

Graphical abstract
Keywords: Unsaturated acyclic pyrimidine nucleoside analogues, Z- and E-isomers, Cytostatic evaluations, Anti-HIV activity
Abstract
A series of the novel C-5 alkynyl pyrimidine nucleoside analogues (1–14) in which the sugar moiety was replaced by the conformationally restricted Z- and E-2-butenyl spacer between the phthalimido and pyrimidine ring were synthesized by using Sonogashira cross-coupling reaction. Cytostatic activity evaluation of the novel compounds showed that E-isomers exhibited, in general, better cytostatic activities than the corresponding Z-isomers. E-isomer 14 exhibited the best cytostatic effect against all evaluated malignant cell lines, particularly against hepatocellular carcinoma (Hep G2, IC50 = 4.3 μM). However, this compound was also cytotoxic to human normal fibroblasts (WI 38). Its Z-isomer 7 showed highly specific antiproliferative activity against Hep G2 (IC50 = 18 μM) and no cytotoxicity to WI 38. Moreover, compounds 3, 4 and 14 expressed some marginal inhibitory activity against HIV-1 and HIV-2.
1. Introduction
Many nucleoside analogues with interesting biological properties have arisen by substitution at the 5-position of the uracil base in the 2′-deoxyuridine series.1 Pyrimidine nucleosides containing C-5 alkynyl groups have been shown to possess significant antiviral and/or anticancer properties.2 The 5-alkynyluracil nucleosides with a longer alkynyl chain at the C-5 position expressed appreciable antiviral activity in contrast to the corresponding alkyl derivatives that showed decreasing antiviral activity with increasing C-5 side chain length.2, 3, 4, 5 Introduction of very rigid allenic moiety as a linker between the heterocyclic base and hydroxymethyl group led to compounds such as adenallene and cytallene which effectively inhibited the replication of HIV.6 Furthermore, thymine with a 2-butenyl spacer was the first acyclic nucleoside analogue exhibiting potent inhibition of thymidine kinase 2 (TK-2) which catalyzes phosphorylation of certain antiviral drugs.7
We have reported that some pyrimidines and purines containing γ-(Z)-ethylidene-2,3-dibenzylbutenolide exhibited antiproliferative effects against malignant human tumour cell lines.8, 9, 10 The C-5 alkynyl substituted pyrimidine derivatives of l-ascorbic acid had some, although slight, inhibitory potency against cytomegalovirus (CMV) and varicella-zoster virus (VZV).11 The 5-propynyl uracil derivative of l-ascorbic acid showed pronounced cytostatic activity against cervical carcinoma (HeLa).12 We have also reported that the (Z)-4-amino-2-butenyladenine nucleoside analogue showed selective activity against HIV-1.13 These results prompted us to synthesize a series of the novel (Z)- and (E)-2-butenyl-N-phthalimido pyrimidines containing an alkynyl longer side chain at C-5 (1–14) (Fig. 1 ) and evaluate their cytostatic and anti-HIV potency in cell culture.
Figure 1.
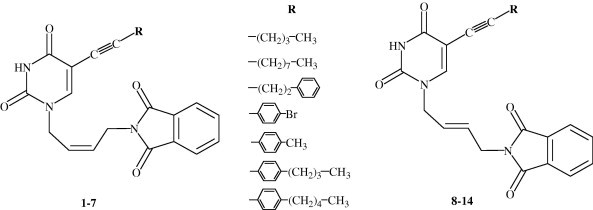
Unsaturated acyclic C-5 alkynyl substituted pyrimidine nucleoside analogues 1–14.
2. Results and discussion
2.1. Compounds preparation
The key precursors (Z)- (IIa) and (E)-1-[4′-(N-phthalimido)-2′-butenyl]-5-iodouracil (IIb) for cross-coupling with alkynes were synthesized by condensation of the (Z)-4-chloro-2-butenyl-N-phthalimide (Ia) and (E)-4-bromo-2-butenyl-N-phthalimide (Ib) with 5-iodouracil.13 The (Z)-4-chloro-2-butenyl (Ia) and (E)-4-bromo-2-butenyl (Ib) derivatives of N-phthalimide were synthesized by using a classical Gabriel reaction.14 Different substituents at C-5 position of the pyrimidine ring in 1–7 and 8–14 were introduced by the reaction of the 5-iodouracil derivatives IIa and IIb under optimized Sonogashira Pd(0)-catalyzed reactions.15, 16, 17, 18, 19, 20 It is important to note that this reaction was performed at room temperature in order to avoid the formation of bicyclic furopyrimidine co-products produced by heating the reaction mixture. Reaction of the (Z)- and (E)-1-[4′-(N-phthalimido)-2′-butenyl]-5-iodouracil with various terminal alkynes in the presence of palladium(0) catalyst and copper(I) iodide as co-catalyst in DMF at room temperature gave 5-substituted Z- and E-uracil derivatives 1–7 and 8–14 (Scheme 1 ).
Scheme 1.
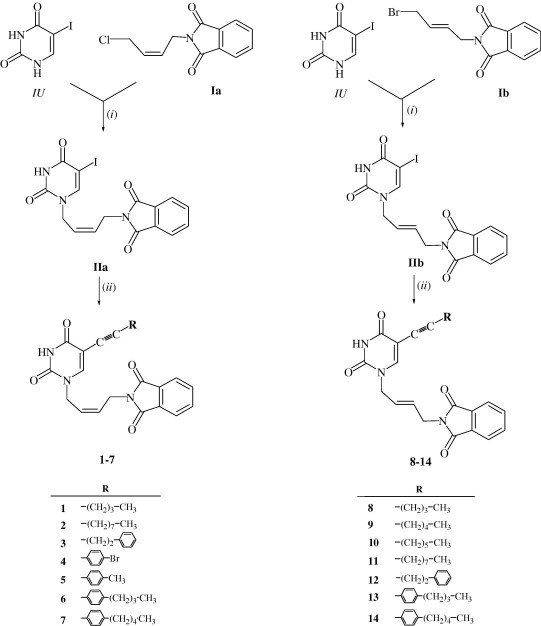
Synthesis of the unsaturated acyclic C-5 substituted pyrimidine nucleoside analogues. Reagents and conditions: (i) NaH, DMF, argon atmosphere, rt; (ii) R-C CH, i-Pr2EtN, (PPh3)4Pd, CuI, DMF, rt.
2.2. 1H and 13C NMR studies
1H and 13C NMR data given in Table 1 and Section 5 are in full agreement with the proposed structures 1–14 (Fig. 2 ).
Table 1.
1H NMR chemical shifts (δ/ppm) and H–H coupling constants (J/Hz) of Z- and E-nucleoside analogues (1–7 and 8–14)
| Compound | CH3 | H4′ | H1′ | H2′ | H3′ | H3″ | ϕH2,H3,H4 | Phth H4,H5 | H6 | H3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.90 (t, J = 7.1) | 4.38 (m) | 4.6 (m) | 5.5–5.7 (m) | 2.40 (t, J = 6.7) | — | 7.8–7.9 (m) | 8.07 (s) | 11.67 (s) | — | |
| 2 | 0.84 (t, J = 6.7) | 4.36 (d, J = 6.6) | 4.51 (d, J = 7.0) | 5.60 (td, J = 7.0; 10.6) | 5.63 (td, J = 6.6; 10.6) | 2.36 (t, J = 6.7) | — | 7.8–7.9 (m) | 7.96 (s) | 11.55 (s) | 10.6 |
| 3 | — | 4.37 (d, J = 7.1) | 4.52 (d, J = 6.9) | 5.61 (td, J = 6.9; 10.6) | 5.65 (td, J = 7.1; 10.6) | 2.65 (m) | 7.2–7.3 (m) | 7.8–7.9 (m) | 7.94 (s) | 11.57 (s) | 10.6 |
| 4 | — | 4.40 (d, J = 5.3) | 4.56 (d, J = 4.7) | 5.6–5.7 (m) | — | 7.53 (d, J = 8.5) | 7.8–7.9 (m) | 8.24 (s) | 11.73 (s) | — | |
| 7.42 (d, J = 8.6) | |||||||||||
| 5 | 1.23 (s) | 4.40 (m) | 4.54 (m) | 5.7–5.8 (m) | — | 7.3–7.4 (m) | 7.8–7.9 (m) | 8.27 (s) | 11.43 (s) | — | |
| 6 | 0.81 (t, J = 7.4) | 4.31 (d, J = 5.3) | 4.48 (d, J = 5.4) | 5.6–5.7 (m) | — | 7.30 (d, J = 8) | 7.8–7.9 (m) | 8.11 (s) | 11.60 (s) | — | |
| 7.16 (d, J = 8) | |||||||||||
| 7 | 0.84 (t, J = 6.7) | 4.39 (d, J = 6.8) | 4.56 (d, J = 6.8) | 5.64 (td, J = 6.8; 11.1) | 5.66 (td, J = 6.8; 11.1) | — | 7.23 (m) | 7.85 (m) | 8.18 (s) | 11.69 (s) | 11.1 |
| 7.38 (m) | |||||||||||
| 8 | 0.87 (t, J = 7.0) | 4.17 (d, J = 5.0) | 4.28 (d, J = 6.0) | 5.66 (td, J = 6.0; 15.7) | 5.73 (td, J = 5.0; 15.7) | 2.35 (t, J = 6.7) | — | 7.8–7.9 (m) | 7.91 (s) | 11.33 (s) | 15.7 |
| 9 | 0.86 (t, J = 7.0) | 4.15 (m) | 4.30 (m) | 5.6–5.7 (m) | 2.34 (t, J = 6.9) | — | 7.8–7.9 (m) | 7.91 (s) | 11.41 (s) | — | |
| 10 | 0.84 (m) | 4.14 (m) | 4.52 (m) | 5.6–5.7 (m) | 2.68 (m) | — | 7.8–7.9 (m) | 8.47 (s) | 11.50 (s) | — | |
| 11 | 0.85 (s) | 4.15 (m) | 4.30 (m) | 5.6–5.7 (m) | 2.34 (t, J = 6.8) | — | 7.8–7.9 (m) | 7.91 (s) | 11.33 (s) | — | |
| 12 | — | 4.18 (d, J = 5.1) | 4.24 (d, J = 5.5) | 5.66 (td, J = 5.5; 15.5) | 5.72 (td, J = 5.1; 15.5) | 2.63 (m) | 7.1–7.3 (m) | 7.8–7.9 (m) | 7.82 (s) | 11.52 (s) | 15.5 |
| 13 | 0.78 (t, J = 7.3) | 4.09 (d, J = 4.9) | 4.18 (d, J = 5.1) | 5.6–5.7 (m) | — | 7.25 (d, J = 7.9) | 7.7–7.8 (m) | 7.97 (s) | 11.55 (s) | — | |
| 7.12 (d, J = 7.9) | |||||||||||
| 14 | 0.85 (t, J = 6.9) | 4.19 (d, J = 5.5) | 4.28 (d, J = 5.7) | 5.69 (td, J = 5.7; 15.7) | 5.75 (td, J = 5.5; 15.7) | — | 7.35 (m) | 7.8–7.9 (m) | 8.07 (s) | 11.64 (s) | 15.7 |
| 7.22 (m) | |||||||||||
Additional data: 1: H4″ and H5″ (1.4–1.5, m); 2: H4″–H9″ (1.2–1.5, m); 3: H4″ (2.81, m); 6: H1‴ (2.52, t, J = 7.7), H2‴ (1.47, p, J = 7.6), H3‴ (1.22, hex, J = 7.3); 7: H4‴ (1.28, m), H3‴ (1.27, m), H2‴ (1.56, m), H1‴ (2.58, t, J = 7.6); 8: H5″ (1.42, m), H4″ (1.45, m); 9: H4″–H6″ (1.1–1.5, m); 10: H4″–H7″ (1.2–1.5, m); 11: H4″–H9″ (1.2–1.4, m); 12: H4″ (2.79, m); 13: H1‴ (2.48, t, J = 7.6), H2‴ (1.44, p, J = 7.5), H3‴ (1.18, hex, J = 7.3); 14: H3‴ (1.26, m), H4‴ (1.32, m), H2‴ (1.56, m), H1‴ (2.58, t, J = 7.6).
Figure 2.
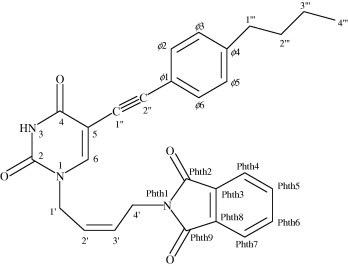
Atom numbering given in Table 1 is exemplified on compound 6. Note that chemical shifts are equal for ϕ2 and ϕ6, ϕ3 and ϕ5, Phth2 and Phth9, Phth3 and Phth 8, Phth 4 and Phth7, Phth5 and Phth6 due to symmetry.
Signals of 1H and 13C resonances were observed which were characteristic for each of the moieties constituting structures of N1, C5-substituted pyrimidine nucleoside analogues (1–14). H1′ and H4′ protons exhibit distinct chemical shifts between 4.09 and 4.60 ppm, whereas H2′ and H3′ protons along C C double bond were observed between 5.50 and 5.80 ppm (Table 1). Chemical shifts of phenyl protons are between 7.1 and 7.53 ppm, while phthalimido protons are between 7.8 and 7.9 ppm. H6 chemical shifts were found in the range from 7.82 to 8.47 ppm and H3 protons in the range from 11.33 to 11.73 ppm. The configuration along C2′ C3′ double bond was evaluated through coupling constants (Table 1). The values of all coupling constants involved in the six spin system of N1-substituent including had to be determined through simulation due to small Δδ/J ratios. As an example of our simulations, multiplets of H2′ and H3′ in 14 are compared to the experimental spectrum in Figure 3 . Vinyl and couplings were below signal half width (<1 Hz). Smaller coupling constants of 10.6, 10.6 and 11.1 Hz in 2, 3 and 7, respectively, are in accordance with Z-isomers along C2′ C3′ double bonds. On the other hand, larger coupling constants of 15.7, 15.5 and 15.7 Hz in 8, 12 and 14, respectively, are in conformity with E-isomers.
Figure 3.
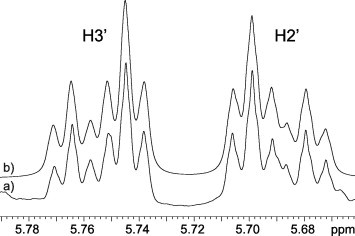
Part of 1H NMR spectra of 14 showing H3′ and H2′ multiplets. (a) Experimental spectrum acquired in DMSO-d6 at 298 K; (b) simulated spectrum (A2BCD2 spin system).
E-Isomers exhibit larger chemical shift difference between H2′ and H3′ (Δδ 0.06–0.07 ppm) than Z-isomers (Δδ 0.02–0.04 ppm). The other significant 1H and 13C chemical shift differences between E- and Z-isomers were observed, and are presented in Figure 4 . Perusal of Figure 4 shows that there is a clear distinction in several 1H and 13C chemical shifts with respect to Z- and E-configurations along the C2′ C3′ double bond.
Figure 4.
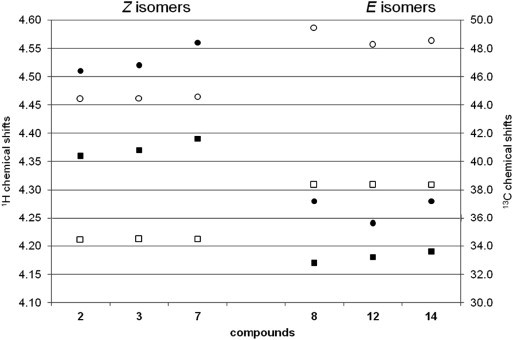
Comparison of 1H and 13C chemical shift in Z- and E-isomers. 1H and 13C chemical shifts axes are indicated on the left and the right side of the plot, respectively. 1H and 13C chemical shifts are labelled with filled (•H1′, ■ H4′) and blank symbols (○ C1′, □ C4′), respectively.
In our further analysis, we have focused on conformational properties of novel pyrimidine-2,4-dione nucleoside analogues. and coupling constants showed unrestricted rotation along the involved single bonds. In full agreement, protons of both methylene groups are isochronous. Evaluation of NOE enhancements between H6 and protons of N1 substituents did not show any interaction that would indicate preference for any of conformational states. In addition, NOE data did not indicate close spatial proximity of moieties attached to N1 and C5 of pyrimidine-2,4-dione.
3. Biological results
3.1. Cytostatic activity
Compounds 1–14 were evaluated for their cytostatic activities against malignant human tumour cell lines: murine leukemia (L1210), human T-lymphocyte (Molt4/C8 and CEM), cervical carcinoma (HeLa), pancreatic carcinoma (MiaPaCa-2), colorectal adenocarcinoma (SW 620), breast epithelial adenocarcinoma (MCF-7) and hepatocellular carcinoma (Hep G2), as well as normal diploid human fibroblasts (WI 38) (Table 2 ). Cytostatic activities of compounds 1–14 were compared with those of 5-fluorouracil (5-FU). The novel C-5 alkynyl substituted olefinic pyrimidine compounds (1–14) showed better antiproliferative activities on the malignant tumour cells than their structurally related C-5 unsubstituted uracil and C-5 fluoro-substituted uracil derivatives containing 4′-(N-phthalimido)-2′-butenyl side chain.13
Table 2.
Inhibitory effects of compounds 1–14 on the growth of malignant tumour cell lines
| Compound | IC50a (μM) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| L1210 | Molt4/C8 | CEM | HeLa | MCF-7 | MiaPaCa-2 | SW 620 | Hep G2 | WI 38 | |
| 1 | 109 | 41 | 100 | 33 | 45 | >100 | 82 | 17 | 28 |
| 2 | 45 | 36 | 39 | 59 | 64 | 68 | 67 | 39 | 49 |
| 3 | 143 | 146 | 168 | >100 | >100 | >100 | >100 | >100 | >100 |
| 4 | 448 | 89 | 80 | 87 | >100 | >100 | >100 | 89 | 47 |
| 5 | 245 | 266 | 238 | >100 | >100 | >100 | >100 | 68 | >100 |
| 6 | 31 | 31 | 23 | 85 | 43 | 56 | >100 | 40 | 17 |
| 7 | 28 | 30 | 102 | >100 | >100 | >100 | >100 | 18 | >100 |
| 8 | 236 | 171 | 192 | 89 | 8.8 | 77 | >100 | 7.9 | 35 |
| 9 | >500 | >500 | >500 | 91 | 44 | 60 | 58 | 9 | 40 |
| 10 | 43 | 35 | 28 | 34 | 24 | 30 | 32 | 14 | 40 |
| 11 | >250 | >250 | >250 | >100 | 66 | >100 | 87 | 43 | 53 |
| 12 | 44 | 39 | 38 | 29 | 13 | 27 | 11 | 7.3 | 31 |
| 13 | 42 | 29 | 31 | 94 | 44 | 79 | >100 | 9.1 | 40 |
| 14 | 39 | 17 | 23 | 27 | 5.6 | 26 | 30 | 4.3 | 25 |
| 5-FUb | 0.69 | 20 | 9.23 | 16 | 4.5 | 6.5 | 8.7 | 9.2 | 10 |
IC50, 50% inhibitory concentration, or compound concentration required to inhibit tumour cell proliferation by 50%.
The cytostatic effects of 5-fluorouracil against all tumour cell lines except for Hep G2 were published.1
In the series of Z-izomers of C-5 alkynyl 1-[4′-(N-phthalimido)-2′-butenyl] pyrimidine derivatives 1–7, compounds containing a hexynyl (1), octynyl (2) and p-butylphenylethynyl (6) side chain at C-5 exhibited moderate cytostatic effects against all evaluated cell lines, particularly against hepatocellular carcinoma (Hep G2; IC50 = 16.6–40.2 μM) (Fig. 5 ). However, these compounds were also cytotoxic against normal human fibroblasts (WI 38; IC50 = 16.7–49 μM) (Fig. 6 ).
Figure 5.
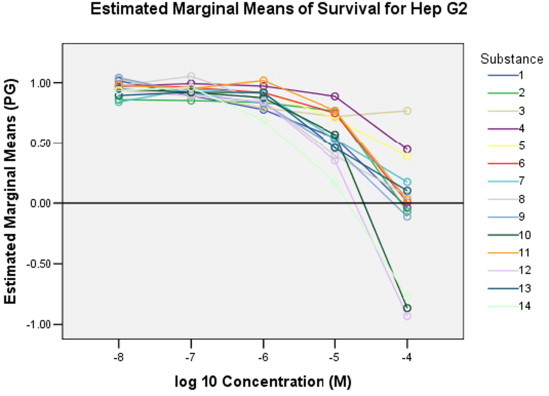
Dose–response curves of the tested compounds 1–14 on hepatocellular carcinoma (Hep G2).
Figure 6.
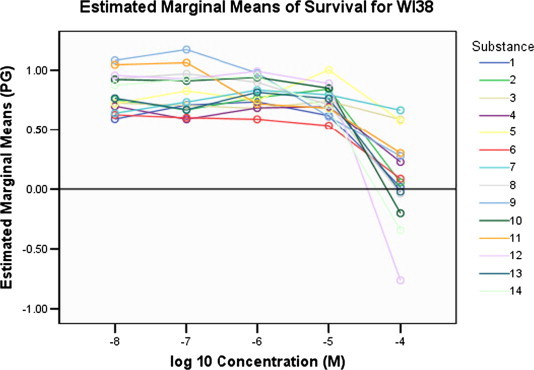
Dose–response curves of the tested compounds 1–14 on the normal diploid fibroblasts (WI 38).
On the contrary, compound 7 with the C-5 p-pentylphenylethynyl side chain showed selective antiproliferative effect on the growth of hepatocellular carcinoma (Hep G2, IC50 = 18 μM). However, this compound had lesser cytostatic activity against Hep G2 cell line than standard 5-fluorouracil (5-FU). Among E-isomers of C-5 substituted alkynyl 1-[4′-(N-phthalimido)-2′-butenyl]pyrimidine derivatives 8–14, compound 14 containing the p-pentylphenylethynyl side chain at C-5 exhibited the best antiproliferative effect on all the evaluated cell lines (IC50 in the range of 4.3–39 μM) and normal fibroblasts (IC50 = 25 μM). Besides, compounds with a C-5 hexynyl (8), heptynyl (9) and phenylbutynyl (12) side chain showed cytostatic activity, particularly against hepatocellular carcinoma (Hep G2; IC50 = 7.3–9 μM) (Fig. 5). Thus, compounds 7 and 14 emerged as the most interesting lead compounds with cytostatic activities that could be used for further structural optimization and biological studies on Hep G2 cells.21, 22, 23
3.2. Antiviral activities
Compounds 1–14 were evaluated for their inhibitory activities against HIV-1 and HIV-2 in human T-lymphocyte (CEM) cells (Table 3 ). Compounds 3 and 4 showed some specific albeit slight activity against HIV-1, while compound 14 exhibited moderate activity against both HIV-1 and HIV-2. None of the other viruses were sensitive to the inhibitory activity of the tested compounds except for compounds 2 (EC50 = 2.5–4 μM) and 5 (EC50 = 20–21 μM) against influenza A (H3N2), for compound 14 against Coxsackie B4 virus (EC50 = 12 μM) and vaccinia virus (EC50 = 12 μM) and for compound 7 for varicella-zoster virus (EC50 = 9.4–8.7 μM). However, the EC50 values of the compounds were close to their toxicity threshold, and therefore, it is unclear whether the observed activity is due to a specific antiviral effect, or indirectly, to a cytotoxic/cytostatic activity of the tested compounds.
Table 3.
Anti-HIV-1 and HIV-2 activities of compounds 1–14 in human T-lymphocyte (CEM) cells
| Compound | EC50 (μM) |
|
|---|---|---|
| HIV-1 | HIV-2 | |
| 1 | ⩾50 | ⩾50 |
| 2 | >50 | >50 |
| 3 | 35 | ⩾50 |
| 4 | 10 | >50 |
| 5 | >250 | >250 |
| 6 | >10 | >10 |
| 7 | >2 | >2 |
| 8 | ⩾50 | ⩾50 |
| 9 | >50 | >50 |
| 10 | ⩾10 | ⩾10 |
| 11 | >50 | >50 |
| 12 | ⩾10 | ⩾10 |
| 13 | >10 | >10 |
| 14 | 14 | 3.1 |
EC50, effective concentration or concentration required to protect CEM cells against the cytopathogenicity of HIV by 50%.
4. Conclusions
The novel (Z)- (1–7) and (E)- (8–14) unsaturated acyclic C-5-alkynyl uracil derivatives were prepared by using Sonogashira coupling of (Z)- and (E)-1-[4′-(N-phthalimido)-2′-butenyl]-5-iodouracil with various terminal alkynes.
By comparison of the cytostatic activities of the Z- and E-isomers of C-5 alkynyl olefinic pyrimidine derivatives, we can infer that the E-isomers (8–14) showed better cytostatic activities. Furthermore, introduction of the phenyl ring in longer side chain caused an increasing cytostatic effect. Thus, the C-5 pentylphenylethynyl substituted olefinic pyrimidine derivative 14 showed the most pronounced inhibitory activity against all the tested tumour cell lines (IC50 = 4.3–39 μM), including human normal fibroblasts. On the contrary, its Z-isomer (7) showed selective inhibitory activity against the Hep G2 cell line (IC50 = 18 μM). Thus, the results of the cytostatic examinations indicated that compounds 7 and 14 are suitable candidates for further biological studies on lead drug and structural optimization.
5. Experimental
5.1. General methods
Melting points were determined on a Kofler micro hot-stage apparatus (Reichert, Wien) and are uncorrected. The electron impact mass spectra were recorded with an EXTREL FT MS 2002 instrument with ionizing energy of 70 eV. 1H and 13C NMR spectra were recorded on Varian Gemini 300 MHz, Varian Unity Inova 300 MHz and Varian NMR System 800 MHz NMR spectrometers. All the data were recorded at 298 K. NMR samples of all compounds were prepared in DMSO-d 6. Chemical shifts were referenced to the residual solvent signal of DMSO at δ 2.50 ppm for 1H and δ 39.50 ppm for 13C. Individual resonances were assigned on the basis of their chemical shifts, signal intensities, multiplicity of resonances, H–H coupling constants involved as well as with the use of a series of 2D experiments (gHSQC and gHMBC). Chemical shifts and coupling constants for H1′–H2′–H3′–H4′ spin system were obtained by spin simulation using VNMRJ 2.1B software. Precoated Merck Silica Gel 60F-254 plates were used for thin layer chromatography (TLC) and the spots were detected under UV light (254 nm). Column chromatography (CLC) was performed using silica gel (0.063–0.2 mm) Fluka; glass column was slurry-packed under gravity. Compounds purity was analyzed by HPLC with DAD detector.
5.2. Compounds preparation
(Z)-4-Chloro-2-butenyl-N-phthalimide (Ia) and (E)-4-bromo-2-butenyl-N-phthalimide (Ib) were prepared as described previously.13
5.2.1. (Z)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-iodouracil (IIa)
To a stirred mixture of 5-iodouracil (1 g, 4.2 mmol) and 60% NaH (101 mg, 4.2 mmol) in DMF (40 mL) after 1 h (Z)-4-chloro-2-butenyl-N-phthalimide (Ia, 824 mg, 3.5 mmol) was added at room temperature. The reaction mixture was stirred under argon atmosphere at 40 °C for 3 h, then at room temperature overnight and concentrated to dryness. Purification of the residue by column chromatography (petroleum ether/ethyl acetate = 1:1) afforded IIa as white crystals (735 mg, 48.1%, mp = 218–219 °C) after recrystallization from MeOH.
1H NMR: δ 11.59 (s, 1H, NH), 8.12 (s, 1H, H-6), 7.76–7.80 (m, 4H, Phth), 5.52–5.57 (m, 2H, H-2′, H-3′), 4.43 (d, 2H, J = 6.0 Hz, H-1′), 4.29 (d, 2H, J = 5.9 Hz, H-4′) ppm.
13C NMR: δ 167.49 (C2-Phth), 160.93 (C4), 150.56 (C2), 149.42 (C6), 134.40 (C5-Phth), 131.67 (C3-Phth), 128.10 (C3′), 127.26 (C2′), 123.03 (C4-Phth), 68.71 (C5), 44.36 (C1′), 34.45 (C4′) ppm. MS m/z 438.1 (M+•). Anal. (C16H12IN3O4) C, H, N.
5.2.2. (E)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-iodouracil (IIb)
To a stirred mixture of 5-iodouracil (1 g, 4.2 mmol) and 60% NaH (101 mg, 4.2 mmol) in DMF (40 mL), (E)-4-bromo-2-butenyl-N-phthalimide (Ib, 980 mg, 3.5 mmol) was added. The reaction mixture was stirred under argon atmosphere for 4 h at 50 °C and then at room temperature overnight. The solvent was evaporated, saturated NH4Cl (100 mL) was added and the solution was extracted with EtOAc (3× 70 mL). Combined organic extracts were dried over MgSO4. The solvent was evaporated and the residue purified by column chromatography (CH2Cl2/MeOH = 60:1), which afforded IIb as white crystals (810 mg, 52.9%, mp = 221–222 °C) after recrystallization from MeOH.
1H NMR: δ 11.57 (s, 1H, NH), 8.02 (s, 1H, H-6), 7.77–7.80 (m, 4H, Phth), 5.57–5.65 (m, 2H, H-2′, H-3′), 3.97–4.21 (m, 4H, H-1′, H-4′) ppm.
13C NMR: δ 167.42 (C2-Phth), 160.93 (C4), 150.38 (C2), 149.41 (C6), 134.55 (C5-Phth), 131.60 (C3-Phth), 128.06 (C3′), 126.35 (C2′), 123.09 (C4-Phth), 68.36 (C5), 48.24 (C1′), 38.27 (C4′) ppm. MS m/z 438.2 (M+•). Anal. (C16H12IN3O4) C, H, N.
5.2.3. General procedure for the preparation of (Z)- and (E)-1-[4′-(N-phthalimido)-2′-butenyl]-5-alkynyluracil (1–14)
To a stirred solution of Z- or (E)-1-[4′-(N-phthalimido)-2′-butenyl]-5-iodouracil (200 mg, 0.46 mmol) in anhydrous dimethylformamide (15 mL) were added diisopropylethylamine (0.16 mL, 0.92 mmol), the acetylene derivative (1.37 mmol), tetrakis(triphenylphosphine)palladium(0) (53 mg, 0.046 mmol) and copper(I) iodide (17.4 mg, 0.09 mmol). The mixture was stirred for 20 h at room temperature under nitrogen, then evaporated to dryness and purified by column chromatography (initial eluent: CH2Cl2, followed by CH2Cl2/MeOH = 60:1).
5.2.4. (Z)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-hexynyluracil (1)
The procedure was carried out using hexyne (0.16 mL, 1.37 mmol) which gave brown powder of 1 (34 mg, 19.6%, mp = 175–176 °C).
13C NMR: δ 168.03 (C2-Phth), 161.52 (C4), 151.33 (C2), 149.98 (C6), 134.92 (C5-Phth), 132.15 (C3-Phth), 131.88 (C3′), 129.15 (C2′), 123.53 (C4-Phth), 94.18 (C5), 88.17 (C2″), 68.88 (C1″), 44.85 (C1′), 35.10 (C4′), 34.93 (C4″), 29.48 (C5″), 21.11 (C3″), 13.90 (CH3) ppm.
MS m/z 392.2 (M+•). Anal. (C22H21N3O4) C, H, N.
5.2.5. (Z)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-decynyluracil (2)
The procedure was carried out using decyne (0.25 mL, 1.37 mmol) which gave white-yellow powder of 2 (83 mg, 41.8%, mp = 167–168 °C).
13C NMR: δ 167.54 (C2-Phth), 162.29 (C4), 149.93 (C2), 147.50 (C6), 134.44 (C5-Phth), 131.68 (C3-Phth), 128.16 (C3′), 127.21 (C-2′), 123.05 (C4-Phth), 98.50 (C5), 93.20 (C2″), 72.71 (C1″), 44.40 (C1′), 34.45 (C4′), 31.24 (C-8″), 28.61, 28.52, 28.26, 28.19, (C4″–C7″), 22.07 (C9″), 18.77 (C3″) 13.93 (CH3) ppm.
MS m/z 448.2 (M+•). Anal. (C26H29N3O4) C, H, N.
5.2.6. (Z)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-(4″-phenylbutynyl)uracil (3)
The procedure was carried out using 4-phenylbutyne (0.19 mL, 1.37 mmol) which gave white-yellow powder of 3 (128 mg, 65.7%, mp = 174–175 °C).
13C NMR: δ 162.26 (C4), 149.94 (C2), 147.58 (C6), 140.44 (C1-Ph), 134.44 (C5-Phth), 131.67 (C3-Phth), 127.21 (C-2′), 126.18 (C4-Ph), 123.06 (C4-Phth), 98.37 (C5), 92.70 (C2″), 73.25 (C1″), 44.47 (C1′), 34.47 (C4′), 34.37 (C4″), 21.03 (C3″) ppm.
MS m/z 440.1 (M+•). Anal. (C26H21N3O4) C, H, N.
5.2.7. (Z)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-(p-bromophenylethynyl)uracil (4)
The procedure was carried out using p-bromophenylacetylene (248.7 mg, 1.37 mmol) which gave yellow powder of 4 (134 mg, 61.6%, mp = 188–189°C).
13C NMR: δ 168.06 (C2-Phth), 162.27 (C4), 150.35 (C2), 149.44 (C6), 134.93 (C5-Phth), 133.38 (C4-Ph), 132.32 (C2-Ph), 132.15 (C3-Phth), 128.92 (C3′), 127.48 (C2′), 123.54 (C4-Phth), 122.38 (C3-Ph), 122.15 (C1-Ph), 97.83 (C5), 91.26 (C2″), 84.20 (C1″), 45.09 (C1′), 34.97 (C4′) ppm.
MS m/z 491.9 (M+•). Anal. (C24H16BrN3O4) C, H, N.
5.2.8. (Z)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-(p-methylphenylethynyl)uracil (5)
The procedure was carried out using p-methylphenylacetylene (0.17 mL, 1.37 mmol) which gave orange-brown powder of 5 (43 mg, 23.5%, mp = 225–226 °C).
13C NMR: δ 167.54 (C2-Phth), 160.72 (C4), 149.90 (C2), 146.97 (C6), 138.41 (C4-Ph), 134.56 (C5-Phth), 131.67 (C3-Phth), 130.95 (C2-Ph), 129.38 (C3-Ph), 126.94 (C-2′), 127.13 (C-3′), 123.03 (C4-Phth), 119.34 (C1-Ph), 97.22 (C5), 91.94 (C2″), 73.11 (C1″), 45.79 (C1′), 34.48 (C4′), 21.12 (CH3) ppm.
MS m/z 426.3 (M+•). Anal. (C25H19N3O4) C, H, N.
5.2.9. (Z)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-(p-butylphenylethynyl)uracil (6)
The procedure was carried out using p-butylphenylacetylene (0.24 mL, 1.37 mmol) which gave white-yellow powder of 6 (107 mg, 51.5%, mp = 163–165 °C).
13C NMR: δ 167.53 (C2-Phth), 161.89 (C4), 149.87 (C2), 148.33 (C6), 143.11 (C4-Ph), 134.41 (C5-Phth), 131.66 (C3-Phth), 130.95 (C2-Ph), 128.66 (C3-Ph), 128.31 (C-3′), 127.07 (C2′), 123.03 (C4-Phth), 119.66 (C1-Ph), 97.84 (C5), 92.00 (C2″), 81.64 (C1″), 44.56 (C1′), 34.63 (C4′), 34.47 (C1‴), 32.76 (C3‴), 21.67 (C2‴), 13.70 (CH3) ppm.
MS m/z 468.1 (M+•). Anal. (C28H25N3O4) C, H, N.
5.2.10. (Z)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-(p-pentylphenylethynyl)uracil (7)
The procedure was carried out using p-pentylphenylacetylene (0.27 mL, 1.37 mmol) which gave yellow-brown crystals of 7 (101 mg, 47.2%, mp = 175–176 °C).
13C NMR: δ 167.57 (C2-Phth), 161.93 (C4), 149.91 (C2), 148.38 (C6), 143.18 (C4-Ph), 134.44 (C5-Phth), 131.68 (C3-Phth), 130.98 (C2-Ph), 128.70 (C3-Ph), 128.34 (C-3′), 127.10 (C2′), 123.06 (C4-Phth), 119.68 (C1-Ph), 97.85 (C5), 92.03 (C2″), 81.66 (C1″), 44.58 (C1′), 34.49 (C4′), 34.95 (C1‴), 30.83 (C3‴), 30.31 (C2‴), 21.91 (C4‴), 13.88 (CH3) ppm.
MS m/z 482.2 (M+•). Anal. (C29H27N3O4) C, H, N.
5.2.11. (E)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-hexynyluracil (8)
The procedure was carried out using hexyne (0.16 mL, 1.37 mmol) which gave brown powder of 8 (52 mg, 30.1%, mp = 114–115 °C).
13C NMR: δ 169.23 (C2-Phth), 160.91 (C4), 149.60 (C2), 146.12 (C6), 128.26 (C3′), 97.67 (C5), 93.17 (C2″), 72.72 (C1″), 49.46 (C1′), 38.34 (C4′), 30.16 (C4″), 21.30 (C5″), 18.39 (C3″), 13.44 (CH3) ppm.
MS m/z 392.1 (M+•). Anal. (C22H21N3O4) C, H, N.
5.2.12. (E)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-heptynyluracil (9)
The procedure was carried out using heptyne (0.18 mL, 1.37 mmol) which gave brown powder of 9 (39 mg, 21.8%, mp = 67–68 °C).
13C NMR: δ 167.93 (C2-Phth), 161.39 (C4), 150.07 (C2), 146.60 (C6), 134.94 (C5-Phth), 132.08 (C3-Phth), 128.72 (C3′), 127.19 (C2′), 123.59 (C4-Phth), 98.15 (C5), 93.70 (C2″), 73.18 (C1″), 49.94 (C1′), 42.28 (C4′), 30.88 (C-4″), 28.26 (C5″), 22.08 (C6″), 19.14 (C3″), 14.30 (CH3) ppm.
MS m/z 406.4 (M+•). Anal. (C23H23N3O4) C, H, N.
5.2.13. (E)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-octynyluracil (10)
The procedure was carried out using octyne (0.2 mL, 1.37 mmol) which gave brown powder of 10 (34 mg, 18.3%, mp = 58–60 °C).
13C NMR: δ 167.77 (C2-Phth), 161.02 (C4), 154.69 (C2), 142.13 (C6), 134.92 (C5-Phth), 132.04 (C3-Phth), 128.79 (C3′), 126.76 (C2′), 123.56 (C4-Phth), 106.58 (C5), 97.65 (C2″), 69.07 (C1″), 51.85 (C1′), 38.82 (C4′), 31.16 (C4″), 28.35 (C5″), 25.30 (C6″), 22.41 (C7″), 19.31 (C3″), 14.32 (CH3) ppm.
MS m/z 420.2 (M+•). Anal. (C24H25N3O4) C, H, N.
5.2.14. (E)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-decynyluracil (11)
The procedure was carried out using decyne (0.25 mL, 1.37 mmol) which gave yellow powder of 11 (61 mg, 30.1%, mp = 62–63 °C).
13C NMR: δ 167.92 (C2-Phth), 161.38 (C4), 150.07 (C2), 146.57 (C6), 134.93 (C5-Phth), 132.08 (C3-Phth), 128.72 (C3′), 127.21 (C2′), 123.40 (C4-Phth), 98.15 (C5), 93.70 (C2″), 73.17 (C1″), 49.93 (C1′), 42.28 (C4′), 31.70 (C4″), 29.04 (C5″), 28.94 (C6″), 28.69 (C7″), 28.58 (C8″), 22.54 (C9″), 19.18 (C3″), 14.38 (CH3) ppm.
MS m/z 448.4 (M+•). Anal. (C26H29N3O4) C, H, N.
5.2.15. (E)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-(4″-phenylbutynyl)uracil (12)
The procedure was carried out using 4-phenylbutyne (0.19 mL, 1.37 mmol) which gave yellow powder of 12 (58 mg, 29.8%, mp = 84–86 °C).
13C NMR: δ 167.49 (C2-Phth), 162.22 (C4), 149.73 (C2), 147.60 (C6), 140.41 (C1-Ph), 134.47 (C5-Phth), 123.13 (C4-Phth), 98.25 (C5), 92.72 (C2″), 73.20 (C1″), 48.25 (C1′), 38.35 (C4′), 34.32 (C4″), 21.00 (C3″) ppm.
MS m/z 440.2 (M+•). Anal. (C26H21N3O4) C, H, N.
5.2.16. (E)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-(p-butylphenylethynyl)uracil (13)
The procedure was carried out using p-butylphenylacetylene (0.24 mL, 1.37 mmol) which gave yellow powder of 13 (143 mg, 68.9%, mp = 171–172 °C).
13C NMR: δ 167.95 (C2-Phth), 162.40 (C4), 150.17 (C2), 148.83 (C6), 143.61 (C4-Ph), 134.91 (C5-Phth), 132.08 (C3-Phth), 131.45 (C2-Ph), 129.15 (C3-Ph), 128.59 (C3′), 126.59 (C2′), 123.58 (C4-Phth), 120.08 (C1-Ph), 98.21 (C5), 92.50 (C2″), 82.07 (C1″), 48.98 (C1′), 38.83 (C4′), 35.12 (C3″), 33.27 (C3‴), 22.17 (C2‴), 14.21 (CH3) ppm.
MS m/z 468.1 (M+•). Anal. (C28H25N3O4) C, H, N.
5.2.17. (E)-1-[4′-(N-Phthalimido)-2′-butenyl]-5-(p-pentylphenylethynyl)uracil (14)
The procedure was carried out using p-pentylphenylacetylene (0.27 mL, 1.37 mmol) which gave yellow powder of 14 (132 mg, 61.7%, mp = 75–76 °C).
13C NMR: δ 167.49 (C2-Phth), 161.93 (C4), 149.70 (C2), 148.37 (C6), 143.19 (C4-Ph), 131.62 (C3-Phth), 130.99 (C2-Ph), 128.69 (C3-Ph), 128.12 (C3′), 126.09 (C-2′), 119.60 (C1-Ph), 97.72 (C5), 92.03 (C2″), 81.59 (C1″), 48.51 (C1′), 38.36 (C4′), 34.93 (C1‴), 30.83 (C3‴), 30.30 (C2‴), 21.90 (C4‴), 13.89 (CH3) ppm.
MS m/z 482.3 (M+•). Anal. (C29H27N3O4) C, H, N.
5.3. Cytostatic activity assays
5.3.1. Antiproliferation assays
The experiments were carried out on nine human cell lines, eight of which are derived from eight cancer types and one normal, fibroblast cell line. The following cell lines were used: murine leukemia (L1210), human T-lymphocytes (Molt4/C8 and CEM), cervical carcinoma (HeLa), breast carcinoma (MCF-7), pancreatic carcinoma (MiaPaCa-2), hepatocellular carcinoma (Hep G2), colon carcinoma (SW 620) and diploid fibroblasts (WI 38).
Murine L1210 and human Molt4/C8 and CEM cells were seeded at 50–75 × 103cells/well in 96-well microtiter plates in the presence of different concentrations of the test compounds. After 2 (L1210) or 3 (Molt4/C8 and CEM) days, the cell number was determined with a Coulter counter (Coulter Electronics, England). The cytostatic concentration was determined as the compound concentration required to reduce L1210, Molt4/C8 or CEM cell growth by 50% relative to the number of cells in the untreated controls (IC50). IC50 values were calculated from graphic plots of the number of cells (percentage of control) as a function of the concentration of the test compounds.
The HeLa, Hep G2, MCF-7, MiaPaCa-2, SW 620 and WI 38 cells were cultured as monolayers and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 2 mM l-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin in a humidified atmosphere with 5% CO2 at 37 °C. As reported previously,24 the panel cell lines were inoculated onto a series of standard 96-well microtiter plates on day 0 at 1 × 104 to 3 × 104 cells/mL, depending on the doubling times of specific cell line. Test agents were then added in five, 10-fold dilutions (10−8 to 10−4 M) and incubated for further 72 h. Working dilutions were freshly prepared on the day of testing. The compounds were prepared as 0.04 M solutions in DMSO, and the solvent was also tested for eventual inhibitory activity by adjusting its concentration to be the same as in the working concentrations. After 72 h of incubation the cell growth rate was evaluated by performing the MTT assay (Sigma) which detects dehydrogenase activity in viable cells. The MTT Cell Proliferation Assay is a colorimetric assay system, which measures the reduction of a tetrazolium component (MTT) into an insoluble formazan product by the mitochondria of viable cells. For this purpose, the substance-treated medium was discarded and MTT was added to each well at a concentration of 20 μg/40 μL. After 4 h of incubation the precipitates were dissolved in 160 μL of DMSO. The absorbance (OD, optical density) was measured on a microplate reader at 570 nm. The absorbancy is directly proportional to the cell viability. The percentage of growth (PG) of the cell lines was calculated according to one or the other of the following two expressions:
If (mean ODtest − mean ODtzero) ⩾ 0 then
If (mean ODtest – mean ODtzero) < 0 then
where mean ODtzero = the average of optical density measurements before exposure of cells to the test compound; mean ODtest = the average of optical density measurements after the desired period of time; and mean ODctrl = the average of optical density measurements after the desired period of time with no exposure of cells to the test compound.
Each test point was performed in quadruplicate in three individual experiments. The results were expressed as IC50, a concentration necessary for 50% of inhibition, which were calculated from dose–response curves using linear regression analysis by fitting the test concentrations that give PG values above and below the respective reference values.
5.4. Antiviral activity assays
Antiviral activity against HIV-1 and HIV-2 was based on the examination of the virus-induced cytopathicity (syncytia formation) in CEM cell cultures, and determined essentially as described previously.25, 26
The antiviral activity of the test compounds against herpes simplex virus type 1 (HSV-1), HSV-2, vaccinia virus, vesicular stomatitis virus (VSV), human cytomegalovirus (HCMV) and varicella-zoster virus (VZV) in HEL cell cultures; VSV, Coxsackie virus B4 and respiratory syncytial virus (RSV) in HeLa cell cultures; parainfluenza virus-3, reovirus-1, Sindbis virus, Coxsackie virus B4 and Punta Toro virus in Vero cell cultures; influenza virus A (H1N1, H3N2) and influenza virus B in MDCK cell cultures; and feline coronavirus (FIPV) and feline herpesvirus in CRFK cell cultures was based on estimating the virus-induced cytopathicity in the cell cultures under conditions and at a time point at which non-treated virus-exposed cell cultures reached a full virus-induced cytopathogenicity effect.
Acknowledgements
Support for this study was provided by the Ministry of Science of the Republic of Croatia (Projects #125-0982464-2925, #125-0982464-2922 and #098-0982464-2393), the ‘Fonds voor Wetenschappelijk Onderzoek’ (FWO) (#G.0188.07) and the ‘Geconcerteerde Onderzoeksacties (GOA)’ of the K.U. Leuven (#05/15). The authors thank Mrs. L. van Berckelaer, L. Ingels, L. Persoons, V. Broeckx, F. Demeyer, A. Camps, S. Carmans and L. Van den Heurck for excellent technical assistance.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2008.03.074.
Supplementary data
References and notes
- 1.De Clercq E. Nucleosides Nucleotides. 1987;6:197. [Google Scholar]
- 2.Herdewijn P. Antiviral Chem. Chemother. 1994;5:131. [Google Scholar]
- 3.Fillastre J.P., Godin M., Legallicier B., Chretien P., Bidault R., Gillotin C., Wooton R., Posner J., Peck R.W. J. Antimicrob. Chemother. 1996;37:965. doi: 10.1093/jac/37.5.965. [DOI] [PubMed] [Google Scholar]
- 4.Beres J., Bentrude W.G., Balzarini J., De Clercq E., Otvos L. J. Med. Chem. 1986;29:494. doi: 10.1021/jm00154a012. [DOI] [PubMed] [Google Scholar]
- 5.De Clercq E., Descamps J., Balzarini J., Giziewicz J., Barr P.J., Robins M.J. J. Med. Chem. 1983;26:661. doi: 10.1021/jm00359a008. [DOI] [PubMed] [Google Scholar]
- 6.Zemlicka J. In: Nucleosides and Nucleotides as Antitumor and Antiviral Agent. Chu C.K., Baker D.C., editors. Plenum Publishing Corp.; New York: 1993. Allenols derived from nucleic acid bases—a new class of anti-HIV agents: chemistry and biological activity; pp. 73–80. [Google Scholar]
- 7.Priego E.M., Balzarini J., Karlsson A., Camarasa M.J., Perez-Perez M.J. Bioorg. Med. Chem. 2004;12:5079. doi: 10.1016/j.bmc.2004.07.036. [DOI] [PubMed] [Google Scholar]
- 8.Raić-Malić S., Hergold Brundić A., Nagl A., Grdiša M., Pavelić K., De Clercq E., Mintas M. J. Med. Chem. 1999;42:2673. doi: 10.1021/jm991017z. [DOI] [PubMed] [Google Scholar]
- 9.Gazivoda T., Plevnik M., Plavec J., Kraljević S., Kralj M., Pavelić K., Balzarini J., De Clercq E., Mintas M., Raić-Malić S. Bioorg. Med. Chem. 2005;13:131. doi: 10.1016/j.bmc.2004.09.052. [DOI] [PubMed] [Google Scholar]
- 10.Raić-Malić S., Svedružić D., Gazivoda T., Marunović A., Hergold-Brundić A., Nagl A., Balzarini J., De Clercq E., Mintas M. J. Med. Chem. 2000;43:4806. doi: 10.1021/jm0009540. [DOI] [PubMed] [Google Scholar]
- 11.Gazivoda T., Šokčević M., Kralj M., Šuman L., Pavelić K., De Clercq E., Andrei G., Snoeck R., Balzarini J., Mintas M., Raić-Malić S. J. Med. Chem. 2007;50:4105. doi: 10.1021/jm070324z. [DOI] [PubMed] [Google Scholar]
- 12.Gazivoda T., Raić-Malić S., Marjanović M., Kralj M., Pavelić K., Balzarini J., De Clercq E., Mintas M. Bioorg. Med. Chem. 2007;15:749. doi: 10.1016/j.bmc.2006.10.046. [DOI] [PubMed] [Google Scholar]
- 13.Krištafor V., Raić-Malić S., Cetina M., Kralj M., Šuman L., Pavelić K., Balzarini J., De Clercq E., Mintas M. Bioorg. Med. Chem. 2006;14:8126. doi: 10.1016/j.bmc.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 14.Norman M.H., Minick D.J., Rigdon G.C. J. Med. Chem. 1996;39:149. doi: 10.1021/jm9502201. [DOI] [PubMed] [Google Scholar]
- 15.Robins M.J., Vinayak R.V., Wood S.G. Tetrahedron Lett. 1990;26:3731. [Google Scholar]
- 16.Takahashi S., Kuroyama Y., Sonogashira K., Hagihara N. Synthesis. 1980:627. [Google Scholar]
- 17.Robins M.J., Barr P.J. J. Org. Chem. 1983;48:1854. [Google Scholar]
- 18.Robins M.J., Barr P.J. Tetrahedron Lett. 1981;22:421. [Google Scholar]
- 19.Bleackey R.C., Jones A., Walker R.T. Tetrahedron. 1976;32:2795. [Google Scholar]
- 20.Janeba Z., Balzarini J., Andrei G., Snoeck R., De Clercq E., Robins M.J. J. Med. Chem. 2005;48:4690. doi: 10.1021/jm050291s. [DOI] [PubMed] [Google Scholar]
- 21.Kraljević S., Stambrook P.J., Pavelić K. EMBO Rep. 2004;5:837. doi: 10.1038/sj.embor.7400236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kraljević S., Sedic M., Scott M., Gehrig P., Schlapbach R., Pavelić K. Cancer Treat. Rev. 2006;32:619. doi: 10.1016/j.ctrv.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Mi Q., Kim S., Hwang B.Y., Su B.N., Chai H., Arbieva Z.H., Kinghorn A.D., Swanson S.M. Anticancer Res. 2006;26:3349. [PubMed] [Google Scholar]
- 24.Mossman T. J. Immunol. Methods. 1983;65:55. [Google Scholar]
- 25.Balzarini J., Pannecouque C., De Clercq E., Pavlov A.Y., Printsevskaya S.S., Miroshnikova O.V., Reznikova M.I., Preobrazhenskaya M.N. J. Med. Chem. 2003;46:2755. doi: 10.1021/jm0300882. [DOI] [PubMed] [Google Scholar]
- 26.Balzarini J., Naesens L., Slachmuylders J., Niphuis H., Rosenberg I., Holy′ A., Schellekens H., De Clercq E. AIDS. 1991;5:21. doi: 10.1097/00002030-199101000-00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.