

Graphical abstract

Keywords: SARS 3CL protease, Inhibitor, Fused ring scaffold, Octahydroisochromene
Abstract
An octahydroisochromene scaffold has been introduced into a known SARS 3CL protease inhibitor as a novel hydrophobic core to interact with the S2 pocket of the protease. An alkyl or aryl substituent was also introduced at the 1-position of the octahydroisochromene scaffold and expected to introduce additional interactions with the protease. Sharpless–Katsuki asymmetric epoxidation and Sharpless asymmetric dihydroxylation were employed to construct the octahydroisochromene scaffold. The introductions of the P1 site His-al and the substituent at 1-position was achieved using successive reductive amination reactions. Our initial evaluations of the diastereo-isomeric mixtures (16a–d) revealed that the octahydroisochromene moiety functions as a core hydrophobic scaffold for the S2 pocket of the protease and the substituent at the 1-position may form additional interactions with the protease. The inhibitory activities of the diastereoisomerically-pure inhibitors (3a–d) strongly suggest that a specific stereo-isomer of the octahydroisochromene scaffold, (1S, 3S) 3b, directs the P1 site imidazole, the warhead aldehyde, and substituent at the 1-position of the fused ring to their appropriate pockets in the protease.
1. Introduction
In 2003, severe acute respiratory syndrome (SARS), a life-threatening pneumonia, spread across Southeast Asia and affected about 8500 patients with >800 fatalities.1, 2, 3 The causative virus is a new betacoronavirus (βCoV) containing a 29.7-kb positive-strand RNA genome. Two large replicative polyproteins, pp1a (486 kDa) and pp1ab (790 kDa), are coded in the genome as precursor proteins to yield functional proteins.4, 5, 6 Among the functional proteins, a 3C-like cysteine protease (SARS 3CLpro) processes the polyprotein to produce itself as well as a variety of proteins necessary to re-construct the virus particles. Thus, SARS 3CLpro is thought to be an ideal target for anti-SARS agents. Numerous studies on SARS 3CLpro inhibitors have been reported,7, 8 but no therapeutic agents are available at present.
In the course of our previous studies on SARS 3CLpro inhibitors,9 a substrate-based peptide aldehyde (1) was shown to be a potent inhibitor (IC50 = 98 nM) for a mutant protease, R188I SARS 3CLpro.10 Analysis of the X-ray crystal structure of 1 complexed with R188I SARS 3CLpro revealed that the P2 site cyclohexyl ring of 1 interacts with the S2 pocket of the protease via a hydrophobic interaction. The distance between the 2-position carbon atom of the cyclohexyl-ring and α-nitrogen atom of cyclohexylalanine (Cha) was 3.48 Å (Fig. 1 ). This result suggested that the cyclohexyl ring may be connected to the peptide chain via a methylene linker, yielding a new inhibitor scaffold containing a hydrophobic decahydroisoquinoline ring. The resulting decahydroisoquinoline inhibitor (2) showed inhibitory activity as expected, but with moderate potency (IC50 = 63 μM).11 Analysis of the X-ray crystal structure of inhibitor 2 complexed with R188I SARS 3CLpro revealed the lack of interactions with the P3 to P4 sites in substrate-based inhibitor 1.
Fig. 1.

The design of decahydroisoquinoline inhibitor 2.
In this study, an octahydroisochromene scaffold was selected as a novel hydrophobic fused-ring in inhibitor 3 instead of decahydroisoquinoline to evaluate the effect of the configuration of the fused-ring structure and novel substituents on the ring system (Fig. 2 ).12 The configuration of the octahydroisochromene ring in 3 can be easily controlled using a stereo-selective ring-closing reaction featuring the nucleophilic opening of an epoxide. The synthesis of the requisite oxygen-containing precursor used in the ring-closing reaction can be achieved using established stereo-selective reactions such as the Sharpless-Katsuki asymmetric epoxidation and Sharpless dihydroxylation reaction. In addition, the effect of the substituent at the 1-position in the fused-ring system of 3 may also be evaluated; the effects of different substituents in this position have not been assessed in our previous studies using the decahydroisoquinoline scaffold in 2.12 Thus, inhibitors containing an octahydroisochromene scaffold with four different substituents at the 1-position were designed and synthesized.
Fig. 2.

The design of octahydroisochromene inhibitor 3 with four different substituents at the 1-position.
2. Results and discussion
2.1. Chemistry
The retrosynthetic analysis of octahydroisochromene inhibitor 3 is shown in Scheme 1 . (S)-2-Amino-3-imidazolyl propanal (His-al), the warhead part of 3, was to be introduced using a reductive amination reaction between His-al and octahydroisochromene derivative 4. The substituent at the 1-position of the octahydroisochromene ring in 4 was also introduced using a reductive amination reaction of the requisite amine and the aldehyde constructed via oxidative cleavage of the 1,2-diol in key intermediate 5. Construction of the octahydroisochromene ring in 5, a key step in the synthesis, may be carried out using an intramolecular nucleophilic addition reaction of the hydroxyl group to the epoxide in cyclohexyl derivative 6. The configuration of the resulting ring structure in 5 can be controlled using a combination of the stereo-chemistry of the nucleophilic secondary hydroxyl group and electrophilic epoxide in 6, which make it possible to selectively form the four possible configurations of the ring system in 5. The configuration of the nucleophilic hydroxyl groups in 6 can be controlled using a Sharpless asymmetric dihydroxylation reaction13 on the olefin in 7 and the configuration of the electrophilic epoxide in 7 can be selectively constructed using a Sharpless-Katsuki asymmetric epoxidation reaction14 on the allylic alcohol obtained upon the functional group conversion of commercially available diol 8.
Scheme 1.

Retrosynthetic analysis of octahydroisochromene inhibitor 3.
Synthesis of key intermediate 5 was achieved starting from a commercially available compound (8) according to the route shown in Scheme 2 . Monoprotection of diol 8 using tert-butyl diphenylsilyl chloride (TBDPSCl) followed by oxidation of the unprotected alcohol in the resulting product via a Ley-Griffith oxidation reaction using tetrapropyl ammonium perruthenate (TPAP) gave the corresponding aldehyde. The product was used with any purification, and reacted with (methoxymethyl)triphenylphosphonium chloride to give 9. The methoxyvinyl group in 9 was then converted to the corresponding aldehyde upon treatment with 10-camphorsulfonic acid. The isolated aldehyde was reacted with methyl triphenylphosphonium chloride to yield a terminal olefin. The TBDPS group in the product was then removed upon treatment with tetra-n-butylammonium fluoride (TBAF) to give 10. The primary alcohol in 10 was oxidized via a Ley–Griffith oxidation reaction and the resulting aldehyde homologated using a Horner–Wadsworth–Emmons (HWE) reaction upon reaction with ethyl 2-(diethoxyphosphoryl)acetate to give 11. The ethyl ester in 11 was reduced with diisobutyl aluminum hydride (DIBAL-H) and the resulting allylic alcohol stereo-specifically oxidized via a Sharpless–Katsuki asymmetric epoxidation reaction using tert-butyl hydroperoxide (TBHP) in the presence of titanium tetraisopropoxide (TTIP) and (S, S)-diethyltartrate ((–)-DET) to yield 12 as a single diastereomer. The terminal olefin in 12 was then oxidized via a Sharpless asymmetric dihydroxylation reaction using potassium osmate (VI) dehydrate (K2OsO2(OH)4) and potassium ferricyanide (K3Fe(CN)6) in the presence of hydroquinine (anthraquinone-1,4-diyl) diether ((DHQ)2AQN). The formation of the expected diol was confirmed by 1H NMR spectroscopy with the simultaneous production of a small amount of the cyclized product (13) formed via the nucleophilic addition of the resulting alcohol. Thus, the mixture was treated with p-toluenesulfonic acid without any further purification to convert the remaining dihydroxylated product into the desired cyclized product (13). However, whether product 13 was formed as a single diasteroisomer or an inseparable mixture of diastereomers could not be determined using 1H NMR spectroscopy. Thus, the product was converted into the final products containing different substituents at the 1-position of the octahydroisochromene scaffold to evaluate whether the fused ring structure would function as a novel inhibitor scaffold.
Scheme 2.

Synthetic route used to prepare compound 13.
Conversion of key intermediate 13 into the final product was carried out according to the route shown in Scheme 3 . The terminal 1,2-diol in 13 was oxidatively cleaved using sodium periodate and the resulting aldehyde subjected to a reductive amination reaction with four different amines in the presence of NaBH3CN. Each aminated product was then methylated with paraformaldehyde to give 14a–d. The primary alcohol in each product was oxidized using N-tert-butylbenzensulfenamide/1-chloropyrrolidine-2,5-dione (NCS) and the resulting aldehydes were reductively condensed with H-His-N(OMe)Me to give their corresponding Weinreb amides (15a–d).15 Each product was finally reduced to their corresponding aldehyde upon treatment with DIBAL-H and purified by LC/MS to give the desired aldehyde product (16a–d), which exhibited a broad single peak in LC.
Scheme 3.

Synthetic route used to prepare inhibitors 16a–d.
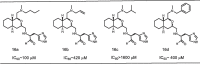
To access whether the octahydroisochromene derivatives containing substituents at the 1-position of the ring system function as a novel inhibitor scaffold, the inhibitory activity of compounds 16a–d toward SARS 3CLpro was preliminary evaluated by the IC50 values obtained using the procedure described below. As summarized in Table 1 , compound 16a bearing an n-butyl group showed moderate inhibitory activity, whereas 16c bearing an isobutyl group showed no inhibitory activity. Compounds 16b and 16d bearing allyl and benzyl substituents, respectively showed weak activity. Although the absolute configurations of 16a to 16d were not identified, these results strongly suggested that the octahydroisochromene scaffold can function as a novel hydrophobic scaffold in the SARS 3CLpro inhibitor and may also interact with the protease at the 1-position of the fused-ring system.
Table 1.
IC50 values obtained for compounds 16a–d.
 |
Confirmation of the configuration of compounds 16a–d was then conducted based on the above findings. Since it was unclear whether key intermediate 13 used for the above syntheses was formed as a single diastereomer or an inseparable mixture of diastereoisomers using 1H NMR spectroscopy, the 13C NMR spectrum of 13 was examined and two signals were detected for the methine carbon atom neighboring an oxygen atom in the octahydroisochromene scaffold (Fig. S-1). This result strongly suggests that triol 13 obtained using the Sharpless asymmetric dihydroxylation reaction using (DHQ)2AQN as a ligand was formed as a 1.5:1 diastereoisomeric mixture (Scheme 2 and entry 1 in Table 2 ). Thus, the reaction conditions used in the asymmetric dihydroxylation were evaluated focusing on the chiral ligand, a key factor in the stereo-selectivity of the reaction.16 Precursor 12 was treated with K2OsO2(OH)4 and K3Fe(CN)6 with or without the chiral ligand as stated above (Scheme 2) and the products immediately converted into triol 13 upon treatment with p-toluenesulfonic acid. The diastereoisomeric ratio of the products was estimated using the methine carbon atom signal in the 13C NMR spectrum. Table 2 shows that in the absence of the chiral ligand, an equal amount of both the possible diastereomers was obtained (entry 4). A slight preference for one diastereomer was observed when hydroquinine (anthraquinone-1,4-diyl) diether ((DHQ)2AQN) was used as the chiral ligand, as in Scheme 2 (Table 2, entry 1). In contrast, a much better preference for one diastereoisomer was observed when 1,4-bis(dihydroquinine)phthalazine ((DHQ)2PHAL) or hydroquinidine-2,5-diphenyl-4,6-pyrimidinediyl diether ((DHQ)2Pyr) was used as the chiral ligand. (DHQ)2Pyr (entry 3) showed a slightly better diastereoisomeric ratio (dr = 7:1) than (DHQ)2PHAL (entry 2, dr = 6:1), which made it technically feasible to isolate the major product. As expected, the diastereoisomeric ratio was inverted when hydroquinidine-2,5-diphenyl-4,6-pyrimidinediyl diether ((DHQD)2Pyr) was used as the chiral ligand (entry 5, dr = 1:15).
Table 2.
Effect of the chiral ligand used in the Sharpless asymmetric dihydroxylation of 12.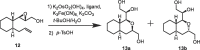
| Entry | Ligand | Ratio of the mixture* | Yield of 13a + 13b |
|---|---|---|---|
| 1 | (DHQ)2AQN | 1.5 : 1 | 96 |
| 2 | (DHQ)2PHAL | 6 : 1 | 85 |
| 3 | (DHQ)2Pyr | 7 : 1 | 84 |
| 4 | – | 1 : 1 | 82 |
| 5 | (DHQD)2Pyr | 1 : 15 | 85 |
The ratio of 13a and 13b was determined by 13C NMR analysis.
To determine the configuration of each diastereomer (13a and 13b), the major product obtained after the asymmetric dihydroxylation reaction using (DHQ)2Pyr as the chiral ligand (Table 2, entry 3) was isolated and purified using preparative thin layer chromatography (PTLC). In the 1H NMR spectrum of the major product, a nuclear Overhauser effect (NOE) was observed between the protons at the 1- and 3-positions of the octahydroisochromene ring and between the protons at the 3-positon and bridgehead α-position, which indicates that these protons adopt a cis-α-configuration (Fig. S-1a). The coupling constant between the proton at the 1-position and the neighboring proton of the secondary alcohol was observed to be 9.7 Hz, which also indicates the cis-configuration of the protons. Taken together, it was estimated that the configuration of the major product was (1-S, 3-R) 13a, as shown in Table 1.17 Although the confirmed structure of 13a was the opposite to that expected for the asymmetric dihydroxylation reaction, it has been occasionally reported depending on the substrate used.18, 19 In this specific substrate, steric hindrance due to the epoxy alcohol interferes with the ordinary interactions with the chiral ligand to direct attack of potassium osmate to the terminal olefin from the less sterically hindered face (Fig. S-1c).
Based on these results, the stereo-selective synthesis of all the possible diastereomers derived from the oxygen atom in the octahydroisochromene ring were then carried out to evaluate the steric effect of the fused ring on their inhibitory activity. In these syntheses, the separation and purification of the major product was achieved in the final step of the synthesis using LC/MS instead of purifying intermediate 13 (Scheme 3). In addition, an n-butyl group was employed as the suitable substituent at the 1-position in accordance with our preliminary evaluation of the inhibitory activity using the diastereo-isomeric mixtures of compounds 16a–d (Table 1).
The synthetic route used to prepare (1-S, 3-R) 3a is shown in Scheme 4 -(i). The allylic alcohol obtained from the reduction of ester 11 was oxidized via a Sharpless-Katsuki asymmetric epoxidation reaction using (–)-DET as the chiral ligand to give 12 as a single diastereoisomer. The terminal olefin in 12 was then oxidized via a Sharpless asymmetric dihydroxylation reaction using (DHQ)2Pyr as the chiral ligand to give 13a as the major product (dr = 7:1). The mixture was then used for the following reactions without separating the major product. Oxidative cleavage of the 1,2-diol in 13a and the subsequent reductive amination reaction using n-butyl amine and paraformaldehyde were conducted as described for compound 14a (Scheme 3). The final coupling with H-His-N(OMe)Me followed by reduction with DIBAL-H was carried out as described for compound 16a (Scheme 3) to yield 3a as the major product. Since the diasteroisomeric ratio of the major product was relatively high (dr = 7:1), purification of the desired 3a could be achieved using LC/MS without difficulty. A diastereoisomer 3b with the configuration of (1-S, 3-S) was synthesized by employing a combination of (–)-DET and (DHQD)2Pyr, which were used in the Sharpless-Katsuki asymmetric epoxidation reaction (yielding a single diastereoisomer) and the Sharpless asymmetric dihydroxylation reaction (dr = 1:15), respectively. The product was then purified using LC/MS with high homogeneity.
Scheme 4.

(i) Synthetic route used to prepare inhibitors (1-S, 3-R) 3a. (ii) Synthetic route used to prepare inhibitors (1-R, 3-R) 3c and (1-R, 3-S) 3d.
Further, diastereoisomers, 3c and 3d, were synthesized with the configurations of (1-R, 3-R) and (1-R, 3-S), respectively; this was achieved by employing synthetic routes as shown in Scheme 4-(ii). For the construction of the configuration of (1-R, 3-R) in 13c, a combination of (+)-DET and (DHQ)2Pyr was used, which were utilized in the Sharpless-Katsuki asymmetric epoxidation reaction (yielding a single diastereoisomer) and the Sharpless asymmetric dihydroxylation reaction (dr = 8:1), respectively. Similarly, the construction for the configuration of (1-R, 3-S) in 13d was performed using a combination of (+)-DET and (DHQD)2Pyr, which were utilized in the Sharpless-Katsuki asymmetric epoxidation reaction (yielding a single diastereoisomer) and the Sharpless asymmetric dihydroxylation reaction (dr > 29:1), respectively. In both syntheses, the diastereo-excess (dr) was exceedingly high, which led to both the products, 13c and 13d, being obtained as single diastereoisomers. Likewise, each product was then converted to 3c or 3d as mentioned above and purified using LC/MS with high homogeneity.
2.2. Inhibitory activity
The inhibitory activities of 16a–d and 3a–d were evaluated based on the IC50 values obtained according to a literature procedure (Table 1, Table 3 ).9 In brief, a peptide substrate, H-Thr-Ser-Ala-Val-Leu-Gln-Ser-Gly-Phe-Arg-Lys-NH2, was incubated with R188I SARS 3CLpro at 37 °C for 2 h in the presence of each inhibitor and the resulting mixture was analyzed using analytical HPLC performed on a C18 reversed-phase column. The IC50 value of 16a was estimated from the sigmoidal plot obtained from the decrease in the digested substrate (Fig. S-2). However, the as-obtained IC50 value (Table 1) was an approximate value and only indicated the inhibition tendency because the obtained plot was not well fitted to the expected sigmoidal curve due to the presence of only 40% of the desired diastereomer (derived from the diastereo-isomeric mixture of 13, dr = 1.5:1). Compound 16b and 16d showed ~50% inhibition at 400 μM and 16d showed weak inhibition even at 1600 μM. In spite of these vague results, they strongly suggest that the octahydroisochromene ring functions as a novel hydrophobic scaffold in the SARS 3CLpro inhibitor. It was also expected that a medium size linear alkyl group at the 1-position, such as n-butyl in 16a, can interact with SARS 3CLpro at a specific site, but bulkier substituents such as the branched group in 16c or planar groups in 16b and 16d exhibit less interactions.
Table 3.
IC50 values obtained for inhibitors 3a–d.
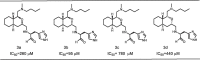 |
The IC50 values observed for the diastereoisomerically pure (3a–d) ranged from 95 to 780 μM, a rather wide range, which clearly indicates the steric effect of the octahydroisochromene scaffold on the inhibitory activity (Table 3). Compound 3b was most active (IC50 = 95 μM) and had a (1S, 3S)-configuration in the ring system. Compound 3c was the least active (IC50 = 780 μM) with an enantiomeric (1R, 3R)-configuration when compared to 3b. The results suggest that converting the stereocenter at the 1-position has a more significant effect on the inhibitory activity than changing the stereochemistry at the 3-position.
2.3. Interaction model for 3a–d with SARS 3CL protease
The interactions between compounds 3a–d and R188I SARS 3CLpro were proposed using a docking model constructed with Molecular Operating Environment (MOE) software. The fused-ring system containing the aldehyde warhead of the decahydroisoquinoline inhibitor complexed with the protease (PDB: 4TWW)11 was used as a template for the docking model. The docking scores obtained for inhibitors 3a, 3b, and 3d were similar (3a, –8.3615 kcal/mol; 3b, –8.3982 kcal/mol; 3d, –8.4354 kcal/mol), whereas the docking score for 3c was rather low (–7.9265 kcal/mol). The docking mode for 3b (IC50 = 95 μM) showed nearly the same interactions with the protease as the parent decahydroisoquinoline inhibitor at its P1 site imidazole as well as those at the P2 site fused-ring system (Fig. 3 a). The warhead aldehyde was also directed toward the active center of the protease, as-observed in the case of the decahydroisoquinolin inhibitor (Fig. 3a). In addition, the n-butyl substituent at the 1-position of the octahydroisochromene scaffold in 3b was assumed to be situated on the surface of the protease near the S1′ to S2′ pockets (Fig. 3b). In contrast, the docking mode for less potent inhibitor 3c (IC50 = 780 μM) showed different interactions at the P1 site imidazole and aldehyde warhead at the active center of the protease, although the interactions with the fused-ring system were expected to be similar to 3b (Fig. 3c). The interactions with the n-butyl substituent at the 1-position of the octahydroisochromene scaffold in 3c were different from those of inhibitor 3b. The different modes of interaction observed for 3b and 3c were derived from the change in the configuration of the octahydroisochromene scaffold, which caused an 88% decrease in the inhibitory activity. To confirm the estimated interactions, X-ray crystallographic analysis of 3b complexed with R188I SARS 3CLpro is now underway in our laboratory.
Fig. 3.

Docking model for 3b and 3c with R188I SARS 3CLpro. (a) and (b) The interaction mode for 3b with a GBVI/WSA dG score of –8.3982 kcal/mol. (c) The interaction mode for 3c with a GBVI/WSA dG score of –7.9265 kcal/mol. Each docking model with R188I SARS 3CLpro was constructed using the X-ray crystal structure obtained for the SARS 3CLpro and decahydroisoquinoline inhibitor (PDB 4TWW) complex as a template. The possible binding mode was obtained using a docking simulation of each inhibitor and SARS 3CLpro using an automated template-guided docking protocol with the Amber10:EHT force field in the MOE 2019.0101 software package (Chemical Computing Group Inc., Montreal, Quebec, Canada).
3. Conclusion
As an alternative non-peptide scaffold to decahydroisoquinoline11, 12, using an octahydroisochromen scaffold has been introduced into a fused-ring type SARS 3CL protease inhibitor as a novel hydrophobic core to interact with the S2 pocket of the protease. Alkyl and aryl substituents were also introduced to the 1-position of the octahydroisochromene scaffold to form additional interactions with the protease. Sharpless-Katsuki asymmetric epoxidation and Sharpless asymmetric dihydroxylation reaction were used to construct the four possible diastereoisomers of the octahydroisochromene scaffold by selecting the appropriate chiral ligand in each asymmetric reaction. The purity and stereochemistry of the synthesized key intermediate 13 were confirmed using 1H NMR, NOE, and 13C NMR spectroscopy. Introductions of the P1 site His-al and substituent at the 1-position was achieved using successive reductive amination reactions.
Inhibitor 16a was prepared as a diastereo-isomeric mixture derived from the Sharpless asymmetric dihydroxylation reaction and showed moderate inhibitory activity toward R188I SARS 3CLpro. In contrast, the diastereoisomeric mixtures of 16b, 16c and 16d, which contain bulky or planar substituents at the 1-position, showed lower inhibitory activity. These results strongly suggested the functionality of the octahydroisochromene as a core hydrophobic scaffold for the S2 pocket of the protease and the additional interactions formed between the 1-position of the scaffold when the substituent is a linear alkyl group of moderate size. Based on these positive findings, the effects of the stereochemistry of the octahydroisochromene scaffold were examined and the results suggest that a specific configuration of the octahydroisochromene, (1S, 3S) 3b, directed the P1 site imidazole and the warhead aldehyde to their counterpart pockets in the protease. In addition, the n-butyl substituent at 1-position of the fused-ring system in 3b was also expected to form some interactions with the protease. In contrast, these functional groups in the enantiomeric octahydroisochromene inhibitor, (1R, 3R) 3c, were probably misdirected causing an 88% decrease in the inhibitory activity. Thus, detailed analysis of these interactions based on X-ray crystal analysis of 3b complexed with R188I SARS 3CLpro to optimize the substituent at the 1-position as well as an alternative warhead group are ongoing in our laboratory.
4. Experimental
4.1. General
Silica gel 70PF 254 Plate-Wako was used for TLC and column chromatography was performed on Wakogel® 60 N (particle size, 63–212 μm) or Wakogel® C-300E (particle size, 45–75 μm). Low-resolution mass spectroscopy (LRMS) was recorded on Shimadzu LCMS-2010EV (ESI) and high-resolution mass spectroscopy (HRMS) was recorded on Shimadzu LCMS-IT-TOF (ESI) or JEOL GC mate II (EI) or JEOL SX-102A (FAB). 1H NMR spectroscopy was recorded on a Bruker AV300 or Ascend500 spectrometer. Chemical shifts (δ) are quoted in parts per million (ppm) and referenced to tetramethylsilane (0 ppm) or the residual solvent peak. 13C NMR spectroscopy was recorded on the same spectrometer at 75 or 125 MHz using the residual solvent peak as an internal reference. Optical rotations were recorded using a Jasco P-2200 Polarimeter.
4.2. ((1S,2S)-2-(((tert-Butyldiphenylsilyl)oxy)methyl)cyclohexyl)methanol
To a solution of diol 8 (14.4 g, 100 mmol) and DIPEA (87 mL, 500 mmol) in CH2Cl2 (200 mL) was added TBDPS-Cl (30 mL, 110 mmol) and the resulting mixture stirred at room temperature for 10 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was purified using silica gel column chromatography (hexane/EtOAc = 30:1) to give the protected alcohol (37.4 g, 98%) as a colorless oil. [α]D 20 + 6.1 (c 0.48, MeOH); 1H NMR (300 MHz, CDCl3) δ: 7.68–7.65 (m, 4H), 7.47–7.36 (m, 6H), 3.65–3.54 (m, 4H), 3.18 (brs, 1H), 1.73–1.65 (m, 3H), 1.48–1.32 (m, 3H), 1.28–0.85 (m, 4H), 1.06 (s, 9H); 13C NMR (75 MHz, CDCl3) δ: 135.6, 133.1, 129.80, 127.78, 127.7, 69.0, 67.2, 44.7, 43.3, 29.9, 29.7, 26.8, 26.09, 26.05, 19.1; HRMS (ESI) calcd for C24H34NaO2Si [M+Na]+: 405.2626. Found: 405.2224.
4.3. (((1S,2R)-2-Allylcyclohexyl)methoxy)(tert-butyl)diphenylsilane
To a solution of the protected alcohol (37.4 g, 98.0 mmol) and TPAP (tetra-n-propyl ammonium perruthenate, 688 mg, 1.96 mmol) in CH2Cl2 (200 mL) was added NMO (N-methylmorphline-N-oxide, 45.9 g, 392 mmol) at room temperature under an argon atmosphere. After stirring for 1 h, the reaction mixture was filtered through a pad of silica gel and the filtrate concentrated in vacuo. The resulting residue was used immediately in the next step without any purification. To a solution of t-BuOK (21.9 g, 196 mmol) in THF (200 mL) was added methoxymethyltriphenylphosphonium chloride (67.1 g, 196 mmol) at 0 °C under an argon atmosphere and the resulting mixture stirred for 1 h. The residue obtained from the TPAP oxidation reaction in THF (50 mL) was added dropwise at 0 ℃ and the resulting mixture stirred for 10 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 30:1) to give 9 (29.7 g, 74%, over 2 steps) as a colorless oil. 9, which was used immediately in the next step. To a solution of (+)-camphorsulfonic acid (CSA; 25.4 g, 109 mmol) in CH2Cl2 (200 mL) was added to a solution of 9 (29.7 g, 73 mmol) in CH2Cl2 (200 mL) and the resulting mixture stirred at room temperature for 2 h. The reaction was quenched with saturated aqueous NaHCO3 and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo to give the crude aldehyde, which was used immediately in the next step without any purification. To a solution of t-BuOK (16.3 g, 146 mmol) in THF (200 mL) was added methyltriphenylphosphonium bromide (52.1 g, 146 mmol) at 0 ℃ under an argon atmosphere and the resulting mixture stirred for 1 h. The crude aldehyde in THF (40 mL) was added dropwise at 0 ℃ and the resulting mixture was stirred for 10 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 30:1) to give the olefin product (20.3 g, 72%, over 2 steps) as a colorless oil. [α]D 20 + 3.1 (c 0.93, MeOH); 1H NMR (300 MHz, CDCl3) δ: 7.69–7.65 (m, 4H), 7.43–7.34 (m, 6H), 5.71 (dddd, J = 16.8, 10.3, 8.0, 6.5 Hz, 1H), 4.94–4.86 (m, 2H), 3.69–3.57 (m, 2H), 2.23–2.18 (m, 1H), 1.88–1.78 (m, 2H), 1.73–1.67 (m, 2H), 1.43–1.17 (m, 4H), 1.06 (s, 9H), 1.06–0.95 (m, 1H); 13C NMR (75 MHz, CDCl3) δ: 137.2, 135.6, 134.0, 129.49, 129.47, 127.56, 127.55, 115.6, 66.3, 43.8, 37.9, 31.3, 30.0, 26.9, 26.3, 26.1, 19.4; HRMS (FAB) calcd for C26H37OSi [M+H]+: 393.2614. Found:393.2623.
4.4. ((1S,2R)-2-Allylcyclohexyl) methanol (10)
To a solution of the olefin (20.3 g, 52.0 mmol) in THF (150 mL) was added TBAF (1.0 M solution in THF; 78 mL, 78 mmol) at room temperature and the resulting mixture stirred for 10 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 20:1) to give 10 (7.30 g, 92%) as a colorless oil. [α]D 20 + 12.9 (c 1.55, MeOH); 1H NMR (300 MHz, CDCl3) δ: 5.80 (dddd, J = 16.8, 10.3, 7.9, 6.5 Hz, 1H), 5.04–4.98 (m, 2H), 3.70 (dd, J = 10.7, 2.9 Hz, 1H), 3.56 (dd, J = 10.8, 5.4 Hz, 1H), 2.33–2.24 (m, 1H), 2.01–1.91 (m, 1H), 1.82–1.71 (m, 3H), 1.26–1.12 (m, 5H), 1.08–0.96 (m, 1H); 13C NMR (75 MHz, CDCl3) δ: 137.0, 115.8, 65.4, 43.7, 37.94, 37.92, 31.6, 29.5, 26.0, 25.8.
4.5. Ethyl (E)-3-((1S,2R)-2-allylcyclohexyl) acrylate (11)
To a solution of 10 (7.30 g, 47.4 mmol) and TPAP (330 mg, 0.940 mmol) in CH2Cl2 (160 mL) was added NMO (22.1 g, 189 mmol) at room temperature under an argon atmosphere. After stirring for 1 h, the reaction mixture was filtered through a pad of silica gel and the filtrate concentrated in vacuo. The resulting residue was used immediately in the next step without any purification. To a solution of NaH (60% dispersion in mineral oil; 2.40 g, 61.6 mmol) in THF (160 mL) was added triethyl phosphonoacetate (13 mL, 66 mmol) at –20 ℃ and the resulting mixture stirred for 20 min. The residue obtained from the TPAP oxidation of 10 in THF (25 mL) was added dropwise to the reaction mixture at –20 ℃ and stirred for 30 min. The reaction was quenched with saturated aqueous NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 30:1) to give 11 (6.50 g, 62% over 2 steps) as a colorless oil. [α]D 20 + 10.3 (c 1.68, MeOH); 1H NMR (300 MHz, CDCl3) δ: 6.81 (dd, J = 15.8, 9.5 Hz, 1H), 5.79 (dd, J = 15.6, 0.6 Hz, 1H), 5.72 (dddd, J = 16.2, 11.0, 8.1, 6.2 Hz, 1H), 5.00 (brs, 1H), 4.97–4.93 (m, 1H), 4.19 (q, J = 7.1 Hz, 1H), 2.22–2.17 (m, 1H), 1.91–1.65 (m, 7H), 1.34–1.21 (m, 4H), 1.29 (t, J = 7.1 Hz, 3H), 1.01–0.88 (m, 1H); 13C NMR (75 MHz, CDCl3) δ: 166.8, 153.5, 136.5, 120.9, 116.2, 60.1, 46.4, 41.0, 39.0, 32.6, 30.9, 25.9, 25.5, 14.3; HRMS (EI) calcd for C14H22O2 [M]+: 222.1620. Found: 222.1615.
4.6. (E)-3-((1S,2R)-2-Allylcyclohexyl)prop-2-en-1-ol
To a solution of 11 (6.50 g, 29.3 mmol) in CH2Cl2 (100 mL) was added DIBAL-H (1.0 M solution in hexane; 88 mL, 88 mmol) at –78 °C under an argon atmosphere and the resulting mixture stirred for 1 h. The reaction was quenched with CH3OH, warmed to room temperature, filtered through a pad of silica gel, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/EtOAc = 6:1) to give the allylic alcohol (4.10 g, 79%) as a colorless oil. [α]D 20 + 11 (c 0.10, MeOH); 1H NMR (300 MHz, CDCl3) δ: 5.81–5.47 (m, 3H), 4.99 (brs, 1H), 4.95–4.92 (m, 1H), 4.10 (d, J = 5.4 Hz, 1H), 2.30–2.22 (m, 1H), 1.82–1.58 (m, 7H), 1.25–1.12 (m, 4H), 0.98–0.84 (m, 1H); 13C NMR (75 MHz, CDCl3) δ: 137.9, 137.3, 128.7, 115.7, 63.9, 46.3, 41.4, 38.9, 33.7, 31.3, 26.2, 25.9.
4.7. ((2R,3R)-3-((1S,2R)-2-Allylcyclohexyl) oxiran-2-yl) methanol (12)
To a solution of diethyl d-tartrate (7.8 mL, 46 mmol) in CH2Cl2 (100 mL) was added titanium(IV) isopropoxide (13 mL, 46 mmol) at 0 °C under an argon atmosphere. After stirring for 20 min, the mixture was cooled to –20 °C and added to t-butyl hydroperoxide (6.0 M solution in toluene; 15 mL, 90 mmol). The allylic alcohol was then added dropwise to the reaction mixture at –20 °C and stirred for 6 h. The reaction was quenched with 10% potassium sodium (+)-tartrate tetrahydrate in water at 0 °C, extracted with CH2Cl2 and the organic layer concentrated in vacuo. 1 M NaOH in diethyl ether was added to the resulting residue at room temperature and stirred for 2 h. The resulting mixture was extracted with diethyl ether, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ EtOAc = 6:1) to give 12 (3.10 g, 68%) as a colorless oil. [α]D 20 + 34 (c 0.50, MeOH); 1H NMR (300 MHz, CDCl3) δ: 5.83–5.69 (m, 1H), 5.03–4.98 (m, 2H), 3.95–3.92 (m, 1H), 3.70–3.63 (m, 1H), 2.99–2.98 (m, 1H), 2.72 (dd, J = 8.7, 2.1 Hz, 1H), 2.33–2.29 (m, 1H), 1.94–1.73 (m, 5H), 1.34–1.13 (m, 4H), 0.99–0.92 (m, 2H); 13C NMR (75 MHz, CDCl3) δ: 136.7, 116.2, 61.4, 59.7, 59.4, 44.6, 39.6, 38.6, 31.2, 29.8, 26.0, 25.4; HRMS (ESI) calcd for C12H20NaO2 [M + Na]+: 219.1361. Found: 219.1357.
Diastereomer of 12: yield 68%; [α]D 20 + 8.7 (c 0.55, MeOH); 1H NMR (300 MHz, CDCl3) δ: 5.89–5.75 (m, 1H), 5.06–4.98 (m, 2H), 3.95–3.90 (m, 1H), 3.64–3.59 (m, 1H), 2.88–2.87 (m, 1H), 2.80 (dd, J = 8.0, 2.3 Hz, 1H), 2.56–2.48 (m, 1H), 2.04–1.94 (m, 1H), 1.82–1.64 (m, 4H), 1.39–1.05 (m, 4H), 1.00–0.87 (m, 2H); 13C NMR (75 MHz, CDCl3) δ: 137.1, 116.0, 61.9, 60.0, 56.1, 44.7, 42.2, 38.5, 31.1, 29.1, 25.8, 25.6; HRMS (ESI) calcd for C12H20NaO2 [M + Na]+: 219.1361. Found: 219.1356.
4.8. (1R)-1-((1S,3RS,4aR,8aS)-3-(Hydroxymethyl)octahydro-1H-isochromen-1-yl) ethane-1,2-diol (13)
To a solution of K3Fe(CN)6 (15.3 g, 46.5 mmol) in t-BuOH/H2O (1:1, 150 mL) at room temperature was added K2CO3 (6.42 g, 46.5 mmol), (DHQ)2AQN (118 mg, 0.137 mmol), and K2OsO2(OH)4 (22.7 mg, 0.0894 mmol). After stirring for 20 min, the resulting mixture was cooled to 0 °C, 12 (3.01 g, 13.1 mmol) in t-BuOH/H2O (1:1, 20 mL) was added, and the resulting mixture stirred for 10 h. The reaction was quenched with aqueous Na2SO3 and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 20:1) to give the triol as a colorless oil, which was used immediately in the next step without any further purification. To a solution of p-toluensulfonic acid monohydrate (836 mg, 4.40 mmol) was added the triol in CH2Cl2 (50 mL) at room temperature and the resulting mixture stirred for 10 h. The reaction was quenched with saturated aqueous NaHCO3 and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 20:1) to give 13 (3.20 g, 96%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ: 4.13–4.07 (m, 1H), 3.87–3.75 (m, 2H), 3.67–3.53 (m, 2H), 3.50–3.29 (m, 2H), 1.79–1.37 (m, 6H), 1.25–1.18 (m, 3H), 1.10–0.88 (m, 3H); 13C NMR (75 MHz, CDCl3, dr = 5:4) δ: 83.8, 78.8, 76.2, 74.2, 71.2, 70.9, 66.1, 62.4, 62.2, 60.4, 43.6, 43.2, 40.5, 36.2, 34.6, 33.5, 33.2, 33.2, 27.3, 27.0, 25.9, 25.8, 25.7; HRMS (ESI) calcd for C12H22NaO4 [M + Na]+: 253.1416. Found: 253.1410.
4.9. ((1S,3RS,4aR,8aS)-1-((Butyl(methyl)amino)methyl)octahydro-1H-isochromen-3-yl) methanol (14a)
To a solution of 13 (800 mg, 3.48 mmol) was added NaIO4 (1.00 g, 4.67 mmol) in THF (8.0 mL) and H2O (8.0 mL) at room temperature. After the mixture was stirred for 10 min, the mixture was washed with brine, extracted with EtOAc, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was used immediately in the next step without any purification. The residue was added to n-butylamine (460 μL, 4.7 mmol) and sodium cyanotrihydroborate (640 mg, 10.2 mmol) in CH2Cl2 (15 mL) at room temperature. After stirring for 1 h, the reaction was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was used immediately in the next step without any purification. The amination product was added to paraformaldehyde (2.70 g) and sodium cyanotrihydroborate (640 mg, 10.2 mmol) in EtOH (14 mL) at room temperature and stirred for 12 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 15:1) to give 14a (468 mg, 59% over 3 steps) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ: 4.04–4.02 (m, 1H), 3.56–3.51 (m, 1H), 3.49–3.36 (m, 1.6H), 3.28–3.21 (m, 0.4H), 2.58–2.57 (m, 1H), 2.52–2.42 (m, 1H), 2.35 (s, 1.2H), 2.33 (s, 1.8H), 2.35–2.15 (m, 3H), 1.84–1.49 (m, 6H), 1.43–1.17 (m, 4H), 1.08–0.82 (m, 9H); 13C NMR (75 MHz, CDCl3) δ: 79.3, 74.9, 74.5, 70.1, 65.6, 60.4, 59.2, 58.6, 58.0, 45.1, 44.8, 43.1, 40.1, 35.6, 34.0, 33.4, 32.9, 27.6, 27.3, 26.3, 25.8, 25.7, 25.6, 20.17, 20.12, 13.68, 13.65; HRMS (ESI) calcd for C16H31NO2 [M+H]+: 270.2434. Found: 270.2432.
Compounds 14b, 14c, and 14d were prepared using the same synthetic procedure. 14b: yield 63%, 3 steps; 1H NMR (300 MHz, CDCl3, 4:1 mixture of two diastereoisomers) δ: 6.12–5.98 (m, 1H), 5.54–5.46 (m, 2H), 4.18–4.07 (m, 2H), 3.96–3.90 (m, 1H), 3.74–3.38 (m, 3.2H), 3.33 (d, J = 9.3 Hz, 0.8H), 3.10 (dd, J = 13.1, 2.0 Hz, 0.8H), 3.03–3.02 (m, 0.2H), 2.94–2.92 (m, 0.2H), 2.84 (dd, J = 12.9, 9.9 Hz, 0.8H), 2.81 (s, 2.4H), 2.75 (s, 0.6H), 1.79–1.69 (m, 2H), 1.62–1.50 (m, 2H), 1.48–1.39 (m, 1H), 1.25–1.22 (m, 3H), 1.03–0.91 (m, 3H); 13C NMR (75 MHz, CDCl3, 4:1 mixture of two diastereoisomers) δ: 128.1, 127.5, 124.9, 124.6, 78.9, 74.3, 74.2, 69.6, 65.4, 60.44, 60.37, 59.1, 57.9, 57.3, 44.9, 44.6, 42.4, 40.5, 39.8, 35.4, 33.9, 33.3, 32.81, 32.75, 27.3, 25.7, 25.6, 25.5; HRMS (ESI) calcd for C15H28NO2 [M+H]+: 254.2122. Found: 254.2113. 14c: yield 66%, 3 steps; 1H NMR (300 MHz, CDCl3, 3:2 mixture of two diastereoisomers) δ: 4.22–4.04 (m, 1H), 3.64–3.48 (m, 2H), 3.41 (td, J = 9.4, 3.1 Hz, 0.6H), 3.31 (dd, J = 11.9, 3.2 Hz, 0.4H), 3.13 (dd, J = 13.4, 2.0 Hz, 0.4H), 3.09–2.75 (m, 3.6H), 2.87 (s, 1.2H), 2.79 (s, 1.8H), 2.17–2.06 (m, 1H), 1.80–1.41 (m, 5H), 1.31–1.24 (m, 3H), 1.14–0.88 (m, 3H) , 1.13 (d, J = 6.6 Hz, 1.2H), 1.08 (t, J = 6.9 Hz, 3H), 1.04 (d, J = 6.6 Hz, 1.8H); 13C NMR (75 MHz, CDCl3, 3:2 mixture of two diastereoisomers) δ: 79.0, 74.7, 74.5, 69.7, 66.5, 65.5, 64.4, 60.3, 59.7, 58.3, 45.0, 44.8, 44.1, 42.8, 40.0, 35.5, 34.0, 33.4, 32.92, 32.86, 27.4, 27.2, 25.8, 25.77, 25.69, 25.65, 25.5, 24.9, 24.5, 21.3, 21.0, 20.93, 20.90; HRMS (EI) calcd for C16H31NO2 [M]+: 269.2355. Found: 269.2355. 14d: yield 62%, 3 steps; 1H NMR (300 MHz, CDCl3, 1:1 mixture of two diastereomers) δ: 7.32–7.24 (m, 5H), 4.03–3.96 (m, 1H), 3.69–3.16 (m, 6H), 2.55–2.48 (m, 2H), 2.26 (s, 1.5H), 2.25 (s, 1.5H), 1.75–1.38 (m, 6H), 1.28–1.17 (m, 3H), 1.07–0.78 (m, 3H); 13C NMR (75 MHz, CDCl3, 1:1 mixture of two diastereomers) δ: 138.6, 138.2, 129.4, 129.2, 128.2, 127.1, 127.0, 79.3, 78.0, 72.90, 72.87, 66.2, 63.1, 62.5, 60.9, 59.8, 59.5, 45.0, 44.5, 43.5, 43.0, 40.6, 36.0, 34.6, 33.6, 33.4, 32.8, 28.0, 27.8, 26.2, 26.1, 26.0, 25.9; HRMS (ESI) calcd for C19H30NO2 [M+H]+: 304.2277. Found: 304.2273.
4.10. (2S)-2-((((1S,3RS,4aR,8aS)-1-((Butyl(methyl)amino)methyl)octahydro-1H-isochromen-3-yl)methyl)amino)-3-(1H-imidazol-4-yl)-N-methoxy-N-methylpropanamide (15a)
To a solution of 14a (30.0 mg, 0.12 mmol) in CH2Cl2 was added benzenesulfenamide (20 μL, 0.11 mmol), 1,8-dizazbicycle[5.4.0]-7-undecene (100 μL, 0.64 mmol), and N-chlorosuccinimide (44.0 mg, 0.66 mmol) at 0 °C in the presence of Molecular Sieves 4A and the resulting mixture was stirred for 2 h. The reaction was quenched with saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was used immediately in the next step without any purification. To a solution of the product in CH2Cl2 (3 mL) was added H-His-N(OMe)Me (65.0 mg, 0.328 mmol), CH3COOH (1 drop from syringe), and cyanotrihydroborate (20.7 mg, 0.329 mmol) at room temperature. After the mixture was stirred for 1 h, the reaction was quenched with saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was washed with saturated aqueous NH4Cl and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by LC-MS [linear gradient of B from 10 to 35% over 30 min (B: 0.05% CH3COOH in CH3CN; A: 0.05% CH3COOH in H2O)] to give 15a (18.0 mg, 18%, 2 steps) as a yellow oil. 1H NMR (300 MHz, CDCl3, 3:2 mixture of two diastereomers) δ: 7.54 (s, 0.4H), 7.51 (s, 0.6H), 6.79 (s, 1H), 4.03–3.96 (m, 0.6H), 3.74–3.72 (m, 1H), 3.65 (s, 3H), 3.50–3.45 (m, 1.4H), 3.23 (s, 1.8H), 3.22 (1.2H), 3.23–3.22 (m, 1H), 2.98–2.86 (m, 2.6H), 2.75–2.57 (m, 3.4H), 2.52–2.42 (m, 4H), 2.37 (s, 1.8H), 2.34 (s, 1.2H), 1.78–1.46 (m, 6H), 1.39–1.26 (m, 5H), 1.08–0.85 (m, 3H), 0.91 (t, J = 7.5 Hz, 3H); HRMS (ESI) calcd for C24H44N5O3 [M+H]+: 450.3446. Found: 450.3439.
Compounds 15b, 15c, and 15d were prepared using the same synthetic procedure. 15b: yield 14%, 2 steps; 1H NMR (300 MHz, CDCl3, 4:1 mixture of two diastereomers) δ: 7.55 (s, 0.2H), 7.52 (s, 0.8H), 6.81 (s, 1H), 5.87 (ddt, J = 17.0, 10.3, 6.7 Hz, 1H), 5.21–5.13 (m, 2H), 4.04–3.98 (m, 1H), 3.75–3.63 (m, 1H), 3.68 (s, 2.4H), 3.65 (s, 0.6H), 3.50–3.43 (m, 1.6H), 3.40–3.34 (m, 0.4H), 3.24 (s, 2.4H), 3.22 (s, 0.6H), 3.21–3.17 (m, 1H), 3.09 (dd, J = 13.5, 6.6 Hz, 1H), 2.97–2.85 (m, 2H), 2.79–2.63 (m, 2H), 2.58 (dd, J = 13.5, 1.5 Hz, 1H), 2.50–2.38 (m, 3H), 2.34 (s, 2.4H), 2.33 (s, 0.6H), 1.74–1.53 (m, 4H), 1.47 (dd, J = 12.5, 1.5 Hz, 1H), 1.39 (dd, J = 3.9, 1.5 Hz, 0.8H), 1.39 (dd, J = 4.1, 1.4 Hz, 0.2H), 1.25–1.16 (m, 3H), 1.10–0.82 (m, 2H); HRMS (EI) calcd for C23H39N5O3 [M]+: 433.3053. Found: 433.3054. 15c: yield 25%, 2 steps; 1H NMR (500 MHz, CDCl3, 55:45 mixture of two diastereomers) δ: 7.56 (s, 0.45H), 7.55 (s, 0.55H), 6.81 (s, 1H), 3.99–3.97 (m, 0.55H), 3.81–3.75 (m, 1H), 3.64 (s, 3H), 3.49–3.42 (m, 1H), 3.27–3.20 (m, 0.45H), 3.224 (s, 1.6H), 3.216 (s, 1.4H), 2.95 (dd, J = 15.0, 4.0 Hz, 1H), 2.91–2.86 (m, 1H), 2.74–2.66 (m, 1.55H), 2.62–2.39 (m, 3.45H), 2.35 (s, 1.6H), 2.33 (s, 1.4H), 2.30–2.17 (m, 2H), 1.82–1.70 (m, 5.35H), 1.61–1.55 (m, 1.1H), 1.51–1.45 (m, 0.55H), 1.41–1.38 (m, 1H), 1.26–1.20 (m, 3H), 1.08–0.86 (m, 8H); HRMS (EI) calcd for C24H43N5O3 [M]+: 449.3366. Found: 449.3364. 15d; yield 17%, 2 steps; 1H NMR (500 MHz, CDCl3, 2:1 mixture of two diastereomers) δ: 7.51–7.45 (m, 1H), 7.34–7.23 (m, 5H), 6.80 (s, 1H), 5.41–5.38 (m, 2H), 3.78–3.70 (m, 1H), 3.65–3.62 (m, 4H), 3.52–3.50 (m, 1H), 3.31–3.20 (m, 1H), 3.23 (s, 1H), 3.22 (s, 2H), 2.92–2.89 (m, 1H), 2.70–2.62 (m, 1H), 2.57–2.46 (m, 1H), 2.43–2.39 (m, 1H), 2.31 (s, 3H), 2.07–1.95 (m, 1H), 1.75–1.38 (m, 5H), 1.25–0.82 (m, 6H); HRMS (EI) calcd for C27H41N5O3 [M]+: 483.3209. Found: 483.3214.
4.11. (2S)-2-((((1S,3RS,4aR,8aS)-1-((Butyl(methyl)amino)methyl)octahydro-1H-isochromen-3-yl) methyl)amino)-3-(1H-imidazol-4-yl) propanal (16a)
To a solution of 15a (4.5 mg, 0.00932 mmol) in CH2Cl2 (1.5 mL) was added DIBAL-H (1.0 M solution in hexane, 200 μL, 0.20 mmol) at –10 °C under an argon atmosphere and the resulting mixture stirred for 30 min. The reaction was quenched with CH3OH and concentrated in vacuo. The residue was filtered and concentrated in vacuo. The resulting residue was purified by LC-MS [linear gradient of B from 5 to 35% over 30 min (B: 0.05% CH3COOH in CH3CN; A: 0.05% CH3COOH in H2O)] to give 16a (450 μg, 10%) as a yellow solid. 1H NMR (500 MHz, CDCl3, 2:1 mixture of two diastereomers) δ: 8.85–8.84 (m, 0.6H), 8.75 (m, 0.4H), 8.25 (m, 0.6H), 8.08 (s, 0.4H), 7.27–7.24 (m, 1H), 4.71 (m, 1H), 4.34 (m, 0.4H), 3.93 (m, 0.6H), 3.81 (m, 1H), 3.69–3.62 (m, 3H), 3.45–3.44 (m, 1H), 3.35–3.29 (m, 1H), 3.21–3.12 (m, 3H), 2.98–2.90 (m, 1H), 2.94 (s, 2H), 2.93 (s, 1H), 1.84–1.65 (m, 6H), 1.46–1.29 (m, 6H), 1.67–0.89 (m, 7H); HRMS (ESI) calcd for C22H39N4O2 [M+H]+: 391.3075. Found: 391.3028.
Compounds 16b, 16c, and 16d were prepared using the same synthetic procedure. 16b: yield 6%; 1H NMR (300 MHz, CDCl3) δ: 8.86 (m, 0.6H), 8.76 (m, 0.4H), 8.22–8.19 (m, 1H), 7.25–7.24 (m, 1H), 6.03–5.94 (m, 1H), 5.63–5.60 (m, 2H), 4.75–4.73 (m, 1H), 4.34–4.32 (m, 0.4H), 4.23 (m, 0.6H), 3.92–3.82 (m, 2H), 3.74–3.56 (m, 3H), 3.37 (m, 1H), 3.24–3.22 (m, 1H), 3.19–3.18 (m, 2H), 2.94 (s, 1.2H), 2.92 (s, 1.8H), 2.69–2.61 (m, 1H), 1.86–1.66 (m, 4H), 1.47–1.25 (m, 3H), 1.17–0.79 (m, 5H); HRMS (ESI) calcd for C21H35N4O2 [M+H]+: 375.2762. Found: 375.2755. 16c: yield 9%; 1H NMR (300 MHz, CDCl3) δ: 8.62 (s, 1H), 8.07 (s, 1H), 7.41–7.38 (m, 1H), 4.77 (m, 1H), 4.37–4.32 (m, 2H), 3.96–3.81 (m, 2H), 3.75–3.63 (m, 3H), 3.26 (m, 3H), 2.98 (s, 3H), 2.98–2.92 (m, 1H), 2.24–2.15 (m, 2H), 1.85–1.64 (m, 6H), 1.42–1.29 (m, 4H), 1.12–1.03 (m, 7H); HRMS (ESI) calcd for C22H39N4O2 [M+H]+: 391.3075. Found: 391.3068. 16d: yield 10%; 1H NMR (500 MHz, CDCl3) δ: 8.33 (s, 1H), 8.07 (s, 1H), 7.54–7.52 (m, 5H), 7.28 (s, 1H), 4.78–4.74 (m, 1H), 4.51 (d, J = 13.5 Hz, 1H), 4.33–4.29 (m, 1H), 3.87 (m, 1H), 3.22–3.09 (m, 2H), 2.93 (s, 3H), 2.84–2.64 (m, 4H), 2.00–1.95 (m, 2H), 1.76–1.61 (m, 4H), 1.48–1.29 (m, 5H), 1.13–0.88 (m, 3H); HRMS (ESI) calcd for C25H37N4O2 [M+H]+: 425.2918. Found: 425.2911.
4.12. (R)-1-((1S,3R,4aR,8aS)-3-(Hydroxymethyl)octahydro-1H-isochromen-1-yl)ethane-1,2-diol (13a)
To a solution of K3Fe(CN)6 (15.3 g, 46.5 mmol) in t-BuOH/H2O (1:1, 150 mL) was added K2CO3 (6.42 g, 46.5 mmol), (DHQ)2Pyr (121 mg, 0.137 mmol) and K2OsO2(OH)4 (22.7 mg, 0.0894 mmol) at room temperature. After stirring for 20 min, the mixture was cooled to 0 °C and 12 (3.06 g, 15.6 mmol) in t-BuOH/H2O (1:1, 20 mL) added, and the resulting mixture stirred for 10 h. The reaction was quenched with aqueous Na2SO3 and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 20:1) to give the triol as a colorless oil, which was used immediately in the next step without any further purification. To a solution of p-toluensulfonic acid monohydrate (836 mg, 4.40 mmol) was added the triol in CH2Cl2 (50 mL) at room temperature and the resulting mixture stirred for 10 h. The reaction was quenched with saturated aqueous NaHCO3 and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 20:1) to give 13a (2.87 g, 80%) as a colorless oil. [α]D 20 + 11.6 (c 1.08, MeOH); 1H NMR (300 MHz, CDCl3) δ: 4.47 (brs, 1H), 4.22 (brs, 1H), 4.14–4.03 (m, 2H), 3.91–3.80 (m, 2H), 3.68–3.65 (m, 1H), 3.59 (dd, J = 9.9, 3.0 Hz, 1H), 3.51–3.36 (m, 1H), 3.32 (d, J = 8.4 Hz, 1H), 1.78–1.37 (m, 5H), 1.27–1.17 (m, 3H), 1.13–0.89 (m, 3H). 13C NMR (75 MHz, CDCl3, major isomer dr = 6:1) δ: 76.3, 74.1, 70.1, 62.5, 60.4, 43.6, 36.2, 33.4, 33.1, 27.2, 25.8, 25.7; HRMS (ESI) calcd for C12H22NaO4 [M + Na]+: 253.1416. Found: 253.1414.
(1-S, 3-R)-13b, (1-R, 3-R)-13c, and (1-R, 3-S)-13d were prepared using the same synthetic procedure. 13b: yield 85%, 2 steps; [α]D 20 + 12 (c 0.48, MeOH); 1H NMR (300 MHz, CDCl3) δ: 4.53 (brs, 1H), 4.22 (brs, 1H), 3.86–3.81 (m, 2H), 3.68–3.60 (m, 1H), 3.57–3.48 (m, 3H), 3.45–3.40 (d, J = 7.8 Hz, 1H), 2.24 (brs, 1H), 1.79–1.62 (m, 4H), 1.44–1.39 (m, 1H), 1.29–1.21 (m, 3H), 1.07–0.92 (m, 4H); 13C NMR (75 MHz, CDCl3) δ: 84.1, 78.9, 70.5, 66.1, 62.5, 43.2, 40.4, 34.6. 33.1, 27.0, 25.9, 25.8; HRMS (ESI) calcd for C12H22NaO4 [M + Na]+: 253.1416. Found: 253.1410. 13c: yield 81%, 2 steps; [α]D 20 –16 (c 0.18, MeOH); 1H NMR (300 MHz, CDCl3) δ: 3.96 (dd, J = 8.3, 4.4 Hz, 1H), 3.86 (dd, J = 12.0, 6.3 Hz, 1H), 3.77–3.57 (m, 3H), 3.51 (dd, J = 11.7, 3.0 Hz, 1H), 3.51 (dd, J = 11.7, 7.8 Hz, 1H), 1.80–1.69 (m, 4H), 1.59–1.50 (m, 3H), 1.44–1.37 (m, 1H), 1.25–1.10 (m, 4H), 1.00–0.93 (m, 1H); 13C NMR (75 MHz, CDCl3) δ: 78.9, 72.8, 71.7, 66.0, 64.2, 40.1, 34.7, 32.3, 31.6, 27.6, 26.7, 26.0; HRMS (ESI) calcd for C12H22NaO4 [M + Na]+: 253.1416. Found: 253.1414. 13d: yield 74%, 2 steps; [α]D 20 –12 (c 0.24, MeOH); 1H NMR (300 MHz, CDCl3) δ: 4.37 (brs, 1H), 4.04 (brs, 1H), 3.83 (dd, J = 11.3, 4.7 Hz, 1H), 3.75 (d, J = 10.5 Hz, 1H), 3.68 (dd, J = 9.0, 4.2 Hz, 1H), 3.61–3.50 (m, 2H), 3.46–3.38 (m, 2H), 1.81–1.65 (m, 3H), 1.54–1.41 (m, 3H), 1.36–1.17 (m, 3H), 1.00–0.94 (m, 2H); 13C NMR (75 MHz, CDCl3) δ: 76.9, 73.2, 69.4, 66.3, 64.0, 44.4, 34.8, 34.5, 34.2, 28.6, 27.0, 26.0; HRMS (ESI) calcd for C12H22NaO4 [M + Na]+: 253.1416. Found: 253.1410.
4.13. ((1S,3R,4aR,8aS)-1-((Butyl(methyl)amino)methyl)octahydro-1H-isochromen-3-yl) methanol (17a)
(1-S, 3-R)-17a, (1-S, 3-S)-17b, (1-R, 3-R)-17c, and (1-R, 3-S)-17d were prepared using the same synthetic procedure described for 14a. 17a: yield 46%, 3 steps; [α]D 20 + 35 (c 0.21, MeOH); 1H NMR (300 MHz, CDCl3) δ: 4.06 (d, J = 10.2 Hz, 1H), 4.01 (d, J = 10.2 Hz, 1H), 3.51–3.47 (m, 0.5H), 3.45–3.32 (m, 1.5H), 2.49–2.24 (m, 4H), 2.26 (s, 3H), 1.75–1.68 (m, 3H), 1.61–1.38 (m, 5H), 1.36–1.22 (m, 5H), 1.04–0.85 (m, 3H), 0.91 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 74.2, 70.6, 60.6, 59.5, 58.6, 44.7, 43.1, 35.7, 33.5, 32.9, 27.7, 27.0, 25.9, 25.6, 20.3, 13.8; HRMS (EI) calcd for C16H31NO2 [M]+: 269.2355. Found: 269.2352. 17b: yield 43%, 3 steps; [α]D 20 + 13 (c 0.50, MeOH); 1H NMR (300 MHz, CDCl3) δ: 3.64–3.59 (m, 2H), 3.50–3.45 (m, 1H), 3.37 (td, J = 9.3, 4.0 Hz, 1H), 3.09–2.88 (m, 4H), 2.76 (s, 3H), 1.82–1.64 (m, 4H), 1.55–1.21 (m, 8H), 1.09–0.92 (m, 4H), 0.97 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz CDCl3) δ: 78.2, 77.8, 66.1, 59.6, 57.9, 45.1, 42.4, 40.4, 34.5, 33.3, 28.2, 27.7, 26.1, 25.9, 20.6, 14.0; HRMS (ESI) calcd for C16H32NO2 [M+H]+: 270.2435. Found: 270.2430. 17c: yield 44%, 3 steps; [α]D 20 –17 (c 0.18, MeOH); 1H NMR (300 MHz, CDCl3) δ: 4.07 (dd, J = 6.0, 5.1 Hz, 1H), 4.00 (dd, J = 10.5, 5.7 Hz, 1H), 3.66 (t, J = 12.0 Hz, 1H), 3.38 (dd, J = 12.3, 4.8 Hz, 1H), 2.96 (t, J = 12.8 Hz, 1H), 2.75–2.65 (m, 1H), 2.52–2,46 (m, 1H), 2.43 (s, 3H), 2.23 (dd, J = 12.6, 4.8 Hz, 1H), 1.78–1.64 (m, 4H), 1.63–1.22 (m, 10H), 1.00–0.88 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ: 72.6, 69.6, 65.2, 58.8, 57.0, 43.2, 40.7, 34.1, 32.7, 29.5, 27.8, 27.7, 26.6, 26.0, 20.5, 13.9; HRMS (EI) calcd for C16H31NO2 [M]+: 269.2355. Found: 269.2360. 17d: yield 42%, 2 steps; [α]D 20 –14 (c 0.74, MeOH); 1H NMR (300 MHz, CDCl3) δ: 3.99–3.95 (m, 1H), 3.81–3.77 (m, 2H), 3.55 (dd, J = 6.6, 1.8 Hz, 1H), 3.48 (dd, J = 6.6, 4.2 Hz, 1H), 2.94 (dd, J = 8.1, 6.3 Hz, 1H), 2.76–2.68 (m, 1H), 2.55–2.44 (m, 2H), 2.35 (s, 3H), 2.24 (dd, J = 8.1, 1.8 Hz, 1H), 1.79–1.58 (m, 3H), 1.53–1.44 (m, 4H), 1.42–1.21 (m, 5H), 1.04–0.91 (m, 6H); 13C NMR (75 MHz, CDCl3, 4:1 mixture of two conformers) δ: 78.7, 73.7, 70.3, 66.4, 65.9, 58.8, 57.9, 57.3, 53.0, 45.1, 44.3, 42.4, 40.3, 34.9, 34.32, 34.25, 34.0, 33.1, 28.7, 28.3, 27.5, 27.1, 26.6, 26.1, 25.9, 25.8, 20.6, 20.4, 14.0, 13.8; HRMS (EI) calcd for C16H31NO2 [M]+: 269.2355. Found: 269.2354.
4.14. (S)-2-((((1S,3R,4aR,8aS)-1-((Butyl(methyl)amino)methyl)octahydro-1H-isochromen-3-yl)methyl)amino)-3-(1H-imidazol-4-yl)-N-methoxy-N-methylpropanamide (18a)
(1-S, 3-R)-18a, (1-S, 3-S)-18b, (1-R, 3-R)-18c, and (1-R, 3-S)-18d were prepared using the same synthetic procedure described for 15a. 18a: yield 17%, 2 steps; [α]D 20 + 17 (c 0.55, MeOH); 1H NMR (300 MHz, CDCl3) δ: 7.54 (s, 1H), 6.79 (s, 1H), 3.99–3.96 (m, 1H), 3.84–3.81 (m, 1H), 3.66–3.64 (m, 1H), 3.61 (s, 3H), 3.56–3.48 (m, 1H), 3.13 (s, 3H), 2.98–2.90 (m, 2H), 2.77–2.61 (m, 4H), 2.56–2.32 (m, 3H), 2.49 (s, 3H), 1.74–1.66 (m, 5H), 1.58–1.44 (m, 3H), 1.39–1.17 (m, 5H), 0.96–0.90 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H); HRMS (EI) calcd for C24H43N5O3 [M]+: 449.3366. Found: 449.3361. 18b: yield 13%, 2 steps; [α]D 20 + 36 (c 0.050, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.54 (s, 1H), 6.79 (s, 1H), 3.74–3.73 (m, 1H), 3.64 (s, 3H), 3.48 (td, J = 8.8, 2.0 Hz, 1H), 3.36–3.34 (m, 1H), 3.27–3.20 (m, 1H), 3.22 (s, 3H), 2.92 (dd, J = 15.0, 3.5 Hz, 1H), 2.75–2.67 (m, 3H), 2.59 (d, J = 13.5 Hz, 2H), 2.52–2.44 (m, 4H), 2.36–2.31 (m, 2H), 2.35 (s, 3H), 1.77–1.66 (m, 4H), 1.59 (d, J = 13.5 Hz, 1H), 1.54–1.44 (m, 2H), 1.39–1.16 (m, 4H), 1.33 (q, J = 7.5 Hz, 2H), 1.07–0.86 (m, 2H), 0.92 (t, J = 7.5 Hz, 3H). 18c: yield 19%, 2 steps; [α]D 20 –7.2 (c 0.42, MeOH); 1H NMR (500 MHz, CDCl3, 4:1 mixture of two conformers) δ: 7.55 (s, 0.8H), 7.52 (s, 0.2H), 6.80 (s, 1H), 3.97–3.93 (m, 1H), 3.85 (dd, J = 8.5, 4.0 Hz, 0.8H), 3.80–3.77 (m, 1.2H), 3.66 (s, 2.4H), 3.64 (s, 0.6H), 3.21 (s, 3H), 2.96 (dd, J = 15.0, 3.5 Hz, 0.8H), 2.91 (dd, J = 15.0, 4.0 Hz, 0.2H), 2.78–2.64 (m, 2.4H), 2.58–2.40 (m, 5.2H), 2.37 (s, 2.4H), 2.32 (s, 0.6H), 2.29–2.28 (m, 0.2H), 2.24 (dd, J = 13.3, 3.3 Hz, 0.2H), 1.78–1.57 (m, 6H), 1.52–1.16 (m, 8H), 1.06–0.89 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H); HRMS (EI) calcd for C24H43N5O3 [M]+: 449.3366. Found: 449.3365: 18d; yield 22%, 2 steps; [α]D 20 –3.1 (c 0.72, MeOH); 1H NMR (500 MHz, CDCl3, 4:1 mixture of two conformers) δ: 7.58 (s, 0.2H), 7.53 (s, 0.8H), 6.80 (s, 1H), 4.02–4.00 (m, 0.8H), 3.83–3.76 (m, 1.8H), 3.72–3.67 (m, 0.2H), 3.64 (s, 3H), 3.61–3.56 (m, 0.2H), 3.22 (s, 2.4H), 3.20 (s, 0.6H), 3.00 (dd, J = 13.3, 10.3 Hz, 0.8H), 2.43 (dd, J = 15.0, 4.0 Hz, 1.2H), 2.73–2.64 (m, 2.8H), 2.58–2.46 (m, 2.4H), 2.43 (dd, J = 11.8, 2.8 Hz, 0.8H), 2.37 (s, 3H), 2.30 (dd, J = 13.3, 2.8 Hz, 1H), 1.77–1.65 (m, 4H), 1.53–1.48 (m, 4H), 1.42–1.28 (m, 3H), 1.24–1.20 (m, 2H), 1.05–0.89 (m, 3H), 0.91 (t, J = 7.3 Hz, 3H); HRMS (EI) calcd for C24H43N5O3 [M]+: 449.3366. Found: 449.3369.
4.15. (S)-2-((((1S,3R,4aR,8aS)-1-((Butyl(methyl)amino)methyl)octahydro-1H-isochromen-3-yl)methyl)amino)-3-(1H-imidazol-4-yl)propanal (3a)
(1-S, 3-R)-3a, (1-S, 3-S)-3b, (1-R, 3-R)-3c, and (1-R, 3-S)-3d were prepared using the same procedure described for 16a. 3a: yield 9%; 1H NMR (300 MHz, CDCl3) δ: 7.90 (s, 1H), 7.72–7.69 (m, 1H), 6.96 (m, 1H), 4.68 (m, 1H), 4.30 (m, 2H), 4.15–4.12 (m, 2H), 3.78–3.65 (m, 3H), 3.45–3.43 (m, 1H), 3.34–3.34 (m, 2H), 3.00–2.79 (m, 4H), 1.85–1.53 (m, 6H), 1.48–1.22 (m, 7H), 1.20–0.90 (m, 6H); HRMS (ESI) calcd for C22H39N4O2 [M+H]+: 391.3075. Found: 391.3068. 3b: yield 7%; 1H NMR (500 MHz, CDCl3) δ: 8.79–8.74 (m, 1H), 8.10 (s, 1H), 7.19–7.18 (m, 1H), 4.71 (m, 1H), 3.90 (d, J = 10.3 Hz, 1H), 3.83–3.79 (m, 1H), 3.63–3.61 (m, 3H), 3.44 (m, 1H), 3.28–3.24 (m, 1H), 3.16–3.10 (m, 3H), 2.99–2.91 (m, 1H), 2.97 (s, 3H), 1.83–1.59 (m, 6H), 1.42–1.28 (m, 6H), 1.15–0.88 (m, 7H); HRMS (ESI) calcd for C22H39N4O2 [M+H]+: 391.3075. Found: 391.3077. 3c: yield 6%; 1H NMR (300 MHz, CDCl3) δ: 8.35 (m, 1H), 8.07 (s, 1H), 7.28 (s, 1H), 4.65 (m, 1H), 4.34 (m, 2H), 4.26–4.20 (m, 2H), 3.25–3.19 (m, 3H), 3.15–3.10 (m, 3H), 2.94–2.93 (m, 4H), 1.91–1.62 (m, 7H), 1.62–1.37 (m, 7H), 1.24–1.01 (m, 5H); HRMS (ESI) calcd for C22H39N4O2 [M+H]+: 391.3075. Found: 391.3068. 3d: yield 7%; 1H NMR (500 MHz, CDCl3) δ: 8.20 (s, 1H), 7.98 (m, 1H), 7.21 (s, 1H), 4.63 (m, 1H), 4.34 (m, 1H), 4.21 (m, 1H), 4.04–4.02 (m, 1H), 3.65 (m, 1H), 3.53 (m, 1H), 3.40–3.36 (m, 2H), 3.22–3.20 (m, 2H), 3.11–2.87 (m, 5H), 1.87–1.57 (m, 7H), 1.46–1.23 (m, 6H), 1.08–0.90 (m, 3H), 1.03 (t, J = 7.3 Hz, 3H); HRMS (ESI) calcd for C22H39N4O2 [M+H]+: 391.3074. Found: 391.3068.
4.16. Estimation of the IC50 values
The peptide substrate (H-Thr-Ser-Ala-Val-Leu-Gln-Ser-Gly-Phe-Arg-Lys-NH2;9 111 μM) in a solution of 20 mM Tris-HCl buffer pH 7.5 containing 7 mM DTT (25 μL) was incubated with R188I SARS 3CLpro (56 nM)9 at 37 °C for 2 h in the presence of various concentrations of the inhibitors. The mixture was eluted on an analytical HPLC column [Cosmosil 5C18 (4.6 × 150 mm)] using CH3CN in aqueous 0.1% TFA (10–20% over 30 min) as the eluent and the cleavage rates were calculated from the reduction in the substrate peak area. Each IC50 value was obtained from the sigmoidal dose–response curve (Fig. S-2 for a typical sigmoidal curve). Each experiment was repeated in triplicate and the results reported as the average value.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported, in part, by a Grant-in-aid for Scientific Research 16H05104 given to KA from the Japan Society for the Promotion of Science.
Footnotes
Determination of the configureation of triol 13 using the nOe spectra, typical sigmoidal curves used to obtain IC50 values, and NMR data of the as-synthesized compounds.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2019.115273.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Lee N., Hui D., Wu A. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 2.Drosten C., Günther S., Preiser W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Rota P.A., Oberste M.S., Monroe S.S. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 5.Marra M.A., Jones S.J., Astell C.R. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 6.Thiel V., Ivanov K.A., Putics Á. Mechanisms and enzymes involved in SARS coronavirus genome expression. J General Viol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 7.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.H. An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J Med Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akaji K. Advances in the design of ligands interacting with 3CL protease of novel coronaviruses causing infectious respiratory syndrome. Amino Acids Pept Proteins. 2018;42:228–279. and references cited therein. [Google Scholar]
- 9.Akaji K., Konno H., Onozuka M., Makino A., Saito H., Nosaka K. Evaluation of peptide-aldehyde inhibitors using R188I mutant of SARS 3CL protease as a proteolysis-resistant mutant. Bioorg Med Chem. 2008;16:9400–9408. doi: 10.1016/j.bmc.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akaji K., Konno H., Mitsui H. Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J Med Chem. 2011;54:7962–7973. doi: 10.1021/jm200870n. [DOI] [PubMed] [Google Scholar]
- 11.Shimamoto Y., Hattori Y., Kobayashi K. Fused-ring structure of decahydroisoquinoline as a novel scaffold for SARS 3CL protease inhibitor. Bioorg Med Chem. 2015;23:876–890. doi: 10.1016/j.bmc.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohnishi K., Hattori Y., Kobayashi K., Akaji K. Evaluation of a non-prime site substituent and warheads combined with a decahydroisoquinoline scaffold as a SARS 3CL protease inhibitor. Bioorg Med Chem. 2019;27:425–435. doi: 10.1016/j.bmc.2018.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hentges S.G., Sharpless K.B. Asymmetric induction in the reaction of osmium tetroxide with olefins. J Am Chem Soc. 1980;102:4263–4265. [Google Scholar]
- 14.Katsuki T., Sharpless K.B. The first practical method for asymmetric epoxidation. J Am Chem Soc. 1980;102:5974–5976. [Google Scholar]
- 15.Basha A., Lipton J.L., Weinreb S.M. A mild, general method for conversion of esters to amides. Tetrahedron Lett. 1977;18:4171–4172. [Google Scholar]
- 16.Moitessier N., Henry C., Len C., Chapleur Y. Toward a computational tool predicting the stereochemical outcome of symmetric reactions. 1. Application to Sharpless asymmetric dihydroxylation. J Org Chem. 2002;67:7275–7282. doi: 10.1021/jo0258148. [DOI] [PubMed] [Google Scholar]
- 17.The major product of entry 5 in Table 1 gave a clear nOe signal between the protons at the 1-position and bridgehead α-position. In addition, no nOe signals were observed between the protons at the 1- and 3-positions as well as between the 3-positon and bridgehead α-position (Figure S-1b). However, although unclear, an nOe signal between the protons at the 1-position and methylene group at the 3-position of the fused ring was observed. These results strongly suggest the major product has a (1R, 3S) configuration.
- 18.Corey E.J., Zhang J. Highly effective transition structure designed catalyst for the enantio- and position-selective dihydroxylation of polyisoprenoids. Org Lett. 2001;3:3211–3214. doi: 10.1021/ol016577i. [DOI] [PubMed] [Google Scholar]
- 19.Bredikhina Z.A., Kurenkov A.V., Antonovich O.A., Pashagin A.V., Bredikhin A.A. A rare case of facial selectivity inversion for Sharpless asymmetric dihydroxylation in a series of structurally homogeneous substrates: Synthesis of non-racemic 3-(nitrophenoxy)-propane-1,2-diols. Tetrahedron Assym. 2014;25:1015–1021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.