

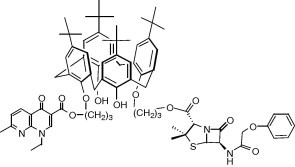
Graphical abstract
Keywords: Calixarene, Penicillin, Nalidixic acid, Prodrug, DMSO, Antibacterial
Abstract
Two well-known antibiotic heterocycles, the ‘quinolone’ nalidixic acid and the β-lactam penicillin V, active at different levels of the bacterial growth process, have been attached via an ether–ester junction to the p-tert-butylcalix[4]arene lower rim, in alternate position. The resulting hydrophobic molecular drug-organisers were fully characterized, and evaluated over two Gram negative and three Gram positive reference strains, using disk diffusion assays with disks impregnated with solution of title compound in pure DMSO. An interesting activity was observed over Staphylococcus aureus ATCC 25923 with the dissymmetrical podand incorporating one penicillin and one nalidixic ester moieties.
1. Introduction
Due to their intrinsic physico-chemical and chemical properties, the calixarenes have often been employed, in the last years, as carriers and spatial organisers of various kinds of active substituent, displaying properties dealing with the recognition of organic substrates or metallic cations.1, 2, 3, 4, 5
Such organising properties could be of great interest in the building of molecular drug-organisers or dispensers, notably as medically valuable structures incorporating probes and drugs; in the last case, a biochemically controlled release should be expected, resulting for such edifices in a drug-carrier or a prodrug behaviour. The resulting oligomeric structures could thus be considered as size-intermediate between discrete drugs and polymeric ones, the latter being emergent over the last 10 years, with the apparition of said polymer therapeutics6 using polymeric drugs,7 hybrid polymer drugs,6, 8, 9 polymeric micelles containing covalently linked drugs.10, 11
As recently reviewed by de Fatima et al,12 and Kalchenko and co-workers13 very few reports, essentially under the form of patents, describe therapeutical activities of calixarenes and derivatives; some of them, hydrophilic, have shown interesting activities against bacteria,14 fungi, cancerous cells and enveloped viruses,15 but also against thrombosis16 or fibrosic diseases.17 Biological studies related to plasmid DNA binding and cell transfection have been recently reported by Ungaro and co-workers.18 In the middle 50’s, the calixarene derivative ‘Macrocyclon’,19a and more recently some parent structures,(b), (c), (d) have been studied in the treatment of tuberculosis and other mycobacterioses. Calixarene-based mimics of vancomycin has also been described.20
In this field, our contribution has been devoted to the design of new anti-HIV21 or anti-corona virus22 agents and, with regards to spreading resistances of pathogenic microorganisms against actual antibiotics,23 antibacterial24, 25, 26, 27, 28, 29, 30 agents.
In parallel, we have approached the synthesis of new compounds thought as molecular drug-organisers or dispensers, based on a calixarene platform, and displaying at the lower rim penicillin, or said ‘quinolone’ moieties attached via a labile bound.
Our first molecular target was an antibiotic penicillin grafted in alternate position of the cone conformer of the 1,3-bis(O-acetyl)-p-tert-butylcalix[4]arene, via amide links involving the external amino group of 6-APA.31 As such a structure should be considered as a pure β-lactam antibiotic (the calixarene subunits playing the role of the external amide function), we have investigated other synthetic approaches involving a labile ester junction. This was applied to nalidixic acid (the first derivative of the said ‘quinolone’ family)32, 33 or penicillin antibiotic derivatives.34 Supposing that a synergistic antibacterial effect could occur through the incorporation of different kinds of known antibiotic species on the same spatial organiser, calixarene derivatives incorporating various penicillin or ‘quinolone’ subunits were also prepared.
In this sense, we present here the synthesis and characterization, then the antibacterial evaluation of their DMSO solutions with disk diffusion assays, of the dissymetric podand 3 bearing one penicillin and one ‘quinolone’ (penicillin V and nalidixic acid) bound distally at the lower rim of p-tert-butylcalix[4]arene by a labile propyl linker, the corresponding bis-nalidixic, bis-penicillin V and bis-hydroxypropyl derivative 5,32 6, and 4 (Scheme 1 ), respectively, then penicillin V, potassium salt (PVK) and nalidixic acid (NA).
Scheme 1.
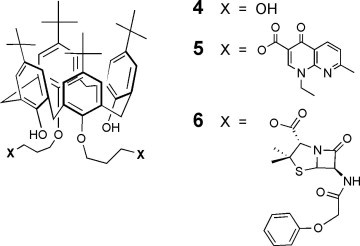
Symmetric calixarene derivatives of the present study.
2. Results and discussion
2.1. Synthesis
Previous results showed first that penicillins esters of O-alkoxy calixarenes were obtained only via reaction of a penicillin salt with a bromoalkyl calixarene,34 forcing introduction of the bromoalkyl arm in a preliminar step; secondly, that adaptation of this pathway to salts of nalidixic acid and parent compounds was uneasy and tedious, a contrario to O-alkylation of calixarene by a bromoalkyl derivative.32 In this sense, the formation of 2 from 1,3-bis(bromopropyl)calixarene was discarded. In addition, the choice was done to introduce the penicillin moiety at the last step, in order to avoid degradation of this pH-sensitive unit. It was thus decided to prepare first the mono-bromopropylcalixarene 1, to alkylate it at the opposite position with bromopropyl nalidixate to obtain 2, then to introduce the penicillin moiety on the bromopropyl arm (Scheme 2 ).
Scheme 2.
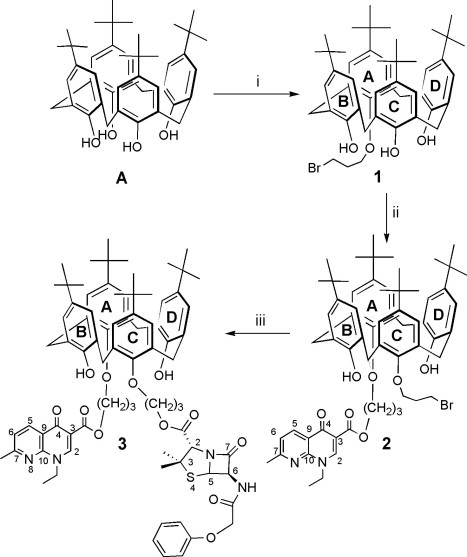
Synthetic pathway leading to podand 3. (i) Br(CH2)3Br, MeCN, 0.6 K2CO3, 7 h, 37%; (ii) bromopropylnalidixate, MeCN, K2CO3, 48 h, 20%; (iii) penicillin V, potassium salt (PVK), DMF, 35 °C, 48 h, 73%. Numbers and letters refer to NMR analyses.
The synthesis of 1 was performed in the conditions favouring the monosubstitution35 by reaction of 4 equiv of 1,3-dibromopropane on p-tert-butylcalix[4]arene A, in the presence of 0.6 equiv of K2CO3 in dry MeCN. 1 was thus obtained with a yield of 37% after purification by chromatography. The reaction of 1 with bromopropylnalidixate, in the presence of 0.6 equiv of K2CO3 in dry MeCN afforded the podand 2. Incorporation of the penicillin V onto the residual brominated arm was performed in dry DMF at 35 °C, under inert atmosphere, by reacting 2 with an excess of potassium salt of penicillin V (PVK). The podand 3 that was formed was thus isolated pure after chromatography with a yield of 73%. The 1,3-bis-propylnalidixic ester 5, prepared as described,32 was chosen as starting material for the synthesis of the 1,3-bis-propyl alcohol 4 (Scheme 3 ). This choice was done after observing some degradation into methylnalidixate over silica gel during chromatography of 5 and other derivatives, when methanol was used as constituent of the mobile phase. This was applied in solution directly onto 5, to give 4 with a yield of 62% after chromatography. The 1,3-bis-propylpenicillinate 6 was prepared in 32% yield by direct reaction of PVK on the 1,3-bis-(3-bromopropyl)-calixarene,36 in dry DMF at 40 °C under Ar.
Scheme 3.
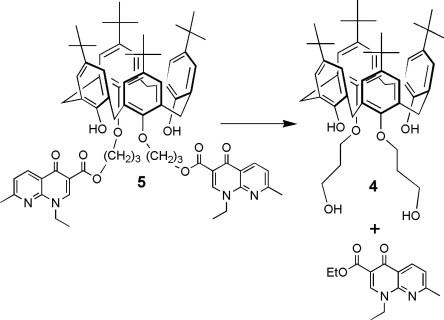
Synthetic pathway leading to podand 4. SiO2, CHCl3, EtOH, rflx, 48 h, 62%.
All compounds were fully characterized, notably by NMR, elemental analysis and ES-mass spectrometry, that gave results consistent with the proposed formulas. The structural complexity of these species necessitated high resolution 2D NMR experiments (COSY, HMQC and HMBC) to assign as far as possible all resonance signals. 1H and 13C NMR showed that the cone conformation was maintained in 1–6, with Ar-CH2Ar resonance signals appearing as AB systems at ca. 3.30–3.50 (equatorial H) and at ca. 4.20–4.40 (axial H) ppm, and at ca. 32.20–33.40 ppm (CH2), respectively. The integrity of the β-lactam structure in the fragile penicillin subunit was verified in compounds 3 and 6, as well as by 1H NMR, with the presence of the expected resonance signals at 5.59 ppm (d, J = 4.3 Hz) for H(5) and 5.71 ppm (dd, J 1 = 9 Hz, J 2 = 4.3 Hz) for H(6), than by IR spectroscopy, through the presence of the pename carbonyl group band at ca 1790 cm−1.
2.2. Biological evaluations
Preliminar biological investigations were attempted in solid and liquid phases, with various Gram positive and Gram negative bacteria. Nevertheless, the lack of solubility in aqueous media of these amphiphilic compounds, useful for studies at the air–water interface,33, 34 was in fact deleterious for bacteriological studies in solution. The amounts of DMSO necessary to co-solubilise podands 3–6 in water was much more higher than the 1–3% admitted in liquid phase antibacterial assays. We verified in this sense that DMSO, depending on concentration in water, could act alone as an antibacterial agent.37 For solid phase, antibiotic disk diffusion assays were performed on Mueller–Hinton agar with sterile 6 mm diameter disks impregnated with different quantities of compound 3, 4, 5 and 6 in volatile organic solvent, and dried prior deposition. No growth inhibition surface around the cellulose disks was observed, that was interpreted either by a lack of activity, or a lack of solubility and diffusibility of title compounds.
For these reasons, this solid phase approach was attempted using disks impregnated with pure DMSO solutions of compounds, taking into account probable antibacterial activities of this solvent. In fact, even if this solvent has been introduced into veterinarian medicine and clinical medicine as an experimental agent in the middle of 20th century,38 we found no clear information concerning the use of pure DMSO as antibacterial agent or as single solvent for drugs to test in liquid or solid phase antibacterial assays.39 In the present work, DMSO was used as pure vehicle to solubilise compounds for impregnation of sterile cellulose disks; this implied the use of disks impregnated with DMSO alone in order to check its own activity on reference bacterial strains used in these assays.
All antibacterial assays were conducted according to CLSI recommendations,40 against three Gram positive reference strains, Staphylococcus aureus ATCC 25923, S. aureus ATCC 29213, and Enterococcus faecalis ATCC 29212, as well as against two Gram negative reference strains E. coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853.
DMSO solutions of nalidixic acid NA and potassium salt of penicillin V (PVK) were preliminary evaluated, pure or as binary mixture, at concentrations equivalent to the maximum dose releasable by calixarene derivatives 3 of the present study.
As shown in Figure 1 , DMSO exhibited an activity only against Pseudomonas aeruginosa ATCC 27853 (image V). The growth inhibition diameters being equivalent for the four disks, one can suppose that only DMSO and not NA and/or PVK displays an activity against this bacterial strain. Against E. coli ATCC 25922, NA exhibits a moderate activity, and PVK a little one; E. faecalis is not sensitive to both antibacterial agents, as expected. The S. aureus ATCC 29213 strain displays a sensitivity to both antibacterial agents, but, with respect to inhibition diameters for penicillin, much lesser than the ATCC strain 25923, that exhibits a very strong sensitivity towards PVK.
Figure 1.
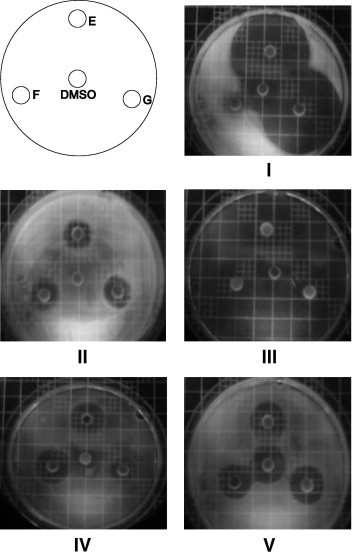
Photographs of Petri dishes. Disk/compound: E = NA (1.88 mg/mL) + PVK (2.83 mg/mL)–F = NA (1.88 mg/mL)–G = PVK (2.83 mg/mL). Images: IS. aureus ATCC 25923, IIS. aureus ATCC 29213, IIIE. faecalis ATCC 29212, IVE. coli ATCC 25922, VP. aeruginosa ATCC 27853.
The calixarene derivatives of the present study were dissolved in pure DMSO, at concentrations corresponding to a maximum release, if hydrolysis occurs, of 20 to 60 μg of penicillin V, or ca. 40 μg of nalidixic acid, from 20 μL of solution applied to 6 mm sterile cellulose disks. The values are given in Table 1 .
Table 1.
Data for sample preparation in the first set of experiments
| Product | 3 | 5 | 4 | 6 |
|---|---|---|---|---|
| (PV/AN) | (AN/AN) | (OH/OH) | (PV/PV) | |
| MW (g mol−1) | 1368.4 | 1193.5 | 765.07 | 1429.84 |
| Weighted mass (mg) | 4.24 | 2.14 | 1.31 | 1.57 |
| DMSO (mL) | 0.424 | 0.428 | 0.403 | 0.620 |
| [ ] (mg/mL) | 10.00 | 5.00 | 3.25 | 2.53 |
| μg NA/20 μL | 37.6 | 41.6 | 0 | 0 |
| μg PV/20 μL | 56.7 | 0 | 0 | 25.5 |
The results given in Figure 2 confirmed first that DMSO (central disk) does not exhibit an antibacterial behavior, except for P. aeruginosa ATCC 27853 (image V), for which all DMSO solutions of compounds display a similar growth inhibition diameter. No inhibition zones were found for S. aureus ATCC 29213 (image II), E. faecalis ATCC 29212 (image III), and E. coli ATCC 25922 (image IV), for all substances. The DMSO solutions of the di-alcohol 4, the bis nalidixate derivative 5 and the bis-penicillinate 6 do not exhibits any activity against all of the reference strains. An important growth inhibition was found only for the dissymmetrical compound 3 that contains one penicillin and one nalidixate arm, against S. aureus ATCC 25923 (disk A, image I), with an inhibition diameter of 31.5 mm, while a very little one was observed for the same strain with bis-penicillinate 6. This strain does not exhibit a natural resistance to nalidixic acid or to penicillin V.
Figure 2.
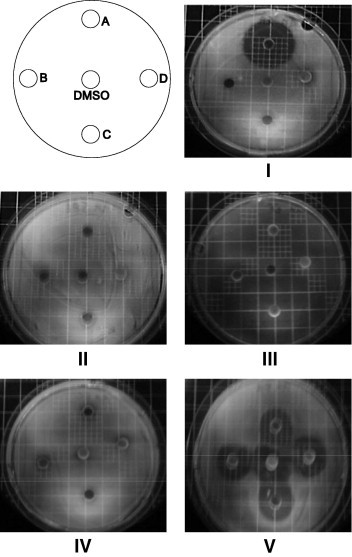
Photographs of Petri dishes. Disk/compounds: A = 3 10.0 × 10−3 g/mL PV/AN–B = 5 5.0 × 10−3 g/mL AN/AN–C = 4 3.25 × 10−3 g/mL OH/OH–D = 6 2.53 × 10−3 g/mL PV/PV. Images: IS. aureus ATCC 25923, IIS. aureus ATCC 29213, IIIE. faecalis ATCC 29212, IVE. coli ATCC 25922, VP. aeruginosa ATCC 27853.
A second series of experiments was conducted with compound 3 (old and fresh solutions), 6 and PVK in order to reach similar released quantities of penicillin V (Table 2 ; Figure 3 )
Table 2.
Data for sample preparation in the second set of experiments
| Product | 3 (PV/AN) old soln. | 5 (AN/AN) fresh soln. | PVK (PV/PV) | 6 (PV/PV) |
|---|---|---|---|---|
| MW (g mol−1) | 1368.4 | 1368.4 | 388.5 | 1429.84 |
| Weighted mass (mg) | 4.24 | 4.44 | 1.00 | 3.10 |
| DMSO (mL) | 0.424 | 0.444 | 0.800 | 0.534 |
| [ ] (mg/mL) | 10.00 | 10.00 | 1.25 | 5.80 |
| μg NA/20 μL | 37.6 | 37.6 | 0.0 | 0.0 |
| μg PV/20 μL | 56.7 | 56.7 | 25 | 56.7 |
Figure 3.
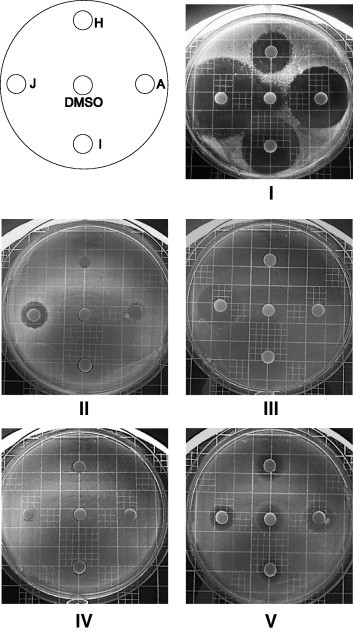
Photographs of Petri dishes. Disk/compounds: A = 3 10.0 10−3 g/mL PV/AN––I = 3 10.0 × 10−3 g/mL PV/AN fresh solution–J = PVK 1.25 × 10−3 g/mL–H = 6 5.8 × 10−3 g/mL PV/PV. Images: IS. aureus ATCC 25923, IIS. aureus ATCC 29213, IIIE. faecalis ATCC 29212, IVE. coli ATCC 25922, VP. aeruginosa ATCC 27853.
The results given in Figure 3 confirm the previous observations: a quasi lack of activity against E. faecalis ATCC 29212 and E. coli ATCC 25922 for 3 and 6, a modest activity of PVK against S. aureus ATCC 29213 and E. faecalis ATCC 29212, and a general, DMSO-related, activity against P. aeruginosa ATCC 27853. Image I shows that the S. aureus ATCC 25923 strain exhibits a high sensitivity to 3, 6 and PVK, and a low one to DMSO, and that the old (A, 1 month, 4 °C) and the fresh (I) solutions of compound 3 give quite similar inhibition diameter (35 mm/A vs 30 mm/I). Adapting the concentration of 6 to reach the theoretical released quantities of PV from 3 results in a visible growth inhibition zone of 22 mm (image IH), corresponding to the double of the one observed in Figure 2, ID, as expected if accepting a linear relation between concentration and inhibition diameter.
Thus, penicillin V-containing calixarene derivatives of this study (compounds 3 and 6) display an antibacterial activity against S. aureus ATCC 25923, more pronounced for the dissymmetric derivative 3.
The results obtained with compound 3 against S. aureus ATCC 25923 could be explained by a specific hydrolysis of the penicillin ester linkage that, surprisingly, does not occur at a similar level with the bis penicillin ester 6. As no growth inhibition zone appears with 3 and its analogue 6 against S. aureus ATCC 29213 strain, while PVK is moderately active, lead us to consider an enzymatic hydrolytic process specific to S. aureus ATCC 25923 strain. In addition, it is well defined that S. aureus ATCC 29213 is a weak betalactamase-producer strain,41 that can also explain the discrepancy observed between these two strains against PVK.
3. Conclusion
In conclusion, we have found that pure DMSO does not exhibit an antibacterial effect against the three Gram positive reference strains S. aureus ATCC 25923, S. aureus ATCC 29213, and E. faecalis ATCC 29212, as well as against the Gram negative reference strain E. coli ATCC 25922; a contrario, an activity is observed against the Gram negative reference strain P. aeruginosa ATCC 27853. It can be used pure as solvent for known drugs and for investigation of antibacterial properties of new substances, but, nevertheless, care must be taken concerning possible synergistic effects.
We have found that only the calixarene derivative 3, integrating one penicillin V and one nalidixic acid subunit, both tethered via a labile ester linkage to the calixarene core, expressed an interesting activity against S. aureus ATCC 25923 strain. This calls for further deeper investigations at the frontier of biology and physico-chemistry, involving drug titration, modelisation of hydrolysis at the air–water interface and in membrane models, in order to understand why 3 and not 6 exhibit an activity, evaluation of the role of the calixarene moiety vs possible synergistic effects, as well as evaluation of antibacterial properties of other lipophilic prodrug-like designed calixarene derivatives.
4. Experimental
4.1. Chemistry
4.1.1. General
Melting points (°C, uncorrected) were determined on an Electrothermal 9200 Capillary apparatus. 1H and 13C NMR spectra were recorded on a Bruker DRX 400 (chemical shifts in ppm). Mass spectra (electronic ionisation–EI, and electrospray–ES) were recorded on a Nermag R-1010C apparatus or a Micromass Platform II apparatus, respectively, at the Service Commun de Spectrométrie de Masse Organique, Nancy. Infrared was performed on a Bruker Vector 22 apparatus (KBr, ν in cm−1) and UV spectra were recorded on a SAFAS UV mc2 apparatus, λ max in nm, ε in mol−1 dm3 cm−1. Elemental analyses were performed at the Service de Microanalyse, Nancy. Merck TLC plates were used for chromatography analysis (SiO2, ref 1.05554; Al2O3, ref 1.05581). All commercially available products were used without further purification unless otherwise specified.
4.1.2. 5,11,17,23-Tetra-(tert-butyl)-25-(3-bromopropyloxy)-26,27,28-trihydroxycalix[4]arene (1)
A suspension of p-tert-butylcalix[4]arene A (2 g, 3.08 × 10−3 mol) and K2CO3 (0.256 g, 1.85 × 10−3 mol) in dry MeCN (100 mL) was refluxed during 30 min. 1,3-dibromopropane (1.7 mL, 11.62 × 10−3 mol) was then added, and reflux was maintained during 7 h. The solvent was evaporated and the solid residue was dissolved in CH2Cl2. The resulting solution was washed with H2O, dried over Na2SO4, concentrated then cooled to 4 °C. The resulting precipitate was filtered off, and the filtrate was chromatographed (SiO2, CH2Cl2/hexane; 1:1) to give 1 (0.9 g, 37%). M.p.:135 °C. IR (KBr): 2960.79(OH). UV–vis (CHCl3): 249 (279); 281 (858). 1H NMR: 1.24 (s, 9 H, Me 3C A or C); 1.27 (s, 18 H, Me 3C B and D); 1.27 (s, 9 H, Me 3C A or C); 2.69 (quint, J = 5.9 Hz, 2H, OCH2CH 2CH2Br); 3.49 (1/2AB, J AB = 13.5 Hz, 4H eq. of ArCH 2Ar); 4.04 (t, J = 6 Hz, 2H, OCH2CH2CH 2Br); 4.25–4.35 (m, 2H ax. of ArCH 2Ar and OCH 2CH2CH2Br); 4.40 (1/2AB, J AB = 13.5 Hz, 2H eq. of ArCH 2Ar); 7.04, 7.11 (AX, J = 2.3 Hz, 4H, ArH B and D); 7.10 (s, 2H, ArH A or C); 7.139 (s, 2H, ArH A or C); 9.48 (s, 2H, OH); 10.12 (s, 2H, OH). 13C NMR (CDCl3): 30.68 (OCH2CH2 CH2Br); 31.64 (Me 3C A or C); 31.89 (Me 3C A or C); 31.93 (Me 3C B and D); 32.56 (ArCH2Ar); 33.25 (OCH2 CH2CH2Br); 33.42 (ArCH2Ar); 34.34 (Me3 C B and D), 34.45 (Me3 C A or C), 34.68 (Me3 C A or C); 74.82 (OCH2CH2CH2Br); 126.11, 126.14, 126.24, 126.98 (C m of Ar); 127.83, 128.35, 128.79 (C o of Ar B, C and D); 133.80 (C o of Ar A); 143.65 (C p of Ar B and D); 144.20 (C p of Ar C); 147.97 (C p of Ar A); 148.86 (C i of Ar B, C and D), 149.27 (C i of Ar A). Anal. calcd for C47H61BrO4, 0.25CH2Cl2 (791.12): C 71.73, H 7.84; found: C 71.67, H 7.84. ES–MS (positive mode): 786.3, 788.5 [1+NH4]+.
4.1.3. 5,11,17,23-Tetra-(tert-butyl)-25-(3-bromopropyloxy)-27-(3-nalidixopropyloxy)-26,28-dihydroxycalix[4]arene (2)
A mixture of monobromopropylcalix[4]arene 1 (0.3 g, 0.39 × 10−3 mol) and K2CO3 (0.033 g, 0.23 × 10−3 mol) in dry MeCN (60 mL) was refluxed under Ar during 30 min. The bromopropyl nalidixate (0.137 g, 0.39 × 10−3 mol) was then added, and reflux was continued during 48 h. The solvent was evaporated to dryness and the residue was dissolved in CH2Cl2. The solution was washed with H2O, dried over Na2SO4, then chomatographed (SiO2, CH2Cl2/CH3OH;10: 0.4) to give 2 (0.08 g; 20%). White powder. M.p.:124–126 °C. IR (KBr): 1637.86 (CO); 1736.03 (COester). UV–vis (CH3CN): 258 (24945); 284 (18952); 330 (12814); 337(13110). 1H NMR (CDCl3): 1.00 (s, 9 H, Me 3C A or C); 1.01 (s, 9 H, Me 3C A or C); 1.27 (s, 18 H, Me 3C B and D); 1.41 (t, J = 7.25 Hz, 3H, CH 3CH2N); 2.49 (quint, J = 5.97 Hz, 2H, OCH2CH 2CH2OCO); 2.61 (quint, J = 5.85 Hz, 4H, OCH2CH 2CH2Br); 2.66 (s, 6 H, CH 3); 3.29, 4.31 (AB, J AB = 12.97 Hz, 4H, ArCH 2Ar); 3.35, 4.32 (AB, J AB = 12.97 Hz, 4H, ArCH 2Ar); 4.10 (t, J = 6 Hz, 2H, OCH2CH2CH 2Br); 4.15 (t, J = 5.34 Hz, 2H, OCH 2CH2CH2Br); 4.22 (t, J = J = 5.72 Hz, 2H, OCH 2CH2CH2OCO); 4.29 (m, 2H, CH3CH 2N); 4.83 (t, J = 6.3 Hz, 2H, OCH2CH2CH 2OCO); 6.84 (s, 2H, ArH A); 6.86 (s, 2H, ArH C); 6.98, 7.05 (AX, JAX = 2.3 Hz, 4H, ArH B and D); 7.25 (d, J = 8.2 Hz, 1H, H(6)), 7.68 (s, 2H, OH); 8.59 (s, 2H, H(2)) ; 8.66 (d, J = 8.01 Hz, H(5)). 13C NMR (CDCl3): 15.57 (CH3CH2N); 25.39 (CH3); 29.61 (OCH2 CH2CH2OCO); 31.44 (OCH2CH2 CH2Br; Me 3C A and C); 32.09 (Me 3C B and D); 32.20 (ArCH2Ar); 33.76 (OCH2 CH2CH2Br); 34.19 (Me3 C B and D), 34.38, 34.40 (Me3 C A and C); 46.87 (CH3 CH2N); 61.72 (OCH2CH2 CH2OCO); 72.66 (OCH2CH2CH2OCO); 73.70 (OCH2CH2CH2Br); 112.47 (C(3)); 121.39 (C(6)); 121.82 (C(9)); 125.47, 125.50 (C m of Ar B and D); 125.96, 126.05 (C m of Ar A and C); 127.95, 128.00 (C o of Ar B and D); 133.15, 133.18 (C o of Ar A and C); 137.16 (C(5)); 141.87 (C p of Ar B and D); 147.37,147.55 (C p of Ar A and C); 148.97, 149.01 (C(2) and C(7)); 149.68 (C i of Ar C); 149.87 (C i of Ar A); 151.08 (C i of Ar B and D); 162.90 (C(10)); 165.79 (COO); 175.11 (C(4)). Anal. calcd for C62H77BrN2O7, 0.2 CH2Cl2 (1059.17): C 70.53, H 7.37, N 2.64; found: C 70.72, H 7.40, N 2.44. ES–MS (positive mode): 1065.5 [2 + Na]+.
4.1.4. 5,11,17,23-Tetra-(tert-butyl)-25-[3-(phenoxyacetyl)penicillopropyloxy]-27-(3-nalidixopropyloxy)-26,28-dihydroxycalix[4]arene (3)
A solution of calixarene 2 (0.2 g, 0.19 × 10−3 mol) and PVK (0.118 g, 0.3 × 10−3 mol) in dry DMF (20 mL) was stirred at 35 °C under Ar during 48 h. The solvent was evaporated under high vacuum at 25 °C and the raw material was dissolved in CH2Cl2. The resulting solution was washed with H2O, dried over Na2SO4, then chromatographed (SiO2, CH2Cl2/CH3OH;10:0.4) to give 3 (0.193 g; 73%). White powder. Mp: 117–118 °C. IR (KBr): 1638.16 (CO quin); 1697.17 (CO amide); 1737.80 (CO ester of quin and PV); 1790.08 (CO lactam). UV–vis (CHCl3): 258 (19083); 290 (10432); 332 (11717). 1H NMR (CDCl3): 1.01 (s, 9 H, Me 3C A or C); 1.02 (s, 9 H, Me 3C A or C); 1.27 (s, 18 H, Me 3C B and D); 1.42 (t, J = 7 Hz, 3H, CH 3CH2N); 1.53 (s, 3H, CH3 of PV); 1.60 (s, 3H, CH3 of PV); 2.42–2.55 (m, 4H, OCH2CH 2CH2OOC-PV and -quin); 2.64 (s, 3H, CH 3); 3.29 (1/2AB, J AB = 13 Hz, 1H eq. of ArCH 2Ar); 3.29 (1/2AB, J AB = 13 Hz, 1H eq. of ArCH 2Ar); 3.34 (1/2AB, J AB = 13 Hz, 1H eq. of ArCH 2Ar); 3.35 (1/2AB, J AB = 13 Hz, 1H eq. of ArCH 2Ar); 4.11 (t, J = 5.8 Hz, 2H, OCH 2CH2CH2OOC-PV); 4.18–4.40 (m, 4H ax. of ArCH 2Ar, CH3CH 2N and OCH 2CH2CH2OOC-quin); 4.53 (s, 1H, H(2) PV); 4.55, 4.60 (AB, J AB = 15 Hz, 2H, C6H5OCH 2); 4.81 (ABm, 2H, OCH2CH2CH 2OOC-PV); 4.84 (t, J = 6 Hz, 2H, OCH2CH2CH 2OOC-quin); 5.59 (d, J = 4.3 Hz,1H, H(5) PV); 5.71 (dd, J1 = 9 Hz, J2 = 4.3 Hz, 1H, H(6) PV); 6.85 (s, 2H, ArH A or C); 6.88 (s, 2H, ArH A or C); 6.95 (d, J = 7.8 Hz, 1H, Cp of C6H5); 6.99 (br s, 2H, ArH B or D); 7.02–7.09 (m, 2H of C6H5 and 2H of ArH B or D); 7.23 (d, J = 8 Hz, 1H, H(6) quin); 7.30–7.40 (m, 2H of C6H5 and C(O)NH); 7.72 (s, 1H, OH B or D); 7.74 (s, 1H, OH B or D); 8.59 (s, 1H, H(2) quin); 8.65 (d, J = 8 Hz, 1H, H(5) quin). 13C NMR (CDCl3): 15.58 (CH3CH2N); 25.43 (CH3-quin); 27.16 (CH3-PV); 29.63 (OCH2 CH2CH2OOC-quin); 29.77 (OCH2 CH2CH2OOC-PV); 31.44 (Me 3C A or C); 32.07 (Me 3C B or D); 32.17 (ArCH2Ar); 32.54 (CH3-PV); 34.20 (Me3 C B or D), 34.41 (Me3 C A or C); 46.87 (CH3 CH2N); 58.51 (C(6) PV); 61.75 (OCH2CH2 CH2OOC-quin); 63.22 (OCH2CH2 CH2OOC-PV); 65.15 (C(3) PV); 67.55 (CH2OC6H5); 68.22 (C(5) PV); 70.91 (C(2) PV); 72.63 (OCH2CH2CH2OOC-PV); 72.81 (OCH2CH2CH2OOC-quin); 112.50 (C(3) quin); 115.16 (C p of C6H5); 121.38 (C(6) quin); 121.87 (C(9) quin); 122.77, 130.24 (C o and Cm of C6H5); 125.48, 125.51 (C m of Ar B and D); 125.98, 126.10 (C m of Ar A and C); 127.80, 128.01 (C o of Ar B and D); 133.08, 133.19, 133.21 (C o of Ar A and C); 137.16 (C(5) quin); 141.97 (C p of Ar B and D); 147.48, 147.69 (C p of Ar A and C); 148.99 (C(7) quin); 149.09 (C(2) quin); 149.77, 149.84 (C i of Ar A and C); 150.98 (C i of Ar B and D); 157.34 (C i of C6H5); 162.88 (C(10) quin); 165.98 (OCH2CH2CH2OOC-quin); 167.91 (OCH2CH2CH2OOC-PV); 168.15 (CONH); 173.36 (COlactam); 175.08 (C(4) quin). Anal. calcd for C78H94O12N4S, 0.8 DMF (1368.4): C 70.50, H 7.27, N 4.91; found: C 70.25, H 7.27, N 4.79. ES–MS (positive mode): 1333.65 (100), 1334.65 (80), 1335.66 (30), 1336.66 (10) [3+H]+.
4.1.5. 5,11,17,23-Tetra-(tert-butyl)-25,27-bis(3-hydroxypropyloxy)-26,28-dihydroxycalix[4]arene (4)
A suspension of silica gel 40–63 μm (0.8 g) in a solution of 5,11,17,23-Tetra-(tert-butyl)-25,27-bis(3-nalidixopropyloxy)-26,28-dihydroxycalix[4]arene 5 32 (0.24 g ; 0.2 × 10−3 mol) in a mixture of dry CHCl3 (19 mL) and EtOH (1 mL) was refluxed during 48 h (TLC monitoring). The silica gel was filtered off, rinsed with 25 mL of a 9:1 mixture of CH2Cl2 and EtOH, and the combined filtrates were evaporated The semi-solid residue was dissolved in CH2Cl2 and evaporated again until a solid material was obtained. The latter was triturated in dry Et2O (5 mL) then filtered. The filtrate was evaporated to dryness, and the residue (0.2 g) was chromatographed (SiO2; CH2Cl2:EtOH 99/1 then 97/3) to separate ethyl nalidixate and the desired diol as a semi-glassy material. The latter was triturated in a 66/33 mixture of pentane and Et2O to give the diol 4 (0.095 g, 62%). White powder. Mp: >250 °C. IR (KBr): 3406 (OH); 2960 (tBu); 1485 (C C aromatic). UV–vis: (CH2Cl2): 284 (7118). 1H NMR (CDCl3): 0.99 (s, 18H, Me 3C A); 1.28 (s, 18H, Me 3C B); 2.20 (q, J = 6 Hz, 4H, OCH2CH 2CH2OH); 3.36, 4.20 (AB, J AB = 13 Hz, 8H, ArCH 2Ar); 4.11 (t, J = 6 Hz, 4H, OCH 2CH2CH2OH); 4.14 (t, J = 6 Hz, 4H, OCH2CH2CH 2OH); 4.37 (t, J = 7 Hz, 2H, OCH2CH2CH2OH); 6.85 (s, 4H, H m A); 7.06 (s, 4H, H m B); 7.74 (s, 2H, OH B). 13C NMR (CDCl3): 30.98 (Me 3C A); 31.66 (Me 3C B); 31.87 (ArCH2Ar); 33.11 (OCH2 CH2CH2OH); 33.81 (Me3 C B); 33.97 (Me3 C A); 61.26 (OCH2CH2 CH2OH); 75.53 (OCH2CH2CH2OH); 125.14 (C m B); 125.66 (C m A); 127.32 (C o B); 132.46 (C o A); 141.84 (C p B); 147.20 (C p A); 149.45 (C i A); 150.33 (C i B). Anal. calcd for C50H68O6 (765.07): C 78.49, H 8.96; found: C 78.66, H 8.91. ES–MS: (pos. mode): 787.5 [M+Na]+; 765.5 [M+H]+.
4.1.6. 5,11,17,23-Tetra-(tert-butyl)-25,27-bis[3-(phenoxyacetyl)penicillopropyloxy]-26,28-dihydroxycalix[4]arene (6)
A solution of 5,11,17,23-tetra-(tert-butyl)-25,27-bis(3-bromopropyloxy)-26,28-dihydroxycalix[4]arene36 (0.2 g, 2.25 × 10−4 mol) and PVK (0.191 g, 4.92 × 10−4 mol) in dry DMF (18 mL) was stirred under argon at 40 °C. The reaction was monitored by chromatography (SiO2; CH2Cl2/Et2O 5:1). After 42 h, the solvent was evaporated to dryness at room temperature, then the residue was dissolved in CH2Cl2 and washed with H2O. The organic phase was dried over Na2SO4 and the solution was concentrated, then chromatographed (SiO2; CH2Cl2/Et2O 5:1) to give 10 (0.1 g; 32%). White powder. Mp: 108 °C. IR (KBr): 1709.31 cm−1 (CO amide); 1745.35 cm−1 (CO lactam); 1788.19 cm−1 (CO ester). UV–vis (CHCl3): 279 (7385). 1H NMR (CDCl3): 1.02 (s, 18 H, Me 3C A); 1.30 (s, 18 H, Me 3C B); 1.53 (s, 6 H, CH 3); 1.60 (s, 6 H, CH 3); 2.394 (quint, J = 6 Hz, 4H, OCH2CH 2CH2OCO); 3.353, 4.223 (AB, J AB = 12.8 Hz, 4H, ArCH 2Ar); 3.360, 4.234 (AB, J AB = 12.8 Hz, 4H, ArCH 2Ar); 4.103 (t, J = 6 Hz, 4H, OCH 2CH2CH2OCO); 4.538 (s, 2H, H(2)); 4.554, 4.609 (strong AB, J AB = 15 Hz, 4H, CH 2OC6H5); 4.751 (ABm, 4H, OCH2CH2CH 2OCO); 5.599 (d, J = J = 4.2 Hz, 2H, H(5)); 5.716 (dd, J 1 = 4.2 Hz, J 2 = 9 Hz, 2H, H(6)); 6.877 (s, 4H, ArH A); 6.954 (d, J = 7.8 Hz, 4H, Ho of C6H5); 7.054 (t, J = 7.1 Hz, 2H, Hp of C6H5); 7.078 (s, 4H, ArH B); 7.346 (t, J = 7.6 Hz, 4H, Hm of C6H5); 7.367 (d, J = 9.2 Hz, 2H, NH); 7.562 (s, 2H, OH). 13C NMR (CDCl3): 27.13 (CH3); 29.78 (OCH2 CH2CH2OCO); 31.42 (Me 3C A); 32.08 (Me 3C B); 32.14 (ArCH2Ar); 32.51 (CH3); 34.23 (Me3 C B); 34.40 (Me3 C A); 58.59 (C(6)); 62.98 (OCH2CH2 CH2OCO); 65.07 (C(3)); 67.60 (CH2OC6H5); 68.22 (C(5)); 70.89 (C(2)); 72.46 (OCH2CH2CH2OCO); 115.18 (C o of C6H5); 122.77 (C p of C6H5); 125.26 (C m of Ar B); 126.12 (C m of Ar A); 127.86 (C o of Ar B); 130.24 (C m of C6H5); 132.96 (C o of Ar A); 142.18 (C p of Ar B); 147.73 (C p of Ar A); 149.66 (C i of Ar A); 150.89 (C i of Ar B); 157.36 (C i of C6H5); 167.91 (COO); 168.17 (CONH); 173.38 (COlactam). Anal. calcd for C82H100N4O14S2 (1429.84): C 68.88, H 7.05, N 3.92; found: C 68.78, H: 6.86, N 3.82. ES-MS (pos. mode): 1451.28 [10+Na]+; 737.23 [10+2 Na]2+/2.
4.2. Antibacterial assay
Antibacterial activities were evaluated on Mueller-Hinton agar by using antibiotic disk diffusion assay. Sterile 6 mm diameter disks were impregnated with 20 μL of DMSO solutions of nalidixic acid NA and potassium salt of penicillin V (PVK) that were prepared at concentrations equivalent to the maximum dose releasable by calixarene derivatives 3, or DMSO solutions of calixarenes prepared at concentrations corresponding to a maximum release, if hydrolysis occurs, of 20–60 μg of penicillin V, or ca. 40 μg of nalidixic acid, from cellulose disks. The plates were incubated at 37 °C, with readings at 24 h of incubation.
Acknowledgments
The authors are grateful to the MRES and to the CNRS for financial support, and thank Mrs E. Eppiger, Mrs B. Fernette for NMR experiments, as well as Mrs S. Adach for elemental analyses and Mr F. Dupire for mass measurements.
References and notes
- 1.Gutsche C.D. In: Monographs in Supramolecular Chemistry Series. Stoddart J.F., editor. Royal Society of Chemistry; Cambridge: 1989. Calixarenes. [Google Scholar]
- 2.Vicens J., Böhmer V., editors. Calixarenes A Versatile Class of Macrocyclic Compounds. Kluwer Academic Publishers; Dordrecht: 1991. [Google Scholar]
- 3.Gutsche C.D. Royal Society of Chemistry; Cambridge: 1998. Calixarenes Revisited. [Google Scholar]
- 4.Asfari Z., Böhmer V., Harrowfield J., Vicens J., editors. Calixarenes 2001. Kluwer Dordrecht; The Netherlands: 2001. [Google Scholar]
- 5.Wieser C., Dieleman D.B., Matt D. Coord. Chem. Rev. 1997;165:93. [Google Scholar]
- 6.Duncan R. Nat. Rev. Drug Disc. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 7.Mandeville W.H., Goldberg D.I. Curr. Pharm. Des. 1997;3:15. [Google Scholar]
- 8.Ringsdorf H. J. Polym. Sci. Polym. Symp. 1975;51:35. [Google Scholar]
- 9.Duncan R., Kopecek J. Adv. Polym. Sci. 1984;57:51. [Google Scholar]
- 10.Yokoyama M., Miyauchi M., Yamada N., Okano T., Sakurai Y., Kataoka K., Inoue S. J. Controlled Release. 1990;11:269. [PubMed] [Google Scholar]
- 11.Bae Y., Fukushima S., Harada A., Kataoka K. Angew. Chem. 2003;115:4788. doi: 10.1002/anie.200250653. Angew. Chem. Int. Ed. 2003, 42, 4641. [DOI] [PubMed] [Google Scholar]
- 12.de Fatima A., Fernandes S.A., Sabino A.A. Curr. Drug Discovery Technol. 2009;6:151. doi: 10.2174/157016309788488302. [DOI] [PubMed] [Google Scholar]
- 13.Rodik R.V., Boyko V.I., Kalchenko V.I. Curr. Med. Chem. 2009;16:1630. doi: 10.2174/092986709788186219. [DOI] [PubMed] [Google Scholar]
- 14.Yo, T., Fujiwara; K., Otsuka, M., JP 10203906, 1998.
- 15.(a) Hwang, K. M.; Qi, Y. M.; Liu, S. Y.; Choy, W.; Chen, J. WO Patent 9403164, 1994.; (b) Harris, S. J. WO Patent 9519974, 1995.; (c) Harris, S. J. WO Patent 0244121, 2002.; (d) Coveney, D.; Costello, B. US 2005113454, 2005.; (e) Kral, V.; Cigler, P.; Konvalinka, J.; Kozisek, M.; Prejdova, J.; Gruener, B.; Plesek, J.; Lepsik, M.; Pokorna, J.; Kraeusslich, H.-G.; Bodem, J. WO 2005073240, 2005.; (f) Dings R.P.M., Chen X., Hellebrekers D.M.E.I., van Eijk L.I., Zhang Y., Hoye T.R., Griffioen A.W., Mayo K.H. J. Natl. Cancer Inst. 2006;98:932. doi: 10.1093/jnci/djj247. [DOI] [PubMed] [Google Scholar]; (g) Motornaya A.E., Alimbarova L.M., Shokova E.A., Kovalev V.V. Pharm. Chem. J. 2006;40:68. [Google Scholar]
- 16.Hwang, K. M.; Qi, Y. M.; Liu, S. Y.; Lee, T. C.; Choy, W.; Chen, J. WO Patent 9403165, 1994.
- 17.Hulmes, D.; Coleman, A.; Aubert-Foucher, E. (CNRS, Fr) WO patent 0007585, 2000.
- 18.(a) Dudic M., Colombo A., Sansone F., Casnati A., Donofrio G., Ungaro R. Tetrahedron. 2004;60:11613. [Google Scholar]; (b) Sansone F., Dudic M., Donofrio G., Rivetti C., Baldini L., Casnati A., Cellai S., Ungaro R. J. Am. Chem. Soc. 2006;128:14528. doi: 10.1021/ja0634425. [DOI] [PubMed] [Google Scholar]; (c) Bagnacani V., Sansone F., Donofrio G., Baldini L., Casnati A., Ungaro R. Org. Lett. 2008;10:3953. doi: 10.1021/ol801326d. [DOI] [PubMed] [Google Scholar]
- 19.(a) Cornforth J.W., D’Arcy Hart P., Nicholls G.A., Rees R.J.W., Stock J.A. Br. J. Pharmacology. 1955;10:73. doi: 10.1111/j.1476-5381.1955.tb00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) D’Arcy Hart P., Armstrong A.J., Brodaty E. Infect. Immun. 1996;64:1491. doi: 10.1128/iai.64.4.1491-1493.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Colston M.J., Hailes H.C., Stavropoulos E., Herve A.C., Herve G., Goodworth K.J., Hill A.M., Jenner P., Hart P.D., Tascon R.E. Infect. Immun. 2004;72:6318. doi: 10.1128/IAI.72.11.6318-6323.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Loiseau F.A., Hill A.M., Hii K.K. Tetrahedron. 2007;63:9947. [Google Scholar]
- 20.Casnati A., Fabbi M., Pelizzi N., Pochini A., Sansone F., Ungaro R. Bioorg. Med. Chem. Lett. 1996;6:2699. [Google Scholar]
- 21.Mourer M., Psychogios N., Laumond G., Aubertin A.-M., Regnouf-de-Vains J.–B. Bioorg. Med. Chem. 2010;18:36. doi: 10.1016/j.bmc.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Geller C., Fontanay S., Mourer M., Massimba Dibama H., Regnouf-de-Vains J.-B., Finance C., Duval R.E. Antivir. Res. 2010;88:343. doi: 10.1016/j.antiviral.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leeb M. Nature. 2004;431:892. doi: 10.1038/431892a. [DOI] [PubMed] [Google Scholar]
- 24.Mourer M., Duval R.E., Finance C., Regnouf-de-Vains J.–B. Bioorg. Med. Chem. Lett. 2006;16:2960. doi: 10.1016/j.bmcl.2006.02.072. [DOI] [PubMed] [Google Scholar]
- 25.Grare M., Mourer M., Regnouf-de-Vains J.–B., Finance C., Duval R.E. Pathol. Biol. 2006;54:470. doi: 10.1016/j.patbio.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 26.Grare M., Mourer M., Fontanay S., Regnouf-de-Vains J.–B., Finance C., Duval R.J. J. Antimicrob. Chemother. 2007;60:575. doi: 10.1093/jac/dkm244. [DOI] [PubMed] [Google Scholar]
- 27.Grare M., Dague E., Mourer M., Regnouf de Vains J.-B., Finance C., Duval J.F.L., Duval R.E., Gaboriaud F. Pathol. Biol. 2007;55:465. doi: 10.1016/j.patbio.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Mourer M., Massimba Dibama H., Fontanay S., Grare M., Duval R.E., Finance C., Regnouf-de-Vains J.-B. Bioorg. Med. Chem. 2009;17:5496. doi: 10.1016/j.bmc.2009.06.040. [DOI] [PubMed] [Google Scholar]
- 29.Grare M., Fontanay S., Massimba Dibama H., Mourer M., Regnouf-de-Vains J.-B., Finance C., Duval R.E. Pathol. Biol. 2010;58:46. doi: 10.1016/j.patbio.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 30.Grare M., Massimba Dibama H., Lafosse S., Ribon A., Mourer M., Fontanay S., Finance C., Regnouf-de-Vains J.-B., Duval R.E. Clin. Microbiol. Infec. 2010;16:432. doi: 10.1111/j.1469-0691.2009.02837.x. [DOI] [PubMed] [Google Scholar]
- 31.Ben Salem A., Regnouf-de-Vains J.–B. Tetrahedron Lett. 2001;42:7033. [Google Scholar]
- 32.Ben Salem A., Regnouf-de-Vains J.–B. Tetrahedron Lett. 2003;44:6769. [Google Scholar]
- 33.Korchowiec B., Orlof M., Sautrey G., Ben Salem A., Korchowiec J., Regnouf de Vains J.B., Rogalska E. J. Phys. Chem. B. 2010;114:10427. doi: 10.1021/jp102471c. [DOI] [PubMed] [Google Scholar]
- 34.Korchowiec B., Ben Salem A., Corvis Y., Regnouf de Vains J.B., Korchowiec J., Rogalska E. J. Phys. Chem. B. 2007;111:13231. doi: 10.1021/jp070970+. [DOI] [PubMed] [Google Scholar]
- 35.Groenen L.C., Ruel B.H.M., Casnati A., Verboom W., Pochini A., Ungaro R., Reinhoudt D.N. Tetrahedron. 1991;47:8379. [Google Scholar]
- 36.Li Z.-T., Ji G.-Z., Zhao C.-X., Yuan S.-D., Ding H., Huang C., Du A.-L., Wei M. J. Org. Chem. 1999;64:3572. doi: 10.1021/jo9824100. [DOI] [PubMed] [Google Scholar]
- 37.Fontanay, S.; Duval, R. E., unpublished results.
- 38.Freeman G.R. The Laryngoscope. 1976;86:921. doi: 10.1288/00005537-197607000-00004. Chemical Abstract Number 1976219214. [DOI] [PubMed] [Google Scholar]
- 39.(a) Correa, W. M.; Correa, C. ; Nogueira, M.; Langoni, H.; Carreira, E. L. C. Arquivos da Escola de Veterinaria da Universidade Federal de Minas Gerais 1981, 33(3), 449–453. Chemical Abstract Number 97:123785. (b) Kligman, Albert M. JAMA, the Journal of the American Medical Association (1965), 193(10;11), 796–804;923–8. AN 1965:483474. (c) Szydlowska, T. Zentralbl Bakteriol [Orig A] 239(2):270–4 (1977) AN 1978057374.
- 40.Clinical and Laboratory Standards Institute. CLSI methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 7th edn. Approved Standard M7-A7. Wayne, PA, USA: CLSI, 2006.
- 41.European Committee on Antimicrobial Susceptibility Testing. EUCAST disk diffusion test for routine antimicrobial susceptibility testing. EUCAST recommended strains for internal quality control (http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Disk_test_documents/EUCAST_QC_tables_1.3.pdf)