

Highlights
-
•
Nineteen compounds were tested for antiviral activity against FCV in vitro.
-
•
The antimalarial mefloquine demonstrated a marked inhibitory effect on FCV.
-
•
Combined treatment with mefloquine and rFelIFN-ω showed additive antiviral effects.
-
•
This is the first report of antiviral effects of mefloquine against a calicivirus.
Keywords: Feline calicivirus, Antiviral, Cats, Mefloquine
Abstract
Feline calicivirus (FCV) is an important viral pathogen of domestic cats causing clinical signs ranging from mild to severe oral ulceration or upper respiratory tract disease through to a severe fatal systemic disease. Current therapeutic options are limited, with no direct acting antivirals available for treatment. This study screened a panel of 19 compounds for potential antiviral activity against FCV strain F9 and recent field isolates in vitro. Using a resazurin-based cytopathic effect (CPE) inhibition assay, mefloquine demonstrated a marked inhibitory effect on FCV induced CPE, albeit with a relatively low selectivity index. Orthogonal assays confirmed inhibition of CPE was associated with a significant reduction in viral replication. Mefloquine exhibited a strong inhibitory effect against a panel of seven recent FCV isolates from Australia, with calculated IC50 values for the field isolates approximately 50% lower than against the reference strain FCV F9. In vitro combination therapy with recombinant feline interferon-ω, a biological response modifier currently registered for the treatment of FCV, demonstrated additive effects with a concurrent reduction in the IC50 of mefloquine. These results are the first report of antiviral effects of mefloquine against a calicivirus and support further in vitro and in vivo evaluation of this compound as an antiviral therapeutic for FCV.
1. Introduction
In domestic cats, feline calicivirus (FCV) is a common and important pathogen (Radford et al., 2007) typically resulting in upper respiratory tract signs or ulcerative oral lesions. Less frequent disease manifestations include a lameness syndrome, pneumonia, feline chronic gingivostomatitis syndrome (FCGS) (Lyon, 2005) and more recently the recognition of FCV-associated virulent systemic disease (FCV-VSD), a condition associated with high morbidity and mortality, even in vaccinated adult cats (Hurley and Sykes, 2003, Pedersen et al., 2000).
Both inactivated and modified live FCV vaccines are available to help control calicivirus-related disease in cats. They are generally effective at reducing the severity and duration of clinical signs, but do not prevent infection or shedding (Radford et al., 2007). The high level of antigenic variability amongst FCV isolates has raised concerns regarding the level of cross protection afforded by older vaccines (Radford et al., 2006), and the apparent limited protection afforded against isolates associated with FCV-VSD (Hurley and Sykes, 2003). Given these concerns and the significant impact of the virus in some cats, a safe and effective antiviral therapeutic would significantly advance feline medicine.
There are currently no direct acting antiviral drugs registered for the treatment of FCV, although the immune modulating drug recombinant feline interferon-ω (rFeIFN-ω), which likely has indirect antiviral properties, has a registered indication for FCV treatment. Efficacy of both human and feline interferons has been demonstrated in vitro against FCV (Fulton and Burge, 1985, Mochizuki et al., 1994, Taira et al., 2005, Truyen et al., 2002) and the use of rFeIFN-ω has been reported to have a positive therapeutic effect in FCV infected cats in experimental and field efficacy trials (Ninomiya et al., 1991, Ohe et al., 2008). Treatment with rFeIFN-ω was also associated with an improvement in clinical signs in cats with refractory FCGS in a double-blinded placebo-controlled study, however FCV shedding was not monitored, making it unclear whether the improvement was due to the antiviral or immunomodulatory properties of interferon (Hennet et al., 2011). Recently, feline calicivirus specific antiviral phosphorodiamidate morpholino oligomers (PMO) were tested in naturally occurring outbreaks of FCV-VSD (Smith et al., 2008) and were highly efficacious, with treatment resulting in improved survival, reduction in shedding, and a more rapid clinical recovery.
The current study screened a panel of compounds for antiviral activity against FCV using a resazurin-based CPE inhibition assay. The antiviral effects of compounds identified during screening were confirmed with plaque reduction and virus yield reduction assays. Effective compounds were tested against a panel of recent FCV field isolates from Australia to confirm their effectiveness against more clinically relevant viruses. Effective compounds were also tested in combination with rFeIFN-ω, currently the only licenced treatment for FCV in Australia.
2. Materials and methods
2.1. Cell culture and viruses
Crandell Rees feline kidney (CRFK) cell line was propagated as outlined previously (McDonagh et al., 2014). The reference feline calicivirus strain F9 was kindly provided by Professor Gilkerson (University of Melbourne). Field isolates of FCV were collected from cats in Sydney and Melbourne and grown on CRFK cells. To confirm FCV as the causative agent of CPE from clinical samples, an indirect immunofluorescence assay was performed using an anti-FCV monoclonal antibody (clone S1-9; Custom Monoclonals International, Sacramento, USA). All viruses were titrated by a carboxymethylcellulose plaque assay on CRFK cells and stored in single use aliquots at −80 °C.
2.2. Test compounds
Nineteen compounds were selected based on reported in vitro antiviral effects against other RNA viruses (McDonagh et al., 2014). To determine an appropriate screening concentration the cytotoxicity of compounds was investigated using sequential resazurin and sulforhodamine B assays as previously reported (McDonagh et al., 2014). Test compound concentrations selected for subsequent antiviral screening were those resulting in cell viability of 80% or greater and are shown in Table 1 . Stock solutions were prepared as outlined previously (McDonagh et al., 2014). The maximum final in-well DMSO concentration used in these studies was 0.33%, which was demonstrated to have no significant effect on cell viability or viral replication (data not shown).
Table 1.
Compounds selected for antiviral screening.
| Compound | Screening concentration |
|---|---|
| Chloroquine diphosphatea | 25 μM |
| Quercetina | 10 μM |
| Curcuminb | 10 μM |
| Rutin trihydrateb | 25 μM |
| Indomethacinb | 10 μM |
| Glycyrrhizic acida | 25 μM |
| Hesperidinb | 50 μM |
| Aurintricarboxylic acida | 2.5 μM |
| Hesperetinb | 50 μM |
| Mefloquine hydrochloridea | 10 μM |
| Artesunatea | 1 μM |
| Ribavirina | 2.5 μM |
| Baicalin hydrateb | 10 μM |
| Hexamethylene amilorideb | 10 μM |
| Cinanserinb | 20 μM |
| Artemisinina | 25 μM |
| Niclosamideb | 0.25 μM |
| Lactoferrina | 0.5 mg ml−1 |
| Recombinant feline interferon ωc | 100 units ml−1 |
Superscripts indicate compound supplier.
Sigma–Aldrich.
Santa Cruz Biotechnology.
Virbac.
2.3. Antiviral screening using CPE inhibition assay
Antiviral screening was performed using a modification of the optimised resazurin-based CPE inhibition assay outlined previously (McDonagh et al., 2014). After 1 h of compound exposure cells were infected with FCV strain F9 at MOI 0.01 (20 μl well−1) for an infection period of 48 h with 50 μl of 1:10 dilution of 4 × stock resazurin in DMEM (final well concentration of resazurin 44 nM) added for the final 3.5 h. The duration of compound exposure in this assay was from 1 h prior to infection through to the assay endpoint, thereby allowing the identification of agents acting at any stage of the viral lifecycle. Plates were removed from the incubator for the final 30 min to allow the plates and media to equilibrate to room temperature. Fluorescent signals were measured with a FLUOstar Omega microplate reader (BMG Labtech, Mornington, VIC, Australia) using a 544 nm excitation filter and 590 nm emission filter with 8 flashes per well in bottom reading mode. Each treatment was performed in triplicate and results represent Mean ± SE of three independent experiments. The percentage inhibition of CPE was calculated by:
where RFUTx is the mean fluorescence intensity of treated infected cells; RFUV(+) is the mean fluorescence intensity in untreated infected cells; and RFUV(−) is the mean fluorescence intensity of untreated uninfected cells. Compounds showing marked, moderate, or mild antiviral effects were defined as those showing 75–100%, 50–74%, and 25–49% inhibition of CPE respectively. Compounds demonstrating marked CPE inhibition were classified as candidate compounds and were selected for further characterisation.
2.4. Titration of effective compounds and determination of selectivity index
Using the optimised FCV CPE inhibition assay, a concentration–response experiment was conducted with serial dilutions of effective compounds identified during screening. To enable calculation of the selectivity index a repeat cytotoxicity screen was performed concurrently. The cytotoxicity screen was performed as per the CPE inhibition assay with the exception that cells were mock infected with DMEM. Each treatment was performed in triplicate and repeated in three independent experiments, with results presented as Mean ± SE. Data analysis were conducted in GraphPad Prism (V5.03 for Windows, GraphPad Software, San Diego, CA, USA), with the 50% inhibitory concentration (IC50) and 50% cytotoxic concentration (CC50) values calculated using the inbuilt non-linear curve fitting functions following log10 transformation of compound concentrations. The selectivity index (SI) for each compound was calculated according to the following formula:
2.5. Confirmatory assays
Plaque reduction and virus yield reduction assays were performed to confirm antiviral effects of candidate compounds identified using a modification of the methods previously described (McDonagh et al., 2014). For the virus yield reduction assay five concentrations of test compound were assessed. Cells were exposed to compounds for 1 h prior to infection with FCV F9 at MOI of 0.05 in 25 μl DMEM. At 12 and 24 h post infection (hpi) cellular morphology was assessed for CPE using an inverted phase contrast microscope (CKX41, Olympus) and supernatant was harvested and stored at −80 °C prior to titration of extracellular virus using the TCID50 assay. The relative viral titre was calculated for each treatment with the value of the untreated control defined as 100%. Each treatment and time point was performed in triplicate and repeated in two independent experiments. Results represent Mean ± SE. Plaque reduction assays were performed using a modification of the plaque assay previously described (McDonagh et al., 2014). Approximately 60 pfu FCV F9 was added per well. The relative plaque number was calculated for each treatment with the value of untreated control defined as 100%. Each treatment was performed in duplicate, and the results represent Mean ± SE of three independent experiments.
2.6. Antiviral efficacy against field isolates of FCV
Using the resazurin-based-CPE inhibition assay, the antiviral efficacy of identified candidate compounds against seven currently circulating field isolates was investigated. Details of the field isolates are shown in Table 2 . Assay conditions were identical to those used for screening except that cells were infected with different field isolates. Untreated cells were also infected with the different isolates as virus controls and uninfected and untreated cells included as positive controls. Each treatment was performed in triplicate wells for each virus, and repeated in three independent experiments. Data expressed as Mean ± SE. To further investigate the antiviral efficacy against different isolates a resazurin-based CPE inhibition concentration–response study was conducted using field isolates 178N and IW1E. Each treatment was performed in triplicate and results represent Mean ± SE of three independent experiments. Calculation of IC50, CC50, and SI values was performed as previously described.
Table 2.
Details of FCV isolates used in experimental studies.
| Strain | Disease manifestation/site of isolation | Location of isolation | Passage |
|---|---|---|---|
| F9 | Vaccine strain | USA | P2a |
| 83E | Conjunctival swab from cat with oro-respiratory disease | Melbourne, Australia | P3 |
| 131M | Oropharyngeal swab from cat with oro-respiratory disease | Melbourne, Australia | P3 |
| 178N | Nasal swab from cat with oro-respiratory disease | Melbourne, Australia | P3 |
| IW1E | Pharyngeal swab from cat with FCGS | Sydney, Australia | P2 |
| IW10 | Pharyngeal swab from cat with oro-respiratory disease | Sydney, Australia | P2 |
| IW16 | Swab from tongue ulcer noted as incidental finding during pre-general anaesthetic examination | Sydney, Australia | P2 |
| IW25 | Oropharyngeal swab from a cat with stomatitis | Sydney, Australia | P2 |
Passage 2 within our laboratory, as original passage number unknown.
2.7. Combination treatment with mefloquine and rFeIFN-ω
Using the optimised resazurin-based CPE inhibition assay, cells were pre-treated with varying combinations of rFeIFN-ω and mefloquine at concentrations from 0 to 1000 units ml−1 and 0 to 12 μM respectively prior to infection with FCV F9 at MOI 0.01. Each treatment was performed in triplicate and repeated in three independent experiments with data presented as Mean ± SE. IC50 values were calculated as previously described.
3. Results
3.1. Identification of mefloquine as an inhibitor of FCV replication in vitro
Mefloquine, at a concentration of 10 μM, was the only compound of the 19 tested that demonstrated marked inhibition (88.6%) of virus induced CPE (Fig. 1 ). All other compounds demonstrated a limited, or no inhibitory effect on CPE. Included among these are four compounds – ribavirin, lactoferrin, chloroquine, and rFeIFN-ω – that had previously demonstrated in vitro efficacy against FCV (Kreutz and Seal, 1995, McCann et al., 2003, Povey, 1978, Truyen et al., 2002). Titration of mefloquine demonstrated a clear concentration–response relationship with almost complete inhibition of CPE at the highest tested concentrations reducing to zero inhibition at lowest tested concentration. Calculated IC50, CC50, and SI values are shown in Fig. 2 .
Fig. 1.
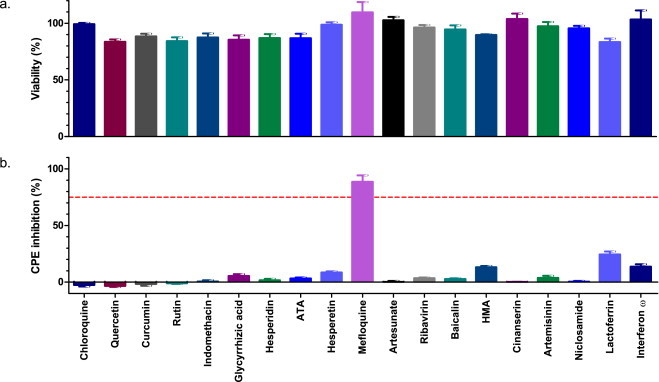
Results of FCV antiviral screening experiment. Cells were pre-treated with various compounds for 1 h prior to infection with FCV F9 at MOI 0.01. Antiviral efficacy was determined 48 hpi using the resazurin-based CPE inhibition assay (b). A concurrent cytotoxicity screen was performed using the same protocol with the exception that cells were mock infected (a). Each treatment was performed in triplicate and repeated in three independent experiments. Results represent Mean ± SE. ATA, aurintricarboxylic acid; HMA, hexamethylene amiloride. Red dotted line = 75% inhibition of CPE.
Fig. 2.
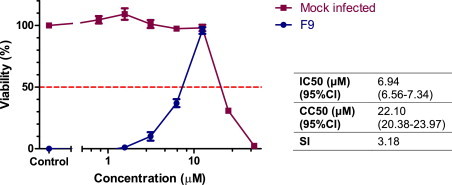
Titration of mefloquine against FCV using the resazurin-based CPE inhibition assay. Each treatment was performed in triplicate and repeated in three independent experiments. Results represent Mean ± SE. Calculated IC50, CC50, and SI values are shown in the accompanying table.
Orthogonal testing confirmed CPE inhibition was associated with marked reductions in the production of progeny virions. Using a virus yield reduction assay mefloquine demonstrated a concentration dependent inhibition of viral replication when assayed at 12 and 24 hpi (Fig. 3 ). At high concentrations, reductions in viral titre at 24 hpi was greater than 3 log10, while at 12 hpi this was reduced to between 1 and 2 log10. Calculated IC50 and SI values at 12 and 24 hpi are shown in Fig. 3. Plaque reduction assays confirmed the findings of the resazurin-based CPE inhibition assay. Pre-treatment with mefloquine resulted in a concentration-dependent reduction in plaque number, with a calculated IC50 of 6.03 μM (95% confidence interval 5.43–6.70) and resulting selectivity index of 3.7. Plaque size and morphology varied considerably in both treated and untreated cells, however in general plaque size was smaller and plaque morphology more consistent in wells treated with higher concentrations of mefloquine.
Fig. 3.
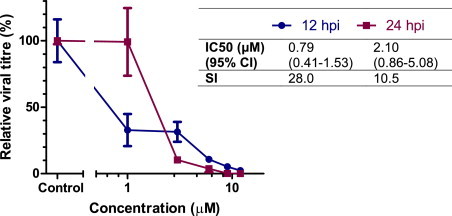
Virus yield reduction assay for mefloquine against FCV. Cells were pre-treated with various dilutions of mefloquine for 1 h prior to infection with FCV F9 at MOI 0.05. Extracellular virus titre was calculated at 12 (blue circles) and 24 hpi (red squares) with a TCID50 end point assay. Titre of untreated control is defined as 100%. Each treatment was performed in triplicate and repeated in two independent experiments. Data represent Mean ± SE. Calculated IC50 and SI values for each time point are shown in the accompanying table.
3.2. Antiviral efficacy against field isolates
Mefloquine demonstrated similar inhibition of virus induced CPE against all tested field isolates to that of the reference strain FCV F9 (Fig. 4 ). To assess any quantitative difference in the efficacy of mefloquine against different field isolates, a concentration–response experiment was conducted using field isolates IW1E and 178N. Mefloquine demonstrated concentration-dependent inhibition of both isolates, with similar shaped concentration–response curves and IC50 values less than half that of FCV F9 (Fig. 5 ).
Fig. 4.
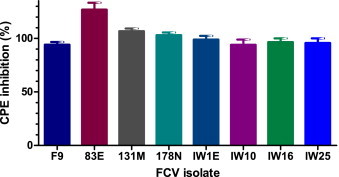
Antiviral efficacy of mefloquine against recent Australian field isolates of FCV. Antiviral efficacy was determined 48 hpi using the resazurin-based CPE inhibition assay. Each treatment was performed in triplicate and repeated in three independent experiments. Results represent Mean ± SE.
Fig. 5.
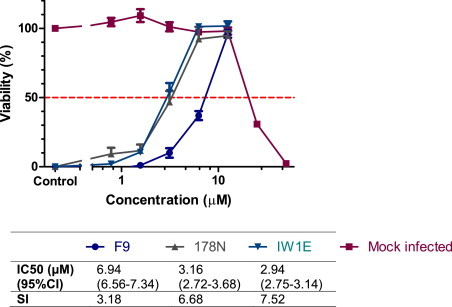
Antiviral titration of mefloquine against Australian field isolates of FCV. Antiviral efficacy was determined 48 hpi using the resazurin-based CPE inhibition assay. Each treatment was performed in triplicate and repeated in three independent experiments. Results represent Mean ± SE.
3.3. Effect of combination treatment with mefloquine and rFeIFN-ω
rFeIFN-ω exerted concentration-dependent additive antiviral effects in vitro when combined with mefloquine (Fig. 6 ). Increasing the rFeIFN-ω concentration from 0 to 500 units ml−1 resulted in a shift of the concentration–response curve to the left with a corresponding decrease in the calculated IC50 value. With this assay the additive effect of rFeIFN-ω on the antiviral efficacy of mefloquine appeared to peak at 500 units ml−1 with greater concentrations not resulting in further reductions in the IC50 value.
Fig. 6.
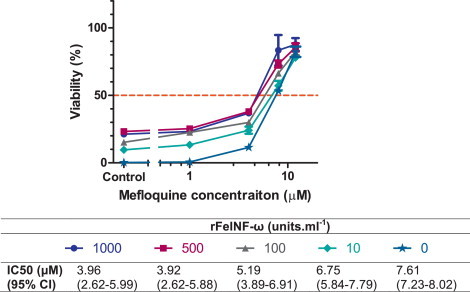
Effect of combination treatment with mefloquine and rFeINF-ω against FCV F9. Cells were pre-treated with varying combinations of rFeINF-ω and mefloquine prior to infection with FCV F9 at MOI 0.01. Antiviral efficacy was determined 48 hpi using the resazurin-based CPE inhibition assay. Each treatment was performed in triplicate and repeated in three independent experiments. Results represent Mean ± SE. IC50 values for mefloquine were calculated for each rFeINF-ω concentration.
4. Discussion
We have demonstrated a marked inhibitory effect of mefloquine on the replication and associated cytopathic effects of FCV in cell culture. Initial screening and confirmatory assays were performed with the culture adapted vaccine strain F9; however subsequent testing against seven current Australian field isolates demonstrated antiviral efficacy remained excellent against potentially more relevant viruses, with concentration–response studies confirming that mefloquine is a more potent inhibitor of field isolates than the reference strain. Combination treatment with rFeIFN-ω resulted in additive effects with a reduction in the IC50 value for mefloquine. These data provide important preliminary information on the potential use of mefloquine or its derivatives as an antiviral therapeutic for the treatment of FCV-associated diseases.
Initial screening and the orthogonal confirmatory assays were performed with FCV F9, a well-characterised strain originally isolated in 1958 (Glenn et al., 1999). FCV F9 was chosen for screening due to its well characterised in vitro growth properties, however it is unclear whether FCV F9 used in the current study is representative of currently circulating viruses, due to natural viral evolution of circulating field viruses over the last half century in face of vaccination with F9 based vaccines, or cell culture adaptation, as can occur with high passage number. It is therefore possible that results obtained against FCV F9 may not be representative of efficacy against circulating strains, particularly given the known genetic heterogeneity of FCV (Coyne et al., 2012). Additional antiviral screening was performed against a panel of seven recent FCV isolates from Australia. Variation in susceptibility to antiviral agents can arise rapidly due to viral evolution in response to selection pressures imposed by the use of an antiviral drug in a population, however it may also pre-exist in treatment naive populations. Ferraris et al. (2005) showed significant variability in the sensitivity of influenza A viruses to the neuraminidase inhibitors zanamivir and oseltamivir, even in viruses isolated prior to the widespread use of these drugs. In contrast, in the current study all tested FCV isolates were susceptible to the inhibitory effects of mefloquine based on the CPE inhibition assay. A concentration–response study demonstrated a greater than two-fold increase in potency for the two field isolates tested compared to the reference virus FCV F9. Although further studies are required to determine whether this is a general feature of field isolates, or whether unique to the two tested isolates, it highlights that consideration must be given to the viral strain used for screening.
Combination antiviral therapy is common practice in significant human viral infections such as HIV and HCV (Feld and Hoofnagle, 2005, Shafer and Vuitton, 1999) and offers several potential theoretical benefits over monotherapy including a more rapid and complete inhibition of replication, a reduction in the likelihood of viral escape, and the potential to reduce the dose of individual drugs, and thereby minimise dose-dependent adverse effects. For HCV, combination therapy of ribavirin and interferon has historically been the standard of care, and demonstrated good clinical success (Feld and Hoofnagle, 2005). As rFeIFN-ω is an approved therapeutic in cats we sought to investigate its use in combination with mefloquine. Interferon monotherapy has previously demonstrated small antiviral effects against FCV in vitro (Mochizuki et al., 1994, Truyen et al., 2002). In vivo efficacy has also been reported in experimental and field studies of FCV (Ninomiya et al. (1991), Uchino et al. (1991) cited in Ohe et al. (2008)). At the concentration used in the current study however, monotherapy with rFeIFN-ω provided limited protection from CPE which may reflect differences in the sensitivity of virus yield reduction assays compared to CPE inhibition assays (McDonagh et al., 2014) as well as differences in the experimental and infection conditions. Combination therapy with rFeIFN-ω and mefloquine however demonstrated additive effects, with increasing concentrations of interferon resulting in a reduction in the IC50 of mefloquine. This combination may prove a useful therapeutic option for FCV infections, particularly if it reduces the dose of mefloquine, and a possible concomitant reduction in the risk of adverse reactions.
The results of this study further expand the known antiviral spectrum of mefloquine with in vitro efficacy previously reported against HIV, JC virus, and feline coronavirus (Brickelmaier et al., 2009, McDonagh et al., 2014, Owen et al., 2005). The mechanism of action of mefloquine against this diverse group of viruses is not known, nor is it clear whether antiviral efficacy is due to a common broad spectrum mechanism of action. Investigations into the likely mechanism of action of mefloquine against FCV were not conducted in the current study. Given the documented effects of mefloquine on endocytosis, and the morphological changes suggestive of alterations in these pathways in CRFK cells (McDonagh et al., 2014) it is tempting to speculate that the antiviral effects against FCV may arise through perturbation of these pathways. Cellular entry of FCV occurs via receptor mediated endocytosis following binding to the cellular receptor fJAM-A (Stuart and Brown, 2006). Treatment with known inhibitors of different steps of viral entry, including inhibitors of endocytosis, revealed FCV entry involves clathrin coated pits and that endosome acidification was required for infection, in agreement with a previous report by Kreutz and Seal (1995) that showed that chloroquine inhibited viral replication. Under the assay conditions of the current study chloroquine provided no protection against virus induced CPE, suggesting that if viral inhibition was arising through the lysosomotropic effects of mefloquine (Glaumann et al., 1992), this effect is considerably more potent for mefloquine than for chloroquine. Alternatively, a mechanism unrelated to its accumulation in lysosomes, such as an action as an adenosine analogue as hypothesised for JC virus (Brickelmaier et al., 2009), may account for the observed antiviral effects.
Although mefloquine is a human approved pharmaceutical with a significant body of literature regarding its pharmacokinetics and safety, its use in cats has not been reported. It is well recognised that due to deficiencies in a number of drug metabolism pathways, the pharmacokinetics of certain drugs in cats can be quite different to those of other species, a fact which should be considered when extrapolating human pharmacokinetic data to this species (Court, 2013). The IC50 value calculated for the field isolates, based on inhibition of CPE, was approximately 3 μM which is lower than plasma concentrations of mefloquine reported in human studies (4–23 μM depending on dosing regimens) (Kollaritsch et al., 2000, Simpson et al., 1999). Extrapolating from available data therefore, it may be possible to achieve in vivo concentrations of mefloquine within the therapeutic range; however the potential for cytotoxicity must be considered given the low SI of the compound in CRFK cells.
5. Conclusion
This study identified mefloquine as a potent inhibitor of FCV in vitro when present at low micromolar concentrations. This is the first report of the antiviral activity of mefloquine against a calicivirus. Testing against a panel of recent Australian FCV isolates demonstrated the antiviral effects of mefloquine against the reference strain FCV F9 extends to more clinically relevant isolates, and there was no evidence of antagonism when used concurrently with rFeIFN-ω. Further investigation and optimisation of this compound for clinical use in treating the more serious manifestations of FCV infection is warranted. Consideration should be given to investigating the effectiveness of mefloquine against other members of the Caliciviridae of medical or veterinary importance.
Acknowledgements
The authors would like to thank Natalie Job and Professor James Gilkerson for providing field isolates from Melbourne. This study was supported by internal funding and the project constituted part of the Ph.D. of the first author.
Contributor Information
Phillip McDonagh, Email: phillip.mcdonagh@sydney.edu.au.
Paul A. Sheehy, Email: paul.sheehy@sydney.edu.au.
Anne Fawcett, Email: anne.fawcett@sydney.edu.au.
Jacqueline M. Norris, Email: jacqui.norris@sydney.edu.au.
References
- Brickelmaier M., Lugovskoy A., Kartikeyan R., Reviriego-Mendoza M.M., Allaire N., Simon K., Frisque R.J., Gorelik L. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrob. Agents Chemother. 2009;53:1840–1849. doi: 10.1128/AAC.01614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court M.H. Feline drug metabolism and disposition: pharmacokinetic evidence for species differences and molecular mechanisms. Vet. Clin. North Am.: Small Anim. Pract. 2013;43:1039. doi: 10.1016/j.cvsm.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne K.P., Christley R.M., Pybus O.G., Dawson S., Gaskell R.M., Radford A.D. Large scale spatial and temporal genetic diversity of feline calicivirus. J. Virol. 2012 doi: 10.1128/JVI.00701-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld J.J., Hoofnagle J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- Ferraris O., Kessler N., Lina B. Sensitivity of influenza viruses to zanamivir and oseltamivir: a study performed on viruses circulating in France prior to the introduction of neuraminidase inhibitors in clinical practice. Antiviral Res. 2005;68:43–48. doi: 10.1016/j.antiviral.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Fulton R.W., Burge L.J. Susceptibility of feline herpesvirus 1 and a feline calicivirus to feline interferon and recombinant human leukocyte interferons. Antimicrob. Agents Chemother. 1985;28:698–699. doi: 10.1128/aac.28.5.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaumann H., Motakefi A.M., Jansson H. Intracellular distribution and effect of the antimalarial drug mefloquine on lysosomes of rat liver. Liver. 1992;12:183–190. doi: 10.1111/j.1600-0676.1992.tb01045.x. [DOI] [PubMed] [Google Scholar]
- Glenn M., Radford A.D., Turner P.C., Carter M., Lowery D., DeSilver D.A., Meanger J., Baulch-Brown C., Bennett M., Gaskell R.M. Nucleotide sequence of UK and Australian isolates of feline calicivirus (FCV) and phylogenetic analysis of FCVs. Vet. Microbiol. 1999;67:175–193. doi: 10.1016/s0378-1135(99)00043-7. [DOI] [PubMed] [Google Scholar]
- Hennet P.R., Camy G.A., McGahie D.M., Albouy M.V. Comparative efficacy of a recombinant feline interferon omega in refractory cases of calicivirus-positive cats with caudal stomatitis: a randomised, multi-centre, controlled, double-blind study in 39 cats. J. Feline Med. Surg. 2011;13:577–587. doi: 10.1016/j.jfms.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley K.F., Sykes J.E. Update on feline calicivirus: new trends. Vet. Clin. North Am.: Small Anim. Pract. 2003;33:759–772. doi: 10.1016/s0195-5616(03)00025-1. [DOI] [PubMed] [Google Scholar]
- Kollaritsch H., Karbwang J., Wiedermann G., Mikolasek A., Na-Bangchang K., Wernsdorfer W. Mefloquine concentration profiles during prophylactic dose regimens. Wien. Klin. Wochenschr. 2000;112:441–447. [PubMed] [Google Scholar]
- Kreutz L.C., Seal B.S. The pathway of feline calicivirus entry. Virus Res. 1995;35:63–70. doi: 10.1016/0168-1702(94)00077-p. [DOI] [PubMed] [Google Scholar]
- Lyon K.F. Gingivostomatitis. Vet. Clin. North Am.: Small Anim. Pract. 2005;35:891–911. doi: 10.1016/j.cvsm.2005.02.001. [DOI] [PubMed] [Google Scholar]
- McCann K.B., Lee A., Wan J., Roginski H., Coventry M.J. The effect of bovine lactoferrin and lactoferricin B on the ability of feline calicivirus (a norovirus surrogate) and poliovirus to infect cell cultures. J. Appl. Microbiol. 2003;95:1026–1033. doi: 10.1046/j.1365-2672.2003.02071.x. [DOI] [PubMed] [Google Scholar]
- McDonagh P., Sheehy P.A., Norris J.M. Identification and characterisation of small molecule inhibitors of feline coronavirus replication. Vet. Microbiol. 2014;174:438–447. doi: 10.1016/j.vetmic.2014.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki M., Nakatani H., Yoshida M. Inhibitory effects of recombinant feline interferon on the replication of feline enteropathogenic viruses in vitro. Vet. Microbiol. 1994;39:145–152. doi: 10.1016/0378-1135(94)90095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya H., Fukutome A., Kabayashi K., Shin Y., Uchino T., Motoyoshi S. Effect of recombinant feline interferon on feline calicivirus infection. XVI World Small Animal Veterinary Association; Vienna, Austria; 1991. [Google Scholar]
- Ohe K., Takahashi T., Hara D., Hara M. Sensitivity of FCV to recombinant feline interferon (rFeIFN) Vet. Res. Commun. 2008;32:167–174. doi: 10.1007/s11259-007-9019-5. [DOI] [PubMed] [Google Scholar]
- Owen A., Janneh O., Hartkoorn R.C., Chandler B., Bray P.G., Martin P., Ward S.A., Hart C.A., Khoo S.H., Back D.J. In vitro synergy and enhanced murine brain penetration of saquinavir coadministered with mefloquine. J. Pharmacol. Exp. Ther. 2005;314:1202–1209. doi: 10.1124/jpet.105.086272. [DOI] [PubMed] [Google Scholar]
- Pedersen N.C., Elliott J.B., Glasgow A., Poland A., Keel K. An isolated epizootic of hemorrhagic-like fever in cats caused by a novel and highly virulent strain of feline calicivirus. Vet. Microbiol. 2000;73:281–300. doi: 10.1016/S0378-1135(00)00183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povey R.C. In vitro antiviral efficacy of ribavirin against feline calicivirus, feline viral rhinotracheitis virus, and canine parainfluenza virus. Am. J. Vet. Res. 1978;39:175–178. [PubMed] [Google Scholar]
- Radford A.D., Coyne K.P., Dawson S., Porter C.J., Gaskell R.M. Feline calicivirus. Vet. Res. 2007;38:319–335. doi: 10.1051/vetres:2006056. [DOI] [PubMed] [Google Scholar]
- Radford A.D., Dawson S., Coyne K.P., Porter C.J., Gaskell R.M. The challenge for the next generation of feline calicivirus vaccines. Vet. Microbiol. 2006;117:14–18. doi: 10.1016/j.vetmic.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Shafer R., Vuitton D. Highly active antiretroviral therapy (HAART) for the treatment of infection with human immunodeficiency virus type 1. Biomed. Pharmacother. 1999;53:73–86. doi: 10.1016/s0753-3322(99)80063-8. [DOI] [PubMed] [Google Scholar]
- Simpson J.A., Price R., ter Kuile F., Teja-Isavatharm P., Nosten F., Chongsuphajaisiddhi T., Looareesuwan S., Aarons L., White N.J. Population pharmacokinetics of mefloquine in patients with acute falciparum malaria. Clin. Pharmacol. Ther. 1999;66:472–484. doi: 10.1016/S0009-9236(99)70010-X. [DOI] [PubMed] [Google Scholar]
- Smith A.W., Iversen P.L., O’Hanley P.D., Skilling D.E., Christensen J.R., Weaver S.S., Longley K., Stone M.A., Poet S.E., Matson D.O. Virus-specific antiviral treatment for controlling severe and fatal outbreaks of feline calicivirus infection. Am. J. Vet. Res. 2008;69:23–32. doi: 10.2460/ajvr.69.1.23. [DOI] [PubMed] [Google Scholar]
- Stuart A.D., Brown T.D.K. Entry of feline calicivirus is dependent on clathrin-mediated endocytosis and acidification in endosomes. J. Virol. 2006;80:7500–7509. doi: 10.1128/JVI.02452-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira O., Suzuki M., Takeuchi Y., Aramaki Y., Sakurai I., Watanabe T., Motokawa K., Arai S., Sato H., Maehara N. Expression of feline interferon-alpha subtypes in Esherichia coli, and their antiviral activity and animal species specificity. J. Vet. Med. Sci. 2005;67:543–545. [Google Scholar]
- Truyen U., Blewaska S., Schultheiss U. Antiviral potency of interferon-omega (IFN-omega) against selected canine and feline viruses. Prakt. Tierarzt. 2002;83:862–865. [Google Scholar]