

Abstract
Natural killer (NK) cells serve as a crucial first line of defense against tumors and a diverse range of pathogens. Recognition of infection by NK cells is accomplished by the activation of receptors on the NK cell surface, which initiate NK cell effector functions. Many of the receptors and ligands involved in NK cell antimicrobial activity have been identified, and we are beginning to appreciate how they function during infection. In addition, NK cells are activated by cytokines (e.g. interleukin 12 and type I interferons), which are products of activated macrophages and dendritic cells. In response to these activating stimuli, NK cells secrete cytokines and chemokines and lyse target cells. Recent studies have focused on the mechanisms by which NK cells recognize and respond to viruses, parasites and bacteria, and on the unique role of NK cells in innate immunity to infection.
Introduction
Natural killer (NK) cells represent a distinct subset of lymphoid cells that have innate immune functions [1]. Derived from the bone marrow, NK cells circulate in the blood and become activated by cytokines or upon encountering target cells that express ligands for NK cell receptors [2]. NK cell receptors are encoded in the germ-line and do not undergo somatic recombination like B- and T-cell antigen receptors; it is the balance of signals from activating and inhibitory receptors that determines the outcome of NK cell activity. Some inhibitory receptors recognize MHC class I, which is present on virtually all healthy cells, and prevent NK cell attack against these cells. Loss of MHC class I from cells owing to infection or transformation can lead to NK cell activation, as proposed by the ‘missing self hypothesis’ [3], provided that an activating receptor is engaged. These activating NK receptors bind to host-derived or pathogen-encoded ligands that are up-regulated on ‘stressed’ or infected cells. Upon activation, NK cells directly lyse target cells by exocytosis of perforin and granzymes and secrete cytokines, such as interferon (IFN)-γ and tumor necrosis factor (TNF)-α, which mediate their immune response to infection. In this manner, NK cells function as important sentinels of the immune system, working as primary responders and alerting the host to the presence of infectious organisms.
In this review, we will highlight recent studies on the mechanisms by which NK cells recognize and respond to viruses, parasites and bacteria, and will underscore the unique role of NK cells in innate immunity to infection (Table 1 ).
Table 1.
NK cells in immunity to pathogens.
| Pathogen | Mechanism of action | References | |
|---|---|---|---|
| Viruses | MCMV | TLR9 and MyD88 activation of DC-induced IFN-α and IL-12 production; this leads to NK cell IFN-γ production and cytotoxicity | [4, 5, 6••, 7••] |
| Perforin and IFN-γ generation by NK cells limits viral replication in the spleen and liver | [8, 9] | ||
| MCMV m157 binds Ly49H, induces NK cell IFN-γ, MIP-1α, MIP-1β, RANTES and ATAC, and controls MCMV in C57BL/6 mice | [17, 18, 19, 20•] | ||
| NK cells mediate resistance in NZW mice by multiple gene products | [21] | ||
| NK cells protect MA/My mice from infection; Ly49P recognizes H-2Dk-restricted ligand in MCMV-infected cells | [22•, 23•] | ||
| KLRG1+ NK cells expand and contract in response to infection | [26] | ||
| HCMV | NK cell IFN-γ, LTα/β and TNF induce IFN-β from infected cells and inhibit HCMV replication | [10] | |
| A truncated form of MICA escapes HCMV down-regulation and activates NK cell NKG2D | [13] | ||
| CD94–NKG2C+ NK cells preferentially expand in response to infected fibroblasts | [25] | ||
| Sendai virus | Viral infection induces IFN-α and MICB gene transcription; this leads to NK cell IFN-γ production | [11] | |
| Influenza A virus | Viral infection induces IFN-α and MICB gene transcription; this leads to NK cell IFN-γ production | [11] | |
| HIV | Neonatal NK cells suppress replication of CCR5-trophic viruses | [29] | |
| NK cells from viremic patients produce more IFN-γ and TNF-α than NK cells from aviremic patients | [30] | ||
| MHV | NK cell recruited to the CNS after intracerebral MHV infection enhance survival and decrease viral titers | [33] | |
| Ebola virus | Injection of Ebola virus-like particles confers NK cell-mediated protection against Ebola virus infection in mice | [34] | |
| Parasites | P. falciparum | Human NK cells produce IFN-γ in response to infected RBC | [36, 37•, 38•, 39] |
| NK cells form stable conjugates with infected RBC | [39] | ||
| P. berghei ANKA | Reduced pathogenesis in BALB/c mice maps to the NKC | [40] | |
| T. cruzi | NK cells enhance survival of infected C57BL/6 mice and reduce parasitemia | [42] | |
| NK cells directly lyse parasites | [41] | ||
| Bacteria | Shigella flexneri | NK cell IFN-γ controls infection | [43] |
| M. tuberculosis | NK cells kill infected monocytes by a mechanism that involves NKp46 and NKG2D | [44] |
Natural killer cells and viral infections
Natural killer cell–dendritic cell interactions and Toll-like receptor 9
NK cells can respond to infection either directly or indirectly. They respond directly by recognizing virus-infected cells, and indirectly by interacting with dendritic cells (DCs), which express Toll-like receptors (TLRs) and secrete cytokines in response to encounter with microbes.
Toll-like receptor 9 (TLR9) recognizes unmethylated CpG DNA, a component of bacterial and viral DNA, and signals for cellular activation through the adapter protein MyD88. Several groups have recently examined the role of TLR9 and MyD88 in response to mouse cytomegalovirus (MCMV) infection by using MyD88-deficient or TLR9-deficient mice. These immunodeficient mice had elevated amounts of virus in their spleens and livers compared with wild-type mice, which indicates an increased susceptibility to infection and suggests a crucial role for TLR9 and MyD88 in immunity to MCMV [4, 5, 6••, 7••]. Infection by MCMV activated IFN-producing cells and myeloid DCs, resulting in the production of IFN-α and IL-12 through pathways dependent upon MyD88 and TLR9 [5]. Mice that lacked MyD88 or TLR9 had reduced amounts of IFN-α and IFN-β, interleukin (IL)-12, macrophage inflammatory protein 1α (MIP-1α) and IFN-γ in their sera [4, 5, 6••]. Of note, NK cells from MCMV-infected TLR9- and MyD88-deficient mice produced less IFN-γ [4, 5], had impaired proliferation, and displayed reduced cytolytic activity compared with NK cells from MCMV-infected wild-type mice [5]. In addition, MCMV-infected CD11b+ DCs directly activated NK cell IFN-γ production by a mechanism that required IFN-α and the NKG2D receptor on NK cells [6••].
Collectively, these data suggest that IFN-producing cells and DCs are activated by MCMV through a TLR9- and MyD88-dependent pathway to produce type I IFNs and IL-12. These cytokines stimulate NK cell IFN-γ production and cytotoxic activity and ultimately control the viral infection (Figure 1 ). In another report, it was found that sera levels of IFN-α in TLR9-deficient mice were unchanged compared with wild-type mice, indicating that IFN-α production in response to MCMV infection is independent of TLR9 [7••]. Further studies will be necessary to resolve these differences.
Figure 1.
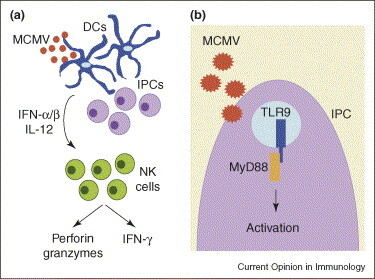
Interaction between DCs and NK cells during MCMV infection. (a) MCMV infection activates DCs to produce IFN-α, IFN-β and IL-12 by a MyD88- and TLR9-dependent pathway, as shown in (b). IFN-α, IFN-β and IL-12 are potent activators of NK cell IFN-γ secretion and cytotoxic activity, which are crucial for NK cell control of infection.
Natural killer cell antiviral effector mechanisms
NK cells use two main effector mechanisms to control MCMV infection: the secretion of IFN-γ and direct lysis of infected cells by exocytosis of granules that contain perforin and granzymes. Using mice deficient in perforin or IFN-γ and mice depleted of IFN-γ by antibody, organ-specific control of MCMV infection by NK cells was observed: perforin mediated viral clearance in the spleen and IFN-γ mediated protection in the liver [8]. A recent study re-evaluated the importance of these two effector mechanisms during MCMV infection and found that mice deficient in perforin or IFN-γ had elevated viral titers in the spleen and the liver three days post-infection compared with wild-type C57BL/6 mice [9]. By six days post-infection, viral titers in the spleens and livers of perforin-deficient mice remained higher than those of wild-type mice, whereas viral titers in IFN-γ-deficient mice were higher only in the livers [9]. The authors noted that the differences observed between their work and the previous study might reflect the background of the mice used in the experiments, because MCMV resistance varies by mouse strain. The original study used perforin-deficient mice on a C57BL/6 × 129 mixed genetic background, whereas the recent study used mice on a C57BL/6 background. It is conceivable that NK cells in different mouse strains use distinct antiviral effector mechanisms in different organs.
Lymphotoxin (LT) and IFN-β have recently been implicated in non-cytolytic NK cell control of human cytomegalovirus (HCMV) infection. When cultured with HCMV-infected fibroblasts, IL-2-activated NK cells produced IFN-γ and induced IFN-β production by the infected cells, which inhibited spread of the virus [10]. IFN-β induction appeared to involve NK cell secretion of LTαβ and TNF, as soluble decoys of both of these molecules prevented HCMV inhibition [10].
IFN-γ production by NK cells might also contribute to protection against influenza A and Sendai viruses. Contact between human NK cells and virus-infected macrophages induced NK cell-mediated IFN-γ production [11]. In addition, gene transcription of IFN-γ and major histocompatibility complex class I-related chain B (MICB), a ligand for the NKG2D receptor, by the NK cells was enhanced by virus-induced IFN-α, which indicates interplay among type I and type II IFNs during influenza A and Sendai virus infections [11].
Human cytomegalovirus infection modulates natural killer cell receptor function
Several studies have examined the effect of CMV infection on ligands for NK cell receptors [12]. Recent experiments analyzing the NKG2D ligand MICA demonstrated that HCMV infection down-regulated a full-length isoform of MICA from the surface of infected fibroblasts, preventing lysis by the human NK cell line NKL [13]. However, a truncated form of MICA, which lacks a cytoplasmic tail owing to a frameshift mutation, was stably expressed on the surface of infected fibroblasts and was efficiently lysed by NKL [13]. Interestingly, the truncated allele was found to be the most common allele in several human populations [14], which suggests that polymorphism in the MICA gene might allow for enhanced viral clearance through NKG2D-dependent activation of NK cells. CD155, the ligand for the NK cell activating receptors DNAM-1 (CD226) and TACTILE (CD96), was targeted for down-regulation by the HCMV glycoprotein UL141, rendering infected cells resistant to NK cell lysis [15•]. Down-regulation of CD155 only occurred during infection of fibroblasts with low-passage strains or clinical isolates of HCMV, and UL141 functioned by preventing maturation of CD155 and retaining it in the endoplasmic reticulum [15•].
An intriguing story has emerged regarding the ability of the HCMV tegument protein pp65 to modulate NK cell killing through the NKp30 receptor. Arnon et al. [16] reported that pp65 binds to NKp30 and inhibits NKp30-mediated NK cell lysis of HCMV-infected targets. Surprisingly, pp65 appeared to cause dissociation of NKp30 from its signaling component, CD3ζ, thereby inhibiting NKp30 activity [16]. The mechanism by which a virion tegument protein, such as pp65, might access the interaction site between NKp30 and CD3ζ on the NK cell plasma membrane remains unclear and will require further investigation.
Natural killer cells mediate host resistance to murine cytomegalovirus by diverse mechanisms
Resistance of C57BL/6 mice to MCMV infection is mediated predominantly by the recognition of m157 — a viral protein expressed on infected cells — by NK cell activating receptor Ly49H [12]. Ligation of Ly49H by m157 during MCMV infection results in the release of cytokines and chemokines, including IFN-γ, MIP-1α, MIP-1β, RANTES and ATAC, which might play a role in containing the infection [17]. A mutant virus that lacks the m157 open reading frame displayed enhanced virulence compared with wild-type MCMV in C57BL/6 mice, which indicates that recognition of m157 by Ly49H governs innate immune control of infection [18]. This idea was reinforced by the emergence of viral escape variants that have mutations in the m157 gene during infection of C57BL/6 mice that lack adaptive immunity [19, 20•]. In this manner, Ly49H+ NK cells might drive the evolution of viruses that can escape from innate immune control.
Like C57BL/6 mice, New Zealand White (NZW) mice are resistant to MCMV infection by use of an NK cell-dependent mechanism; depletion of NK cells with anti-NK1.1 antibody resulted in elevated MCMV titers in the spleen and liver [21]. However, resistance was independent of Ly49H and m157, as NZW mice do not possess a Ly49H gene. Moreover, treatment with an anti-Ly49H antibody, which conferred MCMV susceptibility in C57BL/6 mice, had no effect on viral titers in NZW mice [21]. When MCMV-resistant NZW mice were mated and backcrossed with other MCMV-sensitive mouse strains, resistance was shown to be mediated by multiple gene products [21], which remain to be identified.
NK cells also mediate immune protection against MCMV in the MA/My mouse strain, which lacks the Ly49H receptor. Genetic analysis has revealed that MA/My resistance to MCMV is dependent both on the H-2k haplotype [22•, 23•] and on a locus in the ‘NK gene complex’, a region of mouse chromosome 6 that contains a cluster of genes preferentially expressed by NK cells [22•]. Examination of the activating Ly49 receptors in the MA/My strain identified Ly49P to be the only activating Ly49 capable of responding to MCMV-infected cells [22•]. Although the ligand for Ly49P on MCMV-infected cells has yet to be identified, it appears to be a ligand associated with H-2Dk because antibody blockade of either Ly49P or H-2Dk prevented Ly49P reporter cell recognition of MCMV-infected cells [22•].
The ability of wild mouse populations to control MCMV infection was recently investigated. Viral replication in the spleens and livers of wild outbred mice housed in a specific pathogen-free facility was similar to growth of the virus in ‘MCMV-susceptible’ BALB/c inbred mice [24].
Cytomegalovirus infection promotes the outgrowth of specific natural killer cell populations
NK cells are a heterogeneous population composed of cells that express overlapping subsets of activating and inhibitory receptors. When human peripheral blood lymphocytes were cultured with HCMV-infected fibroblasts, NK cells that express the activating CD94–NKG2C receptor preferentially expanded. Expansion was contingent on viral replication, on direct contact between lymphocytes and infected cells, and on signaling through the CD94–NKG2C–DAP12 receptor complex [25]. Interestingly, a MCMV viral mutant that lacked the US2-11 genes, and therefore could not down-regulate MHC class I, did not drive expansion of this population [25]. Whether the ligand for CD94–NKG2C (i.e. HLA-E) plays a role in proliferation of CD94–NKG2C+ NK cells in response to HCMV-infected fibroblasts awaits further investigation.
In the mouse, MCMV infection induces phenotypic maturation of NK cells, including expression of killer cell lectin-like receptor G1 (KLRG1) [26], which was recently identified to be a receptor for E-cadherin [27, 28]. Tracking of KLRG1+ cells indicated that NK cells activated by MCMV expanded in response to infection and then contracted owing to cell death. A KLRG1− population remained, which could respond to secondary infection [26]. Thus, CMV infection might shape the NK cell repertoire, leading to the selective expansion of specific NK cell subsets.
Human immunodeficiency virus infection and natural killer cells
Neonatal NK cells might play a crucial role in protection from perinatal human immunodeficiency virus (HIV) infection. When cultured with autologous human HIV-infected CD4+ T cells, neonatal NK cells suppressed HIV replication to a greater degree than adult NK cells [29]. Suppression was dependent on a chemokine-mediated mechanism and on infection with a CCR5-dependent virus, because NK cells could not suppress HIV replication by a CXCR4-dependent virus [29].
Highly active antiretroviral therapy can reduce HIV replication and can result in a decrease in viral load in the blood, known as viremia, of responsive patients. Analysis of NK cells in chronically infected viremic and aviremic HIV-1 patients found that the number of CD3–CD56+ NK cells in the blood was dramatically reduced in patients that had ongoing viral replication compared with uninfected or aviremic patients [30]. However, a greater percentage of NK cells from viremic patients, compared with aviremic patients, produced IFN-γ and TNF-α and expressed higher levels of CD107a — a marker of granule exocytosis — after stimulation with the tumor cell line K562 [30]. Thus, NK cell activity is augmented in HIV-1 patients that have ongoing viral replication in the blood, although total numbers of NK cells are reduced.
It has long been appreciated that HIV infection down-regulates expression of MHC class I. A recent study revealed that certain strains of HIV selectively down-regulated HLA-A2 and HLA-B7, but not HLA-C or HLA-E, on infected cells [31]. HLA-C and HLA-E on HIV-infected T cell blasts engaged inhibitory killer-cell Ig-like receptors (KIRs) or CD94–NKG2A receptors, respectively, on NK cells, inhibited killing of the virus-infected cells [31], and blocked antibody-dependent cell-mediated cytotoxicity against HIV-infected cells coated with anti-gp120 [32]. The ability of certain HIV-1 strains to selectively modulate MHC class I on infected cells might determine whether or not NK cells can respond to the infection.
Natural killer cell recruitment to the central nervous system during viral infection
The chemokine CXCL10 might be crucial for innate immunity to coronavirus because it recruits NK cells to the central nervous system (CNS) during infection. Intracerebral infection of RAG1 −/− mice with a CXCL10-expressing mouse hepatitis virus (MHV) resulted in the rapid infiltration of NK cells to the CNS, which correlated with delayed mortality and reduced viral titers compared with animals infected with wild-type virus [33]. Enhanced IFN-γ secretion by NK cells was also observed and was found to be essential for the antiviral effect, possibly by limiting MHV replication [33].
Natural killer cell protection during Ebola virus infection
The role of NK cells was examined in protection against Ebola virus that was induced by treating mice with non-replicating Ebola virus-like particles (VLPs) and then challenging mice with a lethal dose of Ebola virus. VLP immunization enhanced NK cell numbers in the lymph nodes and spleens, increased NK cell secretion of IFN-γ and TNF-α, and induced cytotoxic activity [34]. Depletion of NK cells from mice prior to treatment with VLPs resulted in decreased survival after infection with Ebola virus compared with undepleted mice, and adoptive transfer of NK cells activated by VLPs into naïve mice enhanced their survival after Ebola infection, further implicating NK cells in immunity to Ebola virus [34]. Perforin, but not IFN-γ, was shown to be required for the protective effect. Although not addressed in this report, the VLP-activated NK cells presumably augmented an adaptive immune response that was responsible for protection against challenge with Ebola virus.
Natural killer cell activation during parasitic infection
Natural killer cells respond to Plasmodium falciparum
NK cells play an important role in the host response to infection with Plasmodium falciparum — the causative agent of malaria [35]. IFN-γ production by human NK cells cultured with red blood cells (RBCs) infected with P. falciparum was dependent on IL-12 and IL-18 derived from myeloid cells in the cultures [36]; the amount of IFN-γ generated by NK cells responding to infected RBCs varied by donor [37•, 38•]. It has been proposed that the CD56bright subset of human NK cells is responsible for cytokine production, whereas the CD56dim NK cells are responsible for cytolytic activity. When stimulated by malaria-infected erythrocytes, however, both NK cell subsets produced abundant IFN-γ [37•]. Culture of NK cells with RBC infected by several different strains of P. falciparum also up-regulated CD69 and CD25 activation markers on NK cells from all donors.
The cellular and molecular requirements for NK cell activation by malaria-infected RBCs have recently been examined. Transwell assay experiments revealed that NK cell IFN-γ production required contact of human peripheral blood mononuclear cells (PBMCs) with infected RBCs, and that NK cells produced IFN-γ only in the presence of PBMCs [39]. NK cells formed stable conjugates with infected RBCs [39], and in some cases reorganized the actin cytoskeleton after conjugate formation [37•]. Purified NK cells, however, only produced substantial amounts of IFN-γ in the presence of monocytes [38•]. In addition, IL-18 production by macrophages and MyD88 expression in both NK cells and macrophages were required to induce IFN-γ production by mouse NK cells in response to infected RBCs [38•]. Although NK cells can interact with infected RBCs, robust NK cell activation seems to require secretion of soluble mediators by macrophages or monocytes. Specific NK receptors involved in recognition of infected RBCs have not been defined; however, in humans, KIR molecules might play a role, because associations were found between the KIR genotype and donor susceptibility to malaria [39].
The natural killer complex and Plasmodium berghei pathogenesis
The NK complex (NKC; a region on mouse chromosome 6 containing several genes preferentially expressed by NK cells) appears to contribute to pathogenesis of malaria infection with Plasmodium berghei ANKA. C57BL/6 mice succumb to severe infection with P. berghei ANKA, whereas BALB/c mice are resistant. When BALB/c mice congenic for the C57BL/6 NKC were infected with P. berghei ANKA, parasitemia was unaffected but parasite-specific antibody responses, pro-inflammatory gene expression, and the TH1/TH2 balance were altered [40].
Natural killer cell activation by infection with Trypanosoma cruzi
NK cells might also play a role in protection against Trypanosoma cruzi infection. Depletion of NK cells from mice using the anti-asialo GM1 antibody did not affect survival when a low dose (1 × 104) of T. cruzi trypomastigotes was used for infection [41]; however, when a higher dose was used (5 × 105), NK-depleted mice had compromised survival compared with C57BL/6 mice [42]. Interestingly, increased parasitemia was observed in NK-depleted mice infected with the lower dose of T. cruzi, possibly because NK cells could form intimate interactions with trypomastigotes and contribute to their lysis [41]. Lysis was independent of perforin but dependent upon IL-12 [41], which suggests that monocytes or macrophages activated by T. cruzi might contribute to NK cell activation by producing IL-12. NK cell cytotoxicity and protection against T. cruzi infection was independent of NK T cells [42].
Natural killer cells and bacterial infection
The importance of NK cells in protection against bacterial infection has been controversial and might vary depending upon the site of infection or type of inflammatory response elicited. Mice deficient in the RAG2 and common γ-chain genes (RAG −/− γc −/−) lack B, T and NK cells and can be used to assess the contributions of these cell types in the immune response. After infection with Shigella flexneri, RAG −/−γc −/− mice had severely compromised survival compared with wild-type mice, possibly owing to reduced production of IL-1, IL-6, TNF and IL-12 [43]. Interestingly, RAG −/− mice, which have NK cells, had lower bacterial titers and increased survival compared with RAG −/− γc −/− mice, suggesting that NK cell production of IFN-γ alone might limit infection [43]. If this is the case, it would be expected that depletion of NK cells from RAG −/− mice would lead to an exacerbated disease phenotype.
One mechanism by which NK cells can modulate their activity during infection is by altering receptor expression. Human NK cells cultured with Mycobacterium tuberculosis-infected monocytes up-regulated expression of the activating receptors NKp30, NKp46 and NKG2D and lysed infected monocytes through NKG2D- and NKp46-dependent mechanisms; this is known because antibodies against these receptors inhibited cytotoxicity [44]. Infection of monocytes resulted in up-regulation of the NKG2D ligand ULBP1 through TLR2 activation [44]. Thus, NKG2D and NKp46 might mediate the NK cell response to M. tuberculosis infection. However, depletion of NK cells in mice infected with M. tuberculosis did not affect the bacterial load [45].
Although previously suggested to provide a protective role in response to the intracellular bacterium Listeria monocytogenes (LM), NK cells might not be involved in bacterial clearance [46]. Recent work by Berg et al. [47] has shown that when OT-1 TcR transgenic T cells were transferred into IFN-γ-deficient hosts prior to infection with LM, these T cells secreted IFN-γ and reduced the bacterial burden, despite the fact that these T cells recognized ovalbumin and not Listeria. This suggests a bystander ‘innate’ role for the T cells [47]. In addition, the transferred T cells localized with macrophages and LM lesions in the spleen and liver. By contrast, when NK cells were adoptively transferred into these recipients, the NK cells localized to the splenic red pulp, away from infected foci [47]. In this study, CD8+ T cells were shown to be the more significant player in innate immunity to LM.
Interestingly, recent reports have revealed that type I IFN hinders host control of LM, as mice deficient in this signaling system had reduced bacterial burdens and enhanced survival compared with wild-type mice [48, 49, 50]. Because NK cells are activated by type I IFN, it might be that NK cells worsen disease rather than protect the host from LM infection. This is consistent with the finding that mice that lack the DAP12 adapter protein, which associates with several NK and myeloid cell activating receptors, exhibited enhanced TLR responses and better control of bacterial infection [51].
Conclusions
Although originally identified by their ability to kill tumors, emerging evidence strongly implicates NK cells in host protection against microbial pathogens. In particular, a role for NK cells in host defense against CMV in humans and mice is supported by the existence of viral mutation in response to NK cell-mediated immunity and the evolution of viral proteins that target the NKG2D ligands in CMV-infected cells. Although less well-studied, a developing theme is anti-parasitic and anti-bacterial immunity resulting from the interplay of NK cells with DCs and macrophages. However, NK cells might not always be beneficial in immune responses, as highlighted by recent observations that NK cells might promote the growth of rather than protect against LM. The future promises a better molecular definition of the mechanisms by which NK cells participate in innate immunity against these pathogens.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgements
LLL is an American Cancer Society Research Professor and is funded by National Institutes of Health grants AI068129, AI066897, CA095137 and AI52127.
References
- 1.Cerwenka A., Lanier L.L. Natural killer cells, viruses and cancer. Nat Rev Immunol. 2001;1:41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- 2.Lanier L.L. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 3.Ljunggren H.G., Karre K. Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J Exp Med. 1985;162:1745–1759. doi: 10.1084/jem.162.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabeta K., Georgel P., Janssen E., Du X., Hoebe K., Crozat K., Mudd S., Shamel L., Sovath S., Goode J., et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krug A., French A.R., Barchet W., Fischer J.A., Dzionek A., Pingel J.T., Orihuela M.M., Akira S., Yokoyama W.M., Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 6••.Andoniou C.E., van Dommelen S.L., Voigt V., Andrews D.M., Brizard G., Asselin-Paturel C., Delale T., Stacey K.J., Trinchieri G., Degli-Esposti M.A. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol. 2005;6:1011–1019. doi: 10.1038/ni1244. [DOI] [PubMed] [Google Scholar]; [4, 5, 6••, 7••] show that TLR9 and MyD88 activation of DCs plays a crucial role in immunity against MCMV.
- 7••.Delale T., Paquin A., Asselin-Paturel C., Dalod M., Brizard G., Bates E.E., Kastner P., Chan S., Akira S., Vicari A., et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-α release and initiation of immune responses in vivo. J Immunol. 2005;175:6723–6732. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]; See annotation to [6••].
- 8.Tay C.H., Welsh R.M. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J Virol. 1997;71:267–275. doi: 10.1128/jvi.71.1.267-275.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loh J., Chu D.T., O’Guin A.K., Yokoyama W.M., Virgin H.W. Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J Virol. 2005;79:661–667. doi: 10.1128/JVI.79.1.661-667.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iversen A.C., Norris P.S., Ware C.F., Benedict C.A. Human NK cells inhibit cytomegalovirus replication through a noncytolytic mechanism involving lymphotoxin-dependent induction of IFN-β. J Immunol. 2005;175:7568–7574. doi: 10.4049/jimmunol.175.11.7568. [DOI] [PubMed] [Google Scholar]
- 11.Siren J., Sareneva T., Pirhonen J., Strengell M., Veckman V., Julkunen I., Matikainen S. Cytokine and contact-dependent activation of natural killer cells by influenza A or Sendai virus-infected macrophages. J Gen Virol. 2004;85:2357–2364. doi: 10.1099/vir.0.80105-0. [DOI] [PubMed] [Google Scholar]
- 12.Lodoen M.B., Lanier L.L. Viral modulation of NK cell immunity. Nat Rev Microbiol. 2005;3:59–69. doi: 10.1038/nrmicro1066. [DOI] [PubMed] [Google Scholar]
- 13.Zou Y., Bresnahan W., Taylor R.T., Stastny P. Effect of human cytomegalovirus on expression of MHC class I-related chains A. J Immunol. 2005;174:3098–3104. doi: 10.4049/jimmunol.174.5.3098. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y., Lazaro A.M., Lavingia B., Stastny P. Typing for all known MICA alleles by group-specific PCR and SSOP. Hum Immunol. 2001;62:620–631. doi: 10.1016/s0198-8859(01)00241-5. [DOI] [PubMed] [Google Scholar]
- 15•.Tomasec P., Wang E.C., Davison A.J., Vojtesek B., Armstrong M., Griffin C., McSharry B.P., Morris R.J., Llewellyn-Lacey S., Rickards C., et al. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat Immunol. 2005;6:181–188. doi: 10.1038/ni1156. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates a unique mechanism by which viruses evade NK cell recognition; this occurs through the DNAM-1 activating receptor.
- 16.Arnon T.I., Achdout H., Levi O., Markel G., Saleh N., Katz G., Gazit R., Gonen-Gross T., Hanna J., Nahari E., et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat Immunol. 2005;6:515–523. doi: 10.1038/ni1190. [DOI] [PubMed] [Google Scholar]
- 17.Dorner B.G., Smith H.R., French A.R., Kim S., Poursine-Laurent J., Beckman D.L., Pingel J.T., Kroczek R.A., Yokoyama W.M. Coordinate expression of cytokines and chemokines by NK cells during murine cytomegalovirus infection. J Immunol. 2004;172:3119–3131. doi: 10.4049/jimmunol.172.5.3119. [DOI] [PubMed] [Google Scholar]
- 18.Bubic I., Wagner M., Krmpotic A., Saulig T., Kim S., Yokoyama W.M., Jonjic S., Koszinowski U.H. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J Virol. 2004;78:7536–7544. doi: 10.1128/JVI.78.14.7536-7544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.French A.R., Pingel J.T., Wagner M., Bubic I., Yang L., Kim S., Koszinowski U., Jonjic S., Yokoyama W.M. Escape of mutant double-stranded DNA virus from innate immune control. Immunity. 2004;20:747–756. doi: 10.1016/j.immuni.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 20•.French A.R., Pingel J.T., Kim S., Yang L., Yokoyama W.M. Rapid emergence of escape mutants following infection with murine cytomegalovirus in immunodeficient mice. Clin Immunol. 2005;115:61–69. doi: 10.1016/j.clim.2005.02.008. [DOI] [PubMed] [Google Scholar]; [18, 19, 20•] provide evidence that MCMV evolution is driven by NK cell-mediated immunity.
- 21.Rodriguez M., Sabastian P., Clark P., Brown M.G. Cmv1-independent antiviral role of NK cells revealed in murine cytomegalovirus-infected New Zealand White mice. J Immunol. 2004;173:6312–6318. doi: 10.4049/jimmunol.173.10.6312. [DOI] [PubMed] [Google Scholar]
- 22•.Desrosiers M.P., Kielczewska A., Loredo-Osti J.C., Adam S.G., Makrigiannis A.P., Lemieux S., Pham T., Lodoen M.B., Morgan K., Lanier L.L., et al. Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat Genet. 2005;37:593–599. doi: 10.1038/ng1564. [DOI] [PMC free article] [PubMed] [Google Scholar]; [22•, 23•] demonstrate that NK cell mediated immunity is influenced by MHC and by genes in the NKC (probably the Ly49P receptor).
- 23•.Dighe A., Rodriguez M., Sabastian P., Xie X., McVoy M., Brown M.G. Requisite H2k role in NK cell-mediated resistance in acute murine cytomegalovirus-infected MA/My mice. J Immunol. 2005;175:6820–6828. doi: 10.4049/jimmunol.175.10.6820. [DOI] [PubMed] [Google Scholar]; See annotation to [22•].
- 24.Scalzo A.A., Manzur M., Forbes C.A., Brown M.G., Shellam G.R. NK gene complex haplotype variability and host resistance alleles to murine cytomegalovirus in wild mouse populations. Immunol Cell Biol. 2005;83:144–149. doi: 10.1111/j.1440-1711.2005.01311.x. [DOI] [PubMed] [Google Scholar]
- 25.Guma M., Budt M., Saez A., Brckalo T., Hengel H., Angulo A., Lopez-Botet M. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood. 2005;107:3624–3631. doi: 10.1182/blood-2005-09-3682. [DOI] [PubMed] [Google Scholar]
- 26.Robbins S.H., Tessmer M.S., Mikayama T., Brossay L. Expansion and contraction of the NK cell compartment in response to murine cytomegalovirus infection. J Immunol. 2004;173:259–266. doi: 10.4049/jimmunol.173.1.259. [DOI] [PubMed] [Google Scholar]
- 27.Ito M., Maruyama T., Saito N., Koganei S., Yamamoto K., Matsumoto N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med. 2006;203:289–295. doi: 10.1084/jem.20051986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grundemann C., Bauer M., Schweier O., von Oppen N., Lassing U., Saudan P., Becker K.F., Karp K., Hanke T., Bachmann M.F., et al. Cutting edge: identification of E-cadherin as a ligand for the murine killer cell lectin-like receptor G1. J Immunol. 2006;176:1311–1315. doi: 10.4049/jimmunol.176.3.1311. [DOI] [PubMed] [Google Scholar]
- 29.Bernstein H.B., Kinter A.L., Jackson R., Fauci A.S. Neonatal natural killer cells produce chemokines and suppress HIV replication in vitro. AIDS Res Hum Retroviruses. 2004;20:1189–1195. doi: 10.1089/aid.2004.20.1189. [DOI] [PubMed] [Google Scholar]
- 30.Alter G., Malenfant J.M., Delabre R.M., Burgett N.C., Yu X.G., Lichterfeld M., Zaunders J., Altfeld M. Increased natural killer cell activity in viremic HIV-1 infection. J Immunol. 2004;173:5305–5311. doi: 10.4049/jimmunol.173.8.5305. [DOI] [PubMed] [Google Scholar]
- 31.Bonaparte M.I., Barker E. Killing of human immunodeficiency virus-infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood. 2004;104:2087–2094. doi: 10.1182/blood-2004-02-0696. [DOI] [PubMed] [Google Scholar]
- 32.Ward J.P., Bonaparte M.I., Barker E. HLA-C and HLA-E reduce antibody-dependent natural killer cell-mediated cytotoxicity of HIV-infected primary T cell blasts. AIDS. 2004;18:1769–1779. doi: 10.1097/00002030-200409030-00005. [DOI] [PubMed] [Google Scholar]
- 33.Trifilo M.J., Montalto-Morrison C., Stiles L.N., Hurst K.R., Hardison J.L., Manning J.E., Masters P.S., Lane T.E. CXC chemokine ligand 10 controls viral infection in the central nervous system: evidence for a role in innate immune response through recruitment and activation of natural killer cells. J Virol. 2004;78:585–594. doi: 10.1128/JVI.78.2.585-594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warfield K.L., Perkins J.G., Swenson D.L., Deal E.M., Bosio C.M., Aman M.J., Yokoyama W.M., Young H.A., Bavari S. Role of natural killer cells in innate protection against lethal ebola virus infection. J Exp Med. 2004;200:169–179. doi: 10.1084/jem.20032141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevenson M.M., Riley E.M. Innate immunity to malaria. Nat Rev Immunol. 2004;4:169–180. doi: 10.1038/nri1311. [DOI] [PubMed] [Google Scholar]
- 36.Artavanis-Tsakonas K., Riley E.M. Innate immune response to malaria: rapid induction of IFN-γ from human NK cells by live Plasmodium falciparum-infected erythrocytes. J Immunol. 2002;169:2956–2963. doi: 10.4049/jimmunol.169.6.2956. [DOI] [PubMed] [Google Scholar]
- 37•.Korbel D.S., Newman K.C., Almeida C.R., Davis D.M., Riley E.M. Heterogeneous human NK cell responses to Plasmodium falciparum-infected erythrocytes. J Immunol. 2005;175:7466–7473. doi: 10.4049/jimmunol.175.11.7466. [DOI] [PubMed] [Google Scholar]; [36, 37•, 38•, 39] provide evidence that NK cells recognize and respond to malaria-infected erythrocytes by a mechanism that requires monocyte-derived cytokines.
- 38•.Baratin M., Roetynck S., Lepolard C., Falk C., Sawadogo S., Uematsu S., Akira S., Ryffel B., Tiraby J.G., Alexopoulou L., et al. Natural killer cell and macrophage cooperation in MyD88-dependent innate responses to Plasmodium falciparum. Proc Natl Acad Sci USA. 2005;102:14747–14752. doi: 10.1073/pnas.0507355102. [DOI] [PMC free article] [PubMed] [Google Scholar]; See annotation to [37•].
- 39.Artavanis-Tsakonas K., Eleme K., McQueen K.L., Cheng N.W., Parham P., Davis D.M., Riley E.M. Activation of a subset of human NK cells upon contact with Plasmodium falciparum-infected erythrocytes. J Immunol. 2003;171:5396–5405. doi: 10.4049/jimmunol.171.10.5396. [DOI] [PubMed] [Google Scholar]
- 40.Hansen D.S., Evans K.J., D’Ombrain M.C., Bernard N.J., Sexton A.C., Buckingham L., Scalzo A.A., Schofield L. The natural killer complex regulates severe malarial pathogenesis and influences acquired immune responses to Plasmodium berghei ANKA. Infect Immun. 2005;73:2288–2297. doi: 10.1128/IAI.73.4.2288-2297.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lieke T., Graefe S.E., Klauenberg U., Fleischer B., Jacobs T. NK cells contribute to the control of Trypanosoma cruzi infection by killing free parasites by perforin-independent mechanisms. Infect Immun. 2004;72:6817–6825. doi: 10.1128/IAI.72.12.6817-6825.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duthie M.S., Kahn S.J. NK cell activation and protection occur independently of natural killer T cells during Trypanosoma cruzi infection. Int Immunol. 2005;17:607–613. doi: 10.1093/intimm/dxh239. [DOI] [PubMed] [Google Scholar]
- 43.Le-Barillec K., Magalhaes J.G., Corcuff E., Thuizat A., Sansonetti P.J., Phalipon A., Di Santo J.P. Roles for T and NK cells in the innate immune response to Shigella flexneri. J Immunol. 2005;175:1735–1740. doi: 10.4049/jimmunol.175.3.1735. [DOI] [PubMed] [Google Scholar]
- 44.Vankayalapati R., Garg A., Porgador A., Griffith D.E., Klucar P., Safi H., Girard W.M., Cosman D., Spies T., Barnes P.F. Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol. 2005;175:4611–4617. doi: 10.4049/jimmunol.175.7.4611. [DOI] [PubMed] [Google Scholar]
- 45.Junqueira-Kipnis A.P., Kipnis A., Jamieson A., Juarrero M.G., Diefenbach A., Raulet D.H., Turner J., Orme I.M. NK cells respond to pulmonary infection with Mycobacterium tuberculosis, but play a minimal role in protection. J Immunol. 2003;171:6039–6045. doi: 10.4049/jimmunol.171.11.6039. [DOI] [PubMed] [Google Scholar]
- 46.Teixeira H.C., Kaufmann S.H. Role of NK1.1+ cells in experimental listeriosis. NK1+ cells are early IFN-γ producers but impair resistance to Listeria monocytogenes infection. J Immunol. 1994;152:1873–1882. [PubMed] [Google Scholar]
- 47.Berg R.E., Crossley E., Murray S., Forman J. Relative contributions of NK and CD8 T cells to IFN-γ mediated innate immune protection against Listeria monocytogenes. J Immunol. 2005;175:1751–1757. doi: 10.4049/jimmunol.175.3.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Auerbuch V., Brockstedt D.G., Meyer-Morse N., O’Riordan M., Portnoy D.A. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–533. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Connell R.M., Saha S.K., Vaidya S.A., Bruhn K.W., Miranda G.A., Zarnegar B., Perry A.K., Nguyen B.O., Lane T.F., Taniguchi T., et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carrero J.A., Calderon B., Unanue E.R. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. 2004;200:535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamerman J.A., Tchao N.K., Lowell C.A., Lanier L.L. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol. 2005;6:579–586. doi: 10.1038/ni1204. [DOI] [PMC free article] [PubMed] [Google Scholar]