H., Hughes C., Caterson B., Jose Alcaraz M., Torrent Gibert A., Alaez Verson C.R., McGuigan C. J. Med. Chem. 2012;55:4629. doi: 10.1021/jm300074y. [DOI] [PubMed] [Google Scholar]
- 7.Rubira M.J., Jimeno M.L., Balzarini J., Camarasa M.J., Perez-Perez M.J. Tetrahedron. 1998;54:8223. [Google Scholar]
- 8.(a) Zamyatina A.K.P. Carbohydr. Chem. 2009;35:71–98. [Google Scholar]; (b) Nikolaev A.V., Botvinko I.V., Ross A.J. Carbohydr. Res. 2007;342:297. doi: 10.1016/j.carres.2006.10.006. [DOI] [PubMed] [Google Scholar]; (c) Hansson J., Oscarson S. Curr. Org. Chem. 2000;4:535. [Google Scholar]; (d) Nakamura S., Nambu H.H.S. In: Handbook of Chemical Glycosylation. Demchenko A., editor. Wiley-Vch; Weinheim, Germany: 2008. p. 223. ch 3.4. [Google Scholar]
- 9.(a) Binch H., Stangier K., Thiem J. Carbohydr. Res. 1998;306:409. [Google Scholar]; (b) Timmons S.C., Jakeman D.L. Carbohydr. Res. 2008;343:865. doi: 10.1016/j.carres.2008.01.046. [DOI] [PubMed] [Google Scholar]; (c) Oberthur M., Leimkuhler C., Kahne D. Org. Lett. 2004;6:2873. doi: 10.1021/ol049187f. [DOI] [PubMed] [Google Scholar]
- 10.Bowers S.G., Mahmud T., Floss H.G. Carbohydr. Res. 2002;337:297. doi: 10.1016/s0008-6215(01)00323-8. [DOI] [PubMed] [Google Scholar]
- 11.Illarionov P.A., Torgov V.I., Hancock I.C., Shibaev V.N., Illarionova P.A. Tetrahedron Lett. 1999;40:4247. [Google Scholar]
- 12.(a) Ferrieres V., Blanchard S., Fischer D., Plusquellec D. Bioorg. Med. Chem. Lett. 2002;12:3515. doi: 10.1016/s0960-894x(02)00822-3. [DOI] [PubMed] [Google Scholar]; (b) Euzen R., Ferrieres V., Plusquellec D. J. Org. Chem. 2005;70:847. doi: 10.1021/jo0484934. [DOI] [PubMed] [Google Scholar]
- 13.(a) Hitchcock S.A., Eid C.N., Aikins J.A., Zia-Ebrahimi M., Blaszczak L.C. J. Am. Chem. Soc. 1916;1998:120. [Google Scholar]; (b) Men H.B., Park P., Ge M., Walker S. J. Am. Chem. Soc. 1998;120:2484. [Google Scholar]; (c) Liu H.T., Sadamoto R., Sears P.S., Wong C.H. J. Am. Chem. Soc. 2001;123:9916. doi: 10.1021/ja011708w. [DOI] [PubMed] [Google Scholar]; (d) VanNieuwenhze M.S., Mauldin S.C., Zia-Ebrahimi M., Aikins J.A., Blaszczak L.C. J. Am. Chem. Soc. 2001;123:6983. doi: 10.1021/ja016082o. [DOI] [PubMed] [Google Scholar]
- 14.Naundorf A., Natsch S., Klaffke W. Tetrahedron Lett. 2000;41:189. [Google Scholar]
- 15.(a) Joe M., Lowary T.L. Carbohydr. Res. 2006;341:2723. doi: 10.1016/j.carres.2006.08.020. [DOI] [PubMed] [Google Scholar]; (b) Smellie I.A., Bhakta S., Sim E., Fairbanks A.J. Org. Biomol. Chem. 2007;5:2257. doi: 10.1039/b704788f. [DOI] [PubMed] [Google Scholar]; (c) Zhang Q.B., Liu H.W. J. Am. Chem. Soc. 2001;123:6756. doi: 10.1021/ja010473l. [DOI] [PubMed] [Google Scholar]; (d) Maryanoff B.E., Reitz A.B., Nortey S.O. Tetrahedron. 1988;44:3093. [Google Scholar]
- 16.(a) Komatsu H., Awano H. J. Org. Chem. 2002;67:5419. doi: 10.1021/jo025793h. [DOI] [PubMed] [Google Scholar]; (b) Voorde J.V., Quintiliani M., McGuigan C., Liekens S., Balzarini J. Biochem. Pharmacol. 2012;83:1358. doi: 10.1016/j.bcp.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 17.(a) Penglis A.A.E. Adv. Carbohydr. Chem. Biochem. 1981;38:105. doi: 10.1016/s0065-2318(08)60314-5. [DOI] [PubMed] [Google Scholar]; (b) Tsuchiya T. Adv. Carbohydr. Chem. Biochem. 1990;48:91. doi: 10.1016/s0065-2318(08)60032-3. [DOI] [PubMed] [Google Scholar]
- 18.Cahard D., McGuigan C., Balzarini J. Mini-Rev. Med. Chem. 2004;4:371. doi: 10.2174/1389557043403936. [DOI] [PubMed] [Google Scholar]
- 19.Sabesan S., Neira S. Carbohydr. Res. 1992;223:169. doi: 10.1016/0008-6215(94)00309-4. [DOI] [PubMed] [Google Scholar]
- 20.Lee, J. P., Gha Seung; Lee, Moonsub; Bang, Hyo-Jeong; Lee, Jae Chul; Kim, Cheol Kyong; Choi, Chang-Ju; Kim, Han Kyong; Lee, Hoe Chul; Chang, Young-Kil et al. From PCT Int. Appl. (2006), WO 2006071090 A1 20060706.
- 21.Zalcharova V.M., Serpi M., Krylov I.S., Peterson L.W., Breitenbach J.M., Borysko K.Z., Drach J.C., Collins M., Hilfinger J.M., Kashemirov B.A., McKenna C.E. J. Med. Chem. 2011;54:5680. doi: 10.1021/jm2001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikan M., Bare G.A.L., Sherman J.C. Tetrahedron Lett. 2011;52:1791. [Google Scholar]
- 23.Yamada K.M.N., Hayakawa H. Nucleosides Nucleotides Nucleic Acids. 2009;28:1117. doi: 10.1080/15257770903396741. [DOI] [PubMed] [Google Scholar]
- 24.Chang A.-L.G.G., Wagner C.R. Nucleosides Nucleotides Nucleic Acids. 2001;20:1571. doi: 10.1081/NCN-100105248. [DOI] [PubMed] [Google Scholar]
- 25.Jaworek C.H., Iacobucci S., Calias P., d’Alarcao M. Carbohydr. Res. 2001;331:375. doi: 10.1016/s0008-6215(01)00047-7. [DOI] [PubMed] [Google Scholar]
- 26.Tener G.M. J. Am. Chem. Soc. 1961;83:159. [Google Scholar]
- 27.Gao M., Chen Y., Tan S., Reibenspies J.H., Zingaro R.A. Heteroat. Chem. 2008;19:199. [Google Scholar]
- 28.McGuigan C., Derudas M., Quintiliani M., Andrei G., Snoeck R., Henson G., Balzarini J. Bioorg. Med. Chem. Lett. 2009;19:6264. doi: 10.1016/j.bmcl.2009.09.116. [DOI] [PubMed] [Google Scholar]
- 29.Guzi, T. J. P., D. A.; Labroli, M. A.; Dwyer, M. P.; Paruch, K.; Rosner, K. E.; Shen, R.; Popovici-Muller, J. PCT Int. Appl. WO 2009061781 A1 20090514, 2009.
- 30.Cho J.H., Amblard F., Coats S.J., Schinazi R.F. Tetrahedron. 2011;67:5487. doi: 10.1016/j.tet.2011.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Shortnacy-Fowler A.T., Tiwari K.N., Montgomery J.A., Secrist J.A. Nucleosides Nucleotides Nucleic Acids. 2001;20:1583. doi: 10.1081/NCN-100105249. [DOI] [PubMed] [Google Scholar]; (b) Pathak A.K., Pathak V., Suling W.J., Riordan J.R., Gurcha S.S., Besra G.S., Reynolds R.C. Bioorg. Med. Chem. 2009;17:872. doi: 10.1016/j.bmc.2008.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tiwan K.N., Shortnacy-Fowler A.T., Parker W.B., Waud W.R., Secrist J.A., III Nucleosides Nucleotides Nucleic Acids. 2009;28:657. doi: 10.1080/15257770903091946. [DOI] [PubMed] [Google Scholar]; (d) Hertel L.W., Kroin J.S., Misner J.W., Tustin J.M. J. Org. Chem. 1988;53:2406. [Google Scholar]
- 32.Wirth, D. Eur. Pat. Appl. EP 727433 A1 19960821, 1996.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data on the phosphorylation of the anomeric position of 2,3-di-benzyl-2-deoxyribose-1-phosphate; experimental procedures, 1H, 13C, 19F and 31P NMR spectra of compounds 14a–h, 24a–h and 25a–h; HPLC analysis of compounds 24a–h and 25a–h are given in the supplementary data.