

Abstract
The ‘infectious DNA’ approach, which is based on in vivo transcription of (+)RNA virus genome cDNA cassettes from eukaryotic promoters in transfected cells, became a popular alternative to the classical scheme in the infectious clone methodology. Its use, however, is often limited by the instability of plasmids due to a transcriptional activity of eukaryotic promoters in Escherichia coli resulting in synthesis of products toxic for the bacterial host. Using a highly unstable representative infectious clone of Japanese encephalitis (JE) flavivirus, we tested a new approach in design of such problematic ‘infectious DNA’ constructs, which is based on minimizing unwanted transcription in the bacterial host. A plasmid containing full genome size JE cDNA under control of the minimal cytomegalovirus (CMV) promoter can be propagated in E. coli with growth and stability characteristics similar to that of constructs controlled by the T7 promoter. Transfection of this plasmid into susceptible cells leads to the establishment of a productive infectious cycle. Reinsertion of the CMV enhancer at the 3′-end of the JE cassette substantially increased the specific infectivity without affecting the stability and growth characteristics of the construct. This approach can be useful when stabilization of infectious clones by modification of a viral cDNA cassette is not the feasible or suitable alternative.
Keywords: Flavivirus, Infectious clone, cDNA, Infectious DNA, Japanese encephalitis virus, Minimal CMV promoter, BGHTT, CMV enhancer
1. Introduction
Modern (+)RNA virus studies increasingly rely on the infectious clone methodology (reviewed, Boyer and Haenni, 1994), which allows targeted manipulation of viral genomes. In the classical approach, (+)RNA viruses are recovered from cells transfected with synthetic RNA made by in vitro transcription of infectious clone cDNA templates (Campbell and Pletnev, 2000, Gritsun and Gould, 1995, Kapoor et al., 1995, Polo et al., 1997, Rice et al., 1989, Sumiyoshi et al., 1992). In a layered DNA/RNA approach, also known as ‘infectious DNA’ (Herweijer et al., 1995), (+)RNA viruses are recovered directly after transfection of plasmids carrying viral genome cDNA into susceptible cells. It was first reported for poliovirus (Racaniello and Baltimore, 1981), and has been widely used for plant viruses (reviewed, Boyer and Haenni, 1994). Recently, it was adapted for alphaviruses (reviewed, Schlesinger and Dubensky, 1999). Substantial difficulties, however, were encountered in design of flavivirus ‘infectious DNA’, requiring either modification of the viral genome cassette (Yamshchikov et al., 2001) to prevent unwanted expression of viral genome segments encoding toxic for Escherichia coli products, or deletion of the structural protein region (Varnavski et al., 2000). Substantial stabilization of viral genome cassettes by blocking unwanted expression was also reported for a number of plant viruses (Johansen, 1996, Lopez-Moya and Garcia, 2000, Olsen and Johansen, 2001). In this approach, deleterious effects of spurious transcription from eukaryotic promoters in bacterial cells are eliminated at the translational level by insertion of intron(s) into viral cDNA cassettes. There may be situations, however, when such modification of a viral cassette is not feasible or is not acceptable. For this reason we sought to investigate if the stability of constructs containing an unmodified virus genome cassette can be improved by preventing its deleterious expression at the transcriptional level, i.e. by minimizing spurious transcription from eukaryotic promoters in E. coli.
As a model, we have used a highly unstable infectious clone of Japanese encephalitis flavivirus (JE) (Sumiyoshi et al., 1992, Yamshchikov et al., 2001). JE is a member of the Flavivirus genus of the family Flaviviridae, which also includes yellow fever (YF), West Nile, tick borne encephalitis, dengue, and hepatitis C viruses (Kuno et al., 1998). The flavivirus virion contains single-stranded RNA of positive polarity, which is approximately 11 kb in length and encodes three structural and seven nonstructural proteins (Rice, 1996). Viral RNA is capped at its 5′-end, but has no poly(A) tract at the 3′-end. Both virion RNA and in vitro synthesized capped RNA are infectious when transfected into susceptible cells (Rice et al., 1989). In this report we demonstrate that spurious transcription in E. coli can be substantially reduced by utilizing a minimal eukaryotic promoter (Davis and Huang, 1988) to control transcription of the viral cassette. As an enhancement of this ‘minimal approach’, a substantial increase in the specific infectivity of ‘infectious DNA’ was achieved by placing the enhancer at the 3′-end of the viral genome cassette. While this methodology does not resolve completely the instability problem, it may be considered as an alternative approach when for any reason the viral genome cassette cannot be modified.
2. Materials and methods
2.1. Plasmid construction
DNA manipulations were performed using standard procedures (Sambrook et al., 1989) with commercially available enzymes according to manufacturer's protocols. E. coli strain HB101 was used for cloning and maintenance of recombinant constructs. Sequence analysis of plasmids and of recovered viruses was performed on ABI 310 Genetic Analyzer (Perkin–Elemer) using manufacturer's kits and protocols.
Luciferase encoding plasmids (Fig. 1 A) were derived from pBR322. Plasmids pCMV5′UTRJE/luc and pCMVmp5′UTRJE/luc include the first 69 nucleotides of JE 5′-UTR followed by the CMV promoter or minimal CMV promoter, respectively. Plasmids pCMVmp/luc-3′enhD and pCMVmp/luc-3′enhR were designed by blunt-end cloning of a Bgl II–Pvu I CMV enhancer fragment (from pCIneo; Promega) into Bam HI site at the 3′-end of the luciferase gene in pCMVmp/luc. These plasmids contain the CMV enhancer in the opposite orientations.
Fig. 1.
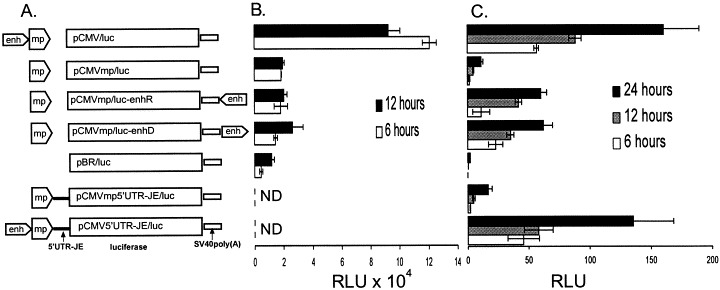
(A) Schematic representations of the plasmids containing the luciferase gene under control of the CMV enhancer/promoter and the minimal CMV promoter. Plasmid maps are not drawn to the scale. CMVenh, CMV enhancer; CMVmp, CMV minimal promoter; 5′UTR-JE, 69 nucleotides of the JE 5′-UTR; SV40poly(A), simian virus 40 (SV40) site of transcription termination/polyadenylation. (B) Expression of the luciferase gene in E. coli DH10B from plasmids shown in panel A. Culture media were diluted 1:50 with the fresh medium and incubated 6 and 12 h. Normalization of luciferase activity was done to amounts of plasmid DNA isolated from 1 ml of culture. (C) Expression of the luciferase gene in BHK cells transfected with plasmids shown in panel A as described in Section 2. Luciferase activity in cell lysates was measured 6, 12 and 24 h post transfection. All samples were run in triplicate. Bars indicate standard deviation.
Plasmids used in the design of the final construct (Fig. 2 ) were described earlier (Yamshchikov et al., 2001). Digestion of pCMVJE5′(943) at the Pvu I (situated between the enhancer and promoter regions in the CMV promoter/enhancer fragment) and Mlu I sites yielded a CMV minimal promoter—JE 5′-end conjoint fragment, which replaced the Sal I–Mlu I fragment in pT7JE5′ resulting in a pCMVmpJE5′ construct. An Eag I–Bam HI CMVmp-JE 5′ half genome fragment from this plasmid was inserted into pJE3′(BGHTT) (Yamshchikov et al., 2001) digested with the same enzymes. The resulting plasmid pCMVmpJE(BGHTT) contained full-length JE genome cDNA under transcriptional control of the CMV minimal promoter and the BGH transcriptional terminator. Plasmids pCMVmpJE(BGHTT)enhD and pCMVmpJE(BGHTT)enhR were derived from pCMVmpJE(BGHTT) by blunt-end ligation of the Bgl II–Pvu I enhancer fragment into the Ssp I site (Fig. 2).
Fig. 2.
Construction of plasmid pCMVmpJE(BGHTT)(enh). (A) Designation of flavivirus proteins is according to Chambers et al. (1990). An asterisk denotes the silent mutation, which appeared during cloning (see text for details). Upper letters indicate relative positions of endonuclease restriction sites used in assembly and analysis of designed constructs: M, Mlu I; B, Bam HI; S, Sal I; P, Pvu I; E, Eag I; A, Asp 718 (KpnI); Ss, Ssp I; T7, T7 promoter, enh, CMV enhancer, mp, CMV minimal promoter, BGHTT, bovine growth hormone transcriptional terminator. The scheme is not drawn exactly to the scale. (B) Sequence at junctions of CMV promoter, 5′- and 3′-ends of the JE genome, and BGHTT.
2.2. Promoter activity assays
Luciferase activity in transfected eukaryotic cells was measured using the Dual-Luciferase reporter assay kit (Promega) according to manufacturer's recommendations. Plasmid pRLnull (Promega) was used for normalization of expression by admixing to luciferase-containing plasmids in proportion 1:10. BHK cells (Bredenbeek et al., 1993) were transfected with plasmid DNA using Superfect (Qiagen) according to the manufacturer. Transfection mix containing 0.275 μg plasmid DNA per well was placed into wells of 24 well plate in triplicate per each time interval. Cells were lysed with 75 μl of the lysis buffer (Promega) at 6, 12 and 24 h post transfection and luciferase activities were quantified as recommended by the manufacturer.
To determine of the CMV promoter activity in bacteria, overnight cultures of E. coli DH10B cells (Gibco-BRL) transformed with plasmids were diluted 1:50 with fresh LB and grown 6 and 12 h. Cells from 1 ml culture were resuspended in 100 μl buffer I (100 mM Tris–HCl, pH 8.0; 100 mM NaCl; 2 mM EDTA) and lysozyme was added to 0.5 μg/ml. After incubation for 1 h at 0 °C, cells were lysed by adding 100 μl of 2× Lysis buffer (Promega). After centrifugation at 14,000 rpm at 4 °C for 10 min, 5 μl of the clear lysate was used for luciferase assay. Normalization of luciferase activity was done to amounts of plasmid DNA isolated from 1 ml of culture. Isolated plasmid DNA was treated with ATP-dependent DNase, RNase A and proteinase K before measuring plasmid concentration using DyNA Quant fluorimeter (Hoefer) according to the manufacturer's protocol.
2.3. Cells culture and viruses
BHK and Vero (ATCC CRL 1586) cells were maintained at 37 °C, in a humidified atmosphere containing 5% CO2 in Dulbecco's Modified Eagle's medium (DMEM) supplemented with 5% fetal calf serum (FCS; HyClone) and 1× Antibiotic–Antimycotic (Gibco-BRL). Wild-type virus JaOArS982 (Sumiyoshi et al., 1987) and attenuated vaccine strain SA14-14-2 (Nitayaphan et al., 1990) were grown in Vero cells.
Virus growth characteristics were determined either after infection of 4×105 BHK cells in 6-well plates with 103 pfu of virus (MOI=0.0025) or after transfection with 1 μg of infectious plasmid. Media of the infected or transfected cells were sampled at various times and assayed for virus titers using BHK cells. For evaluation of the specific infectivity of DNA, BHK cells were transfected with serial 10-fold dilutions of plasmid DNA in the presence of 1 μg carrier DNA (pBR322). In either case, after 1 h for virus adsorption or 3 h for DNA transfection, cells were washed two times with Dulbecco's PBS (DPBS) and overlaid with DMEM supplemented with 1% methylcellulose (Sigma) and 2% FCS (HyClone). Cells were incubated for 3–4 days and stained with 1% crystal violet in 70% methyl alcohol to visualize foci of cytopathology (plaques). All plaque assays were performed in duplicate, and median virus concentration (log pfu/ml) was plotted as function of time. All procedures involving infectious material were performed under the Biological Safety Level 3 containment.
2.4. Detection of viral proteins by indirect immunofluorescence
BHK cells seeded on 12 mm glass coverslips were transfected with DNA–Superfect complexes (0.25 μg of plasmid per 2×105 cells). At daily intervals, cells were fixed in cold methanol–acetic acid (95:5), washed with DPBS containing 1% FCS (DPBS/FCS) and reacted with JE (ATCC VR 1259AF) mouse hyperimmune ascites fluid (at 1:1000 dilution in DPBS/FCS). A goat anti-mouse IgG-fluorescein conjugate (Gibco-BRL) was used as the secondary antibody at 1:200 dilution in DPBS/FCS and cells were observed under fluorescent microscope equipped with a video imaging system.
2.5. Isolation and analysis of RNA
Total cellular RNA from infected and transfected cells was isolated using the RNeasy Mini Kit (Qiagen) with DNase treatment as recommended by the manufacturer. Removal of residual DNA was monitored by PCR of RNA samples using the same primer sets as for RT-PCR assays. Analysis of the 5′-end of JE-specific RNA in transfected and infected cells was performed by primer extension assays (Sambrook et al., 1989) using AMV reverse transcriptase (PanVera) and [32P]-labeled primer JE100R (5′-GTCATGGTTATCTTCCGTTCT-3′), complementary to pos. 80–100 of the JE genome. Primer extension products were analyzed on 6% PAAG containing 7 M urea.
To determine the 5′-end of a primary CMV transcript we used First Choice RLM-RACE kit (Ambion) according to manufacturer's recommendation. Total RNA from cell transfected with pCMV5′UTR-JE/luc was analyzed. Primers luc22R (5′-TTATGTTTTTGGCGTCTTC-3′) and luc87R (5′-GCCTTATGCAGTTGCTCTC-3′) complementary to pos. 4–22 and 69–87 of the luciferase gene were used for RT, amplification and sequencing reaction. The 3′-end of JE specific RNA was determined by using an adapted RACE-PAT assay (Salles et al., 1999). Briefly, total RNA polyadenylated in the presence of polyA polymerase (Gibco-BRL) was reverse transcribed from a dT15-anchor reverse primer (Salles et al., 1999) following by PCR amplification using the same reverse primer and a specific primer corresponding to pos. 10 468–10 485 of the JE genome. Products of RT-PCR were purified using 1% agarose gel and sequenced.
3. Results
3.1. Assessment of the CMV promoter activity in E. coli and BHK cells
Earlier, it has been demonstrated (Johansen, 1996, Lopez-Moya and Garcia, 2000, Olsen and Johansen, 2001, Yamshchikov et al., 2001) that interruption of a viral open reading frame either by frameshift mutations or by insertion of introns leads to substantial stabilization of plasmids containing viral genome cassettes under control of eukaryotic promoters. This suggests that spurious transcription from eukaryotic promoters in E. coli (Antonucci et al., 1989, Davis and Huang, 1988) may substantially contribute to the observed instability of such constructs. As an alternative to the mentioned above modification of viral genome cassettes, we sought to explore if a decrease of such spurious transcription can eliminate the destabilizing effect of eukaryotic promoters on ‘infectious DNA’ constructs. The powerful and versatile CMV promoter, often used to drive expression of engineered constructs in a variety of mammalian cells (Foecking and Hofstetter, 1986), consists of a transcriptionally active minimal promoter region of approximately 100 bp immediately adjacent to the RNA transcription start, and of an about 600 bp upstream enhancer element, which displays only a minute transcriptional activity in mammalian cells (Thomsen et al., 1984). To determine if the enhancer has any effect on the promoter activity in E. coli, we designed plasmids pBR/luc (no promoter), pCMV/luc (full promoter), pCMVmp/luc (no enhancer), and a pair of plasmids pCMVmp/luc-enhD and pCMVmp/luc-enhR containing the enhancer relocated to the end of the reporter gene (Fig. 1A). Comparative evaluation of luciferase production in E. coli transformed with the above plasmids, shown in Fig. 1B, indicated that deletion of the enhancer leads to a 5–6-fold reduction of luciferase gene expression. In contrast to position-independent function of enhancers in mammalian cells (Jonsson et al., 1994), and certain activators in prokaryotic cells (Molina-Lopez and Santero, 1999), the CMV enhancer relocated downstream from the reporter gene had no discernible effect on production of luciferase in E. coli.
We also compared expression of the luciferase gene in BHK cells transfected with the designed plasmids. As indicated in Fig. 1C, luciferase production from the plasmid containing the minimal promoter was 7–10-fold less than from the full promoter construct. In contrast to bacterial expression described above, relocation of the enhancer to the 3′-end of the luciferase gene restored 40–50% of the full CMV promoter activity. Position-dependent modulation of the enhancer activity has been observed (Jonsson et al., 1994).
3.2. Effect of the JE5′UTR on reporter gene expression
Flavivirus genome RNA has a short 5′-untranslated region (5′-UTR), which forms a highly conserved secondary structure (Brinton and Dispoto, 1988). A positive effect of flavivirus 5′-UTR on the translation efficiency of synthetic RNA in cell-free systems has been reported earlier (Preugschat et al., 1990, Ruiz-Linares et al., 1989). Since an enhancing effect of flavivirus 5′-UTR on translation would be beneficial in the devised ‘minimal’ approach, we were interested to determine if such enhancing effect can be observed with JE 5′-UTR in vivo. To address this question, we designed plasmids pCMV5′UTR-JE/luc and pCMVmp5′UTR-JE/luc (Fig. 1A), in which the first 69 nucleotides of JE 5′-UTR have been placed between the luciferase reporter gene and the CMV or minimal CMV promoters, respectively. The effect of 5′-UTR was accessed by measuring luciferase activity in lysates of BHK cells transiently transfected with these plasmids and with plasmids pCMV/luc and pCMVmp/luc (Fig. 1A). Surprisingly, luciferase production from pCMV5′UTR-JE/luc was about 15–20% less than from pCMV/luc, and the presence of JE 5′-UTR had no discernible effect on amounts of luciferase produced from the construct containing the minimal CMV promoter (Fig. 1C). Since for the purposes of the present study we were interested primarily in the activity of the minimal promoter, we did not investigate this question in more detail.
3.3. Construction of infectious DNA
Based on the evidence described above, we attempted to assemble an ‘infectious DNA’ of Japanese encephalitis flavivirus under control of the minimal CMV promoter. Sequence analysis of one isolate of the pCMVJE5′(943) plasmid described earlier (Yamshchikov et al., 2001) revealed a silent substitution at pos. 551 of the JE cDNA fragment resulting in formation of an additional site for restriction endonuclease Sau3A I (GATT → GATC). This mutation has created a convenient marker allowing to distinguish between genomes of the parent and recovered viruses. In contrast to the similar construct controlled by the full CMV promoter (Yamshchikov et al., 2001), a pCMVmpJE5′ plasmid containing 5′-half (1–5576 nt) of the JE genome cDNA controlled by the minimal promoter appeared stable with growth characteristics in E. coli HB101 resembling those of pT7JE5′ (Sumiyoshi et al., 1992, Yamshchikov et al., 2001). Preservation of the open reading frame in the JE fragment was confirmed by its complete sequencing. Similarly, pCMVmpJE(BGHTT) containing full-length JE genome cDNA and the BGH transcriptional terminator (Fig. 2) had the same growth characteristics in E.coli HB101 as the half JE genome-containing plasmid pCMVmpJE5′. To evaluate the stability of the final construct during propagation in E.coli HB101, three independent isolates of pCMVmpJE(BGHTT) were subjected to two transformation-plasmid isolation cycles. Plasmid transformation usually resulted in formation of small uniform colonies, with large colonies observed with a frequency of less than 10−3. Plasmid DNA isolated from small colonies was infectious and had the similar specific infectivity in BHK cells. Plasmids isolated from the large colonies were not infectious and had substantial rearrangements in the viral cDNA cassette. Finally, we have reinserted the enhancer fragment into pCMVmpJE(BGHTT) downstream from the BGHTT fragment. In agreement with the evidence described above, resulting plasmids pCMVmpJE(BGHTT)enhD and pCMVmpJE(BGHTT)enhR had the same stability and growth characteristics as the parent plasmid.
3.4. Virus recovery from plasmid DNA
After transfection of BHK with the full-length pCMVmpJE(BGHTT) construct, individually stained cells were observed on the 1st day after transfection and positive cells progressively increased in number from the 1st to the 4th day after transfection. By day 4 all cells became JE-positive, indicating formation of the virus and the spread of infection. Similar result was obtained for the plasmids pCMVmpJE(BGHTT)enhD and pCMVmpJE(BGHTT)enhR. The specific infectivity of plasmid pCMVmpJE(BGHTT) appeared dependent on a transfection agent and transfection protocol used, and was in the range of 102–103.5 pfu/μg. The presence of the CMV enhancer downstream from BGHTT led to an increase in the specific infectivity to 1–5×105 pfu/μg, which became comparable to that of the full CMV-controlled intron-containing construct pCMVp/eJE(i356i2217)BGHTT reported earlier (Yamshchikov et al., 2001)
Cytopathic effects resembling those resulting from infection of BHK cells with the parent strain JaOArS982 were reproducibly observed 3–4 days after exposure of BHK monolayers to DNase treated media from cells transfected with pCMVmpJE(BGHTT), indicating the presence of a DNase-insensitive infectious agent. The time course of virus accumulation in the media was determined at 12-h intervals after transfection of BHK cells with 1 μg of the plasmid pCMVmpJE(BGHTT). Virus was undetectable in the media during 24 h after transfection of BHK cells (Fig. 3 ). The virus recovered from DNA had the same growth characteristics in BHK cells as the parent JaOArS982 virus, with titers reaching 106.0–6.5 pfu/ml, and producing plaques on monolayers of BHK cells similar by the size and morphology to plaques produced by the parent virus (Fig. 4 ). Plaques produced by either virus (turbid) were distinct from plaques produced by attenuated virus SA14-14-2 (Fig. 4), which produced clear plaques.
Fig. 3.
Time course of virus accumulation after transfection of BHK cells (4×105) with pCMVmpJE(BGHTT) and comparison of virus growth characteristics. Both the parent and recovered viruses were used at MOI=0.0025.
Fig. 4.

Plaque morphology of parent virus JaOArS982, the virus recovered from infectious DNA, and attenuated virus SA14-14-2. After infection, BHK cells were overlaid with 1% methylcellulose. Plaques were visualized with crystal violet after 4 days.
3.5. Molecular characterization of the recovered virus
To verify that virus was recovered from the recombinant DNA, we examined the presence of the T → C mutation at genome pos. 551, which is characteristic for the pCMVmpJE(BGHTT) construct. Total RNA isolated from cells, infected with either the parent virus JaOArS982 or with the virus recovered from cells transfected with pCMVmpJE(BGHTT), was reverse transcribed using a reverse primer complementary to pos. 2306–2330 of the JE genome, followed by PCR amplification of a 780 bp fragment with a pair of primers corresponding to pos. 1–25 and 759–780 of the JE genome and digestion with Sau3A I. The additional Sau3A I site formed as result of the above mutation was present only in the fragment amplified from RNA of the recovered virus, but not in the fragment originating from parent virus RNA (results not shown). This result confirmed that the recovered virus has originated from the plasmid pCMVmpJE(BGHTT); this conclusion was confirmed also by complete sequence analysis of JE-specific RNA isolated from cells infected with the recovered virus.
The 5′-end of the recovered virus genome was analyzed by primer extension of RNA isolated from cells infected with either the parent virus JaOArS982 or with the virus recovered from DNA. Synthetic control RNA with the 5′-end authentic to JE genomic RNA was prepared by in vitro transcription of pT7JE5′ (Sumiyoshi et al., 1992, Yamshchikov et al., 2001) with T7 polymerase. As shown in Fig. 5 , extension products for both viruses demonstrated the same electrophoretic mobility (compare lanes 2 and 3), and both appeared one nucleotide longer than the extension product obtained from the uncapped synthetic transcript (lane 1). This likely resulted from reverse transcription of the 5′-cap structure, which is present in viral genomic RNA. We did not investigate this in more detail. Both viral genomes had identical 3′-ends (data not shown).
Fig. 5.
Primer extension analysis of JE specific RNA with the 5′-[32P]-labeled primer. The sequence ladder GATC was prepared with the same primer and pCMVmpJE(BGHTT). The corresponding sequence is given on the left with the JE genome-specific sequence shown in capital letters. Lane 1, uncapped RNA transcript synthesized from pT7JE5′; lane 2, extension products synthesized from JaOAr982-specific RNA; lane 3, extension products synthesized from recovered virus-specific RNA.
4. Discussion
Eukaryotic promoters have been successfully used for the design of ‘infectious DNA’ of several animal (+)RNA viruses, such as poliovirus (Semler et al., 1984), FMDV (Beard et al., 1999), alphaviruses (reviewed, Schlesinger and Dubensky, 1999), coronavirus (Almazan et al., 2000), as well as plant viruses (reviewed, Boyer and Haenni, 1994, Johansen, 1996, Lopez-Moya and Garcia, 2000). Design of flavivirus ‘infectious DNA’, however, has proven to be a more challenging task, perhaps due to the well known instability of plasmids carrying flavivirus genome cDNA, which is only exacerbated by the transcriptional activity of eukaryotic promoters in E. coli (Antonucci et al., 1989, Davis and Huang, 1988).
Earlier we have reported design of flavivirus ‘infectious DNA’ stabilized by modification of the viral genome cDNA cassette (Yamshchikov et al., 2001). Our attempts to assemble full JE genome cDNA under control of the CMV promoter were unsuccessful; a half genome cassette containing JE structural proteins was also extremely unstable. Similar instability problems were encountered with JE constructs driven by the Rous sarcoma virus (RSV) long terminal repeat. This may have resulted from expression of viral genome segments encoding hydrophobic protein domains, which are characteristic for membrane-embedded structural proteins of enveloped viruses and are toxic for E. coli (Lopez-Moya and Garcia, 2000). A stable Kunjin replicon construct, which did not include genes of the structural proteins, has been reported (Varnavski et al., 2000). The intron-based approach has been successfully used also for stabilization of ‘infectious DNA’ for a number of ‘difficult’ viruses (Johansen, 1996, Lopez-Moya and Garcia, 2000, Olsen and Johansen, 2001). However, since modification of viral genome cassettes may not always be desirable or feasible, we have explored other options allowing use of unmodified viral genome cassettes.
Because transcription from eukaryotic promoters in E. coli (Antonucci et al., 1989, Davis and Huang, 1988) appears to be a major destabilization factor, we have attempted to reduce its deleterious effects by manipulating the promoter to minimize its activity in bacteria. In experiments with luciferase reporter constructs, the minimal CMV promoter was 5–6 times less active than the full CMV promoter both in E. coli and in BHK cells. We expected that while in eukaryotic cells ensuing replication of viral RNA will compensate for the decrease in the transcription rate, substantial reduction in the spurious transcription from the promoter fragment will be beneficial for stabilization of the plasmid during propagation in E. coli. Indeed, replacement of the T7 promoter in pT7JE/Kpn infectious clone (Yamshchikov et al., 2001) with the minimal CMV promoter did not result in destabilization of the plasmid or in change of its growth characteristics. This suggests that in E. coli a substantial part of the spurious transcriptional activity was associated with the enhancer, since its exclusion enabled us to obtain a construct reasonably stable in E. coli with the frequency of stabilizing mutations less than 10−3. Using indirect immunofluorescence, we detected synthesis of virus-specific proteins in cells transfected with the full genome constructs; however we did not detect virus-specific proteins when cells were transfected with the half-genome construct pCMVmpJE5′. This is probably indicative of a low transport rate through nuclear membrane of non-polyadenylated RNA or/and a low transcription rate from the minimal promoter in vivo, which may also explain the substantially lower specific infectivity of the DNA construct driven by such promoter.
Certain regulatory elements of the eukaryotic genes, such as enhancers, can influence gene expression in position- and orientation-independent manner (Jonsson et al., 1994). We reasoned that such capability would be beneficial in our system, since we could increase transcription rate in mammalian cells without affecting the stability of constructs in E. coli. Indeed, plasmids containing the luciferase reporter gene under control of the minimal CMV promoter and the enhancer inserted at the end of the gene directed increased production of luciferase in BHK cells, i.e. only 40–50% of that directed by the full promoter, and expression levels did not depend on the enhancer orientation. In contrast, no effect on luciferase production was observed in E. coli transformed with these plasmids. Similarly, insertion of the CMV enhancer into pCMVmpJE(BGHTT) after the polyadenylation signal resulted in a 10–100-fold increase in the specific infectivity as compared to the parent plasmid. In our hands, the specific infectivity of the designed DNA construct reached 5×105 pfu/μg and became comparable to the specific infectivity of the intron-stabilized plasmid (106 pfu/μg; Yamshchikov et al., 2001). Yet, no substantial difference in the stability or in the growth characteristics of the resulting plasmid has been observed in E. coli.
In this study we have demonstrated an alternative ‘minimal’ approach useful in design of problematic ‘infectious DNA’ constructs of (+)RNA viruses, which is based on manipulation of eukaryotic promoter with the goal to reduce its spurious transcription in E. coli. While this approach cannot resolve instability problems associated with the presence in viral genome cDNA prokaryotic promoter-like elements (Johansen, 1996, Lopez-Moya and Garcia, 2000, Olsen and Johansen, 2001), it simplifies design of ‘infectious DNA’ constructs, in which spurious transcription from an eukaryotic promoter fragment does serve as the major destabilizing factor. Together with the intron-based stabilization approach, it provides a certain flexibility and a choice in the design strategy. The availability of more convenient flavivirus ‘infectious DNA’ should facilitate advances in studies on mechanisms of flavivirus replication and in design of novel flavivirus vaccines.
Acknowledgements
We thank Dr S. Schlesinger for BHK-21 cells, Dr D. Trent for pM343 and pH756 plasmids, and Jennifer Ascaño for help in manuscript preparation. This study was supported by Research Award V22/181/128 to V.F.Y. from the World Health Organization.
References
- Almazan F., Gonzalez J.M., Penzes Z., Izeta A., Calvo E., Plana-Duran J., Enjuanes L. From the cover: engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA. 2000;97:5516–5521. doi: 10.1073/pnas.97.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonucci T.K., Wen P., Rutter W.J. Eukaryotic promoters drive gene expression in Escherichia coli. J. Biol. Chem. 1989;264:17 656–17 659. [PubMed] [Google Scholar]
- Beard C., Ward G., Rieder E., Chinsangaram J., Grubman M.J., Mason P.W. Development of DNA vaccines for foot-and-mouth disease, evaluation of vaccines encoding replicating and non-replicating nucleic acids in swine. J. Biotechnol. 1999;73:243–249. doi: 10.1016/s0168-1656(99)00142-x. [DOI] [PubMed] [Google Scholar]
- Boyer J.C., Haenni A.L. Infectious transcripts and cDNA clones of RNA viruses. Virology. 1994;198:415–426. doi: 10.1006/viro.1994.1053. [DOI] [PubMed] [Google Scholar]
- Bredenbeek P.J., Frolov I., Rice C.M., Schlesinger S. Sindbis virus expression vectors: packaging of RNA replicons by using defective helper RNAs. J. Virol. 1993;67:6439–6446. doi: 10.1128/jvi.67.11.6439-6446.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton M.A., Dispoto J.H. Sequence and secondary structure analysis of the 5′-terminal region of flavivirus genome RNA. Virology. 1988;162:290–299. doi: 10.1016/0042-6822(88)90468-0. [DOI] [PubMed] [Google Scholar]
- Campbell M.S., Pletnev A.G. Infectious cDNA clones of langat tick-borne flavivirus that differ from their parent in peripheral neurovirulence. Virology. 2000;269:225–237. doi: 10.1006/viro.2000.0220. [DOI] [PubMed] [Google Scholar]
- Chambers T.J., Hahn C.S., Galler R., Rice C.M. Flavivirus genome organization, expression, and replication. Ann. Rev. Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- Davis M.G., Huang E.S. Transfer and expression of plasmids containing human cytomegalovirus immediate-early gene 1 promoter–enhancer sequences in eukaryotic and prokaryotic cells. Biotechnol. Appl. Biochem. 1988;10:6–12. [PubMed] [Google Scholar]
- Foecking M.K., Hofstetter H. Powerful and versatile enhancer–promoter unit for mammalian expression vectors. Gene. 1986;45:101–105. doi: 10.1016/0378-1119(86)90137-x. [DOI] [PubMed] [Google Scholar]
- Gritsun T.S., Gould E.A. Infectious transcripts of tick-borne encephalitis virus, generated in days by RT-PCR. Virology. 1995;214:611–618. doi: 10.1006/viro.1995.0072. [DOI] [PubMed] [Google Scholar]
- Herweijer H., Latendresse J.S., Williams P., Zhang G., Danko I., Schlesinger S., Wolff J.A. A plasmid-based self-amplifying Sindbis virus vector. Hum. Gene Ther. 1995;6:1161–1167. doi: 10.1089/hum.1995.6.9-1161. [DOI] [PubMed] [Google Scholar]
- Johansen I.E. Intron insertion facilitates amplification of cloned virus cDNA in Escherichia coli while biological activity is reestablished after transcription in vivo. Proc. Natl. Acad. Sci. USA. 1996;93:12 400–12 405. doi: 10.1073/pnas.93.22.12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson J.J., Converse A., McIvor R.S. An enhancer in the first intron of the human purine nucleoside phosphorylase-encoding gene. Gene. 1994;140:187–193. doi: 10.1016/0378-1119(94)90543-6. [DOI] [PubMed] [Google Scholar]
- Kapoor M., Zhang L., Mohan P.M., Padmanabhan R. Synthesis and characterization of an infectious dengue virus type-2 RNA genome (New Guinea C strain) Gene. 1995;162:175–180. doi: 10.1016/0378-1119(95)00332-z. [DOI] [PubMed] [Google Scholar]
- Kuno G., Chang G.J., Tsuchiya K.R., Karabatsos N., Cropp C.B. Phylogeny of the genus Flavivirus. J. Virol. 1998;72:73–83. doi: 10.1128/jvi.72.1.73-83.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Moya J.J., Garcia J.A. Construction of a stable and highly infectious intron-containing cDNA clone of plum pox potyvirus and its use to infect plants by particle bombardment. Virus Res. 2000;68:99–107. doi: 10.1016/s0168-1702(00)00161-1. [DOI] [PubMed] [Google Scholar]
- Molina-Lopez J.A., Santero E. An artificial enhancer with multiple response elements stimulates prokaryotic transcriptional activation medicated by various regulatory proteins. Mol. Gen. Genet. 1999;262:291–301. doi: 10.1007/s004380051086. [DOI] [PubMed] [Google Scholar]
- Nitayaphan S., Grant J.A., Chang G.J., Trent D.W. Nucleotide sequence of the virulent SA-14 strain of Japanese encephalitis virus and its attenuated vaccine derivative, SA-14-14-2. Virology. 1990;177:541–552. doi: 10.1016/0042-6822(90)90519-w. [DOI] [PubMed] [Google Scholar]
- Olsen B.S., Johansen I.E. Nucleotide sequence and infectious cDNA clone of the L1 isolate of Pea seed-borne mosaic potyvirus. Arch. Virol. 2001;146:15–25. doi: 10.1007/s007050170187. [DOI] [PubMed] [Google Scholar]
- Polo S., Ketner G., Levis R., Falgout B. Infectious RNA transcripts from full-length dengue virus type 2 cDNA clones made in yeast. J. Virol. 1997;71:5366–5374. doi: 10.1128/jvi.71.7.5366-5374.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preugschat F., Yao C.W., Strauss J.H. In vitro processing of dengue virus type 2 nonstructural proteins NS2A, NS2B, and NS3. J. Virol. 1990;64:4364–4374. doi: 10.1128/jvi.64.9.4364-4374.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racaniello V.R., Baltimore D. Cloned poliovirus complementary DNA is infectious in mammalian cells. Science. 1981;214:916–919. doi: 10.1126/science.6272391. [DOI] [PubMed] [Google Scholar]
- Rice C.M. Flaviviridae: the viruses and their replication. In: Fields B.N., Knipe D.M., Howley P.M., Chanock R.M., Melnick J.L., Monath T.P., Roizman B., Strauss S.E., editors. 3rd edition. Vol. 1. Lippincott-Raven; Philadelphia: 1996. pp. 931–960. (Fields Virology). [Google Scholar]
- Rice C.M., Grakoui A., Galler R., Chambers T.J. Transcription of infectious yellow fever RNA from full-length cDNA templates produced by in vitro ligation. New Biol. 1989;1:285–296. [PubMed] [Google Scholar]
- Ruiz-Linares A., Bouloy M., Girard M., Cahour A. Modulations of the in vitro translational efficiencies of Yellow Fever virus mRNAs: interactions between coding and noncoding regions. Nucl. Acids Res. 1989;17:2463–2476. doi: 10.1093/nar/17.7.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salles F.J., Richards W.G., Strickland S. Assaying the polyadenylation state of mRNAs. Methods. 1999;17:38–45. doi: 10.1006/meth.1998.0705. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd edition. Cold Spring Harbor Laboratory Press; New York: 1989. [Google Scholar]
- Schlesinger S., Dubensky T.W. Alphavirus vectors for gene expression and vaccines. Curr. Opin. Biotechnol. 1999;10:434–439. doi: 10.1016/s0958-1669(99)00006-3. [DOI] [PubMed] [Google Scholar]
- Semler B.L., Dorner A.J., Wimmer E. Production of infectious poliovirus from cloned cDNA is dramatically increased by SV40 transcription and replication signals. Nucl. Acids Res. 1984;12:5123–5141. doi: 10.1093/nar/12.12.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumiyoshi H., Hoke C.H., Trent D.W. Infectious Japanese encephalitis virus RNA can be synthesized from in vitro-ligated cDNA templates. J. Virol. 1992;66:5425–5431. doi: 10.1128/jvi.66.9.5425-5431.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumiyoshi H., Mori C., Fuke I., Morita K., Kuhara S., Kondou J., Kikuchi Y., Nagamatu H., Igarashi A. Complete nucleotide sequence of the Japanese encephalitis virus genome RNA. Virology. 1987;161:497–510. doi: 10.1016/0042-6822(87)90144-9. [DOI] [PubMed] [Google Scholar]
- Thomsen D.R., Stenberg R.M., Goins W.F., Stinski M.F. Promoter-regulatory region of the major immediate early gene of human cytomegalovirus. Proc. Natl. Acad. Sci. USA. 1984;81:659–663. doi: 10.1073/pnas.81.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnavski A.N., Young P.R., Khromykh A.A. Stable high-level expression of heterologous genes In vitro and In vivo by noncytopathic DNA-based kunjin virus replicon vectors. J. Virol. 2000;74:4394–4403. doi: 10.1128/jvi.74.9.4394-4403.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamshchikov V., Mishin V., Cominelli F. A new strategy in design of +RNA virus infectious clones enabling their stable propagation in E. coli. Virology. 2001;281:272–280. doi: 10.1006/viro.2000.0793. [DOI] [PubMed] [Google Scholar]