

Abstract
Defense against biothreat agents requires a broad-spectrum approach. Modulation of the innate immune system might fulfill this requirement. Hackett's previous review of innate immune activation as a broad-spectrum biodefense strategy identified several unresolved questions. The current article is a systematic approach to answering those questions with the focused participation of research groups developing this technology. Our team of academic and industry participants reviewed the promising agents and came to the following conclusions. It is feasible to construct a biodefense platform combining synergistic agents that activate the innate immune system against a broad range of pathogens on the basis of conserved microbial components by using a nasal spray for immune activation in the respiratory and gastrointestinal tracts because these are the most likely routes of attack. It might also be possible to include agents that inhibit molecular events leading to septic shock. Innate immune-activating agents designed to activate Toll-like and other receptors will probably provide protection against the biothreat pathogen spectrum for periods ranging from 2 to 14 days for IFNs up to 26 weeks for immunomodulatory oligonucleotides. Initial treatment is proposed on the first index case or biosensor alert. Boost doses would be required. Harmful inflammation is possible, but thus far, only transient fever has been observed. Autoimmune reaction and retroviral activation have not been seen thus far in preclinical and human trials of many of these compounds. Toll-like receptor agonists caused cytokine production in all subjects tested, but genetic polymorphism reduced the response to IFN in African American subjects.
Key words: Biodefense, immunomodulation, innate immunity, Toll-like receptors
Abbreviations used: BTA, Biothreat agent; DC, Dendritic cell; FDA, US Food and Drug Administration; HBV, Hepatitis B virus; HCV, Hepatitis C virus; HMGB1, High mobility group box 1; HPV, Human papillomavirus; HSV, Herpes simplex virus; IMO, Immunomodulatory oligonucleotide; IRM, Immune response modifier; NK, Natural killer; ODN, Oligodeoxynucleotide; TLR, Toll-like receptor
The biodefense problem
A bioterror attack requires an immediate and effective response. Vaccines are limited by the inability to predict the pathogen and resistance to prophylactic vaccination. The inherent delay in adaptive immune response renders it inadequate for protection from bioterror attack. Innate immunity is an underexplored option for biodefense. Recent therapeutic advances suggest that innate immunomodulation holds the potential for improved survival after a bioterror attack with an infectious agent. Numerous products targeting various processes in the innate immune response are either currently available and moving toward human trials or in the initial stages of development.
In-depth investigation into opportunities offered by innate immunity for biodefense is currently lacking.
Innate immune cells use pathogen recognition receptors to recognize pathogen macromolecules to provide an immediate response with broad specificity. The pathogen-associated molecular pattern system of Toll-like receptors (TLRs) comprises cell-surface and endosomic receptors that recognize broadly conserved ligands unique to microorganisms. Currently, one TLR agonist is licensed for use in human subjects for certain viral infections and skin cancers, and other agonists are in advanced stages of clinical development. TLRs might be exploited against bioterrorism agents on the basis of their mechanism of action; however, excessive activation of the innate immune system can result in autoimmune disease and septic shock.1 These issues are the focus of this investigation.
Questions about innate immunity for biodefense
In November 2004, a panel of invited experts met to consider the feasibility of innate immune modulation as biodefense, including the risks in stimulating innate immunity, current resources, the time needed to develop and produce effective agents, and interfacing with regulatory agencies. At the conclusion of the meeting, technical documents describing the application of a particular product or technology to biodefense were requested.
In collaboration with independent reviewers, proposals were evaluated on compound–mechanism of action, potential activity, likelihood of product approval within 5 years, uniqueness, and overall grade. Criteria for evaluating this approach have been published by Hackett1 and were applied to submitted technologies. Questions raised by Hackett include the following:
-
1.
Which innate immune receptors stimulate effective prophylactic responses to the broadest range of bacterial and viral pathogens?
-
2.
How long does protection last?
-
3.
Could innate immune therapy trigger harmful inflammation?
-
4.
Will innate immune stimulation promote autoimmune reactions or retroviral activation?
-
5.
How important a factor is human genetic polymorphism within the innate immune system for innate immune therapy strategies?
Answering the questions: Pros and cons to future paths—Innate immunomodulation
Innate immune cells use pathogen recognition receptors to recognize pathogen macromolecules and promote rapid response. As mentioned above, TLRs are type I transmembrane and endosomically expressed proteins that are evolutionarily conserved and have been identified as the key pathogen recognition receptors in innate immunity. TLRs 1 through 10 are present on human innate immune cells and act through cellular signal cascade to augment host immune response through inflammation. TLR agonists can modify host inflammatory responses, potentially offering increased protection from infection. Currently, TLR agonists are licensed and being used in human subjects in the setting of microbial infection and cancer. They can be tailored to specific diseases and administered through multiple routes. Microbial ligands recognized by TLRs are constitutively expressed and highly conserved structural molecules less prone to rapid genetic mutation, and thus TLR agonists are less likely to select for resistant strains ( Fig 1).
Fig 1.
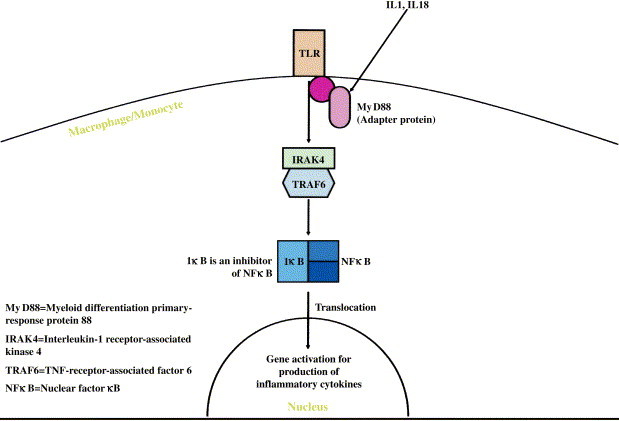
Common pathway used by TLRs for signal transduction.
Specifically, certain TLRs might be exploited against bioterrorism agents on the basis of their mechanism of action. For example, TLR1 blocks TLR2 and TLR4 signaling. TLR2 recognizes bacterial proteoglycans. TLR3 recognizes viral double-stranded RNA. TLR4 recognizes bacterial LPS and lipoteichoic acids. TLR5 recognizes bacterial flagella. TLR6 is required for TLR2 signaling recognition of bacterial proteoglycans. TLR7 and TLR8 recognize viral single-stranded RNA. TLR9 recognizes bacterial CpG DNA. Studies on macrophages show that TLR stimulation can be sustained, and further amplification by other agonists might be possible.
An understanding of naturally occurring cytokines and immune cells presents the possibility of using this knowledge to modulate the body's own defenses. IFN-α and IFN-β (type I IFNs) exhibit antiviral activity. IFN-γ (type II IFN) plays a role in bacterial infections. IFNs perform these roles through their receptors, which result in phosphorylation of kinases crucial to the cytokine cascade. CD4, expressed on macrophages and dendritic cells (DCs), activates and modulates their maturation and activity against pathogens.
An enemy bioterror attack is likely to target either the respiratory or gastrointestinal tracts because they are the most easily accessible from the external environment. However, chronic or systemic activation of the innate immune system as prophylaxis could have harmful or fatal consequences. Activating these receptors for limited time periods through localized delivery (eg, intranasal or oral) might minimize toxicity, with the potential for retreatment as necessary to maintain short-term innate sensitization against bioterror agents.
There are, however, concerns that TLR agonists that activate plasmacytoid DCs (TLR7 and TLR9 agonists) are more likely to induce autoimmune disease. This has been shown in a number of preclinical trials and human topical studies. Furthermore, some studies have suggested a relationship between IFN levels and autoimmune disease. Further studies must demonstrate not only the efficacy but also the safety in the case of TLRs and those products that increase IFN levels.
Imiquimod (Aldara), resiquimod, and other immune response modifiers
3M Pharmaceuticals has developed a class of immune response modifiers (IRMs) that act by stimulating TLR7 and TLR8 and have exhibited antiviral and antineoplastic activity in human subjects. Aldara has been shown to be of benefit in human clinical studies against human papillomavirus (HPV),2 molluscum contagiosum,3 and leishmaniasis.4 Aldara is US Food and Drug Administration (FDA) approved for use in the treatment of actinic keratoses, basal cell carcinoma, and genital warts.
3M Pharmaceuticals plans to use compounds selective for activation of TLR7, TLR8, or both by using an influenza virus challenge with cytokine response and immunity as outcome measures. Similar experiments and goals are planned for select agents (eg, Hantavirus) in collaboration with the US Army Medical Research Institute of Infectious Diseases. Nasal and pulmonary delivery protection against influenza, as well as pharmacokinetics and pharmacodynamics of systemic treatment, will also be explored.
3M has addressed the questions from Hackett's article as follows: TLR7 and TLR8 should have broad activity against numerous pathogens. Potent antiviral activity in mouse and primate models against herpes simplex virus (HSV) 1 and 2, cytomegalovirus, Banzivirus, Rift Valley fever, influenza, and West Nile has been demonstrated. Their IRM models also suggest utility against Leishmania, Listeria, and Mycobacterium species. Imiquimod is currently applied 2 to 3 times per week with active immunomodulatory effects between treatments. IRM models suggest administration 3 to 4 days before exposure provides adequate anti-infective protection. Rare inflammatory side effects have been reported, specifically in those with T cell–mediated autoimmune disorders that subside after therapy is discontinued. Potential systemic side effects, such as fever, are possible in conjunction with cytokine toxicity. Genetic polymorphisms might play a role in response variability. 3M screened blood from more than 100 individuals and demonstrated variable IRM response, but cytokine production was observed in all subjects. Fatigue and influenza-like symptoms have been reported with use. There is concern about the relationship between TLR7 agonists and autoimmune disease; however, in extensive preclinical and clinical trials with IRMs, autoimmunity has not been observed. Aldara (imiquimod 5% cream) has gone through phase III clinical trials and is approved for the treatment of external genital warts, actinic keratosis, and basal cell carcinoma. Other TLR7/TLR8 agonists are in preclinical and clinical development for multiple indications, including cervical HPV infection.
CpG oligodeoxynucleotide or CPG 7909 and other TLR9 stimulants
Coley Pharmaceutical Group has developed a TLR9 agonist, CPG 7909, which activates both innate and adaptive immune systems. Three different classes of CpG oligodeoxynucleotides (ODNs) are distinguished by their structure and immune effects. A-class (also known as type D) CpG ODNs stimulate plasmacytoid DCs to secrete high levels of IFN-α and natural killer (NK) cells to secrete IFN-γ,5 as well as monocyte maturation into functional DCs,6 with little IL-6 or B-cell stimulation. Conversely, B-class (type K) CpG ODNs induce modest IFN-α production, weak NK cell activation, and profound B-cell and monocyte activation, with secretion of IgM, IL-10, and IL-6.7 C-class CpG ODNs have intermediate immune effects7 and are very stable in vivo, with easy formulation. CPG 7909, a B-class ODN, was the first ODN to enter clinical trials in 1999. CPG 10101, a C-class ODN, recently entered clinical trials as monotherapy for chronic HCV infection. A recently developed type of CpG ODN (fma CpG ODN) by Daniela Verthelyi and Serge Beaucage at the FDA has an altered chemistry that will allow for CpG ODNs to be prepared as prodrugs, thereby easing their production (Beaucage and Verthelyi, unpublished data).
Some studies suggest that intranasal or pulmonary delivery might be superior for protection against inhaled pathogens. There is also some evidence suggesting synergy with antibiotic treatment to infection (Mycobacterium avium and clarithromycin) and the possibility of postexposure efficacy. Class CPG 7909 has been tolerated locally and systemically in more than 700 subjects thus far, with flu-like symptoms and transient injection site reactions commonly seen in patients receiving more than 1 mg. C-class CPG 10101 has shown a similar safety profile in clinical trials with more than 80 healthy volunteers or HCV-infected subjects. Possible proinflammatory activity through the intranasal route and autoimmune disease has been described in mice.8 The use of CpG ODNs in most models requires that they be administered 3 to 6 days before infection. Newly developed fma CpG ODNs have backbones with thermolytic protective moieties that prolong the ODN activity. At 37°C, the ODNs become spontaneously deprotected, ultimately allowing ODNs to act as prodrugs.9
Studies in rodents have shown that CpG ODNs have immunoprotective effects against the following pathogens: bacteria (Bacillus anthracis, Francisella tularensis, Listeria monocytogenes, Klebsiella pneumoniae, Mycobacterium species, Brucella abortus, Burkholderia mallei, Burkholderia pseudomallei, Salmonella typhi, Pseudomonas aeruginosa, and Orientia tsutsugamushi), viruses (vaccinia and other orthopox viruses, arena, Ebola, Venezuelan equine encephalitis, Friend leukemia, HSV papillomavirus, cytomegalovirus, respiratory syncytial virus, murine acquired immunodeficiency virus complex, and influenza), fungus (Cryptococcus neoformans), and parasites (malaria, Leishmania major, and toxoplasmosis). CpG DNA innate protection was evident for a period of from approximately 2 days after treatment up to 2 weeks later, with possible repeated administration for extended protection.10 The experience in primates is more restricted. Protection was only demonstrated thus far when CpG ODNs of type D were administered to macaques infected with L major.11 Although theoretic concerns remain regarding the possibility that TLR9 agonist therapy could trigger harmful inflammation, this has not been seen in initial clinical trials in human subjects. Other concerns include genetic polymorphism.12
Lederer, at Harvard, is working with Coley Pharmaceutical Group on a synergistic platform, combining their CpG ODNs with a double-stranded RNA, Ampligen (see later), which stimulates immune reactions through TLR3. CPG 7909 has shown some positive results in several human clinical trials involving a total of more than 900 subjects as an adjuvant for hepatitis B, flu, and cancer vaccines and in the treatment of melanoma, renal cell carcinoma, non-Hodgkin lymphoma, cutaneous T-cell lymphoma, and lung cancer. A C-class CpG ODN, CPG 10101, has induced greater than 1 log reductions in viral load among patients with chronic hepatitis C virus (HCV) infection.
Immunomodulatory oligonucleotides
Hybridon has developed second-generation immunomodulatory oligonucleotides (IMOs) containing synthetic stimulatory motifs, CpR, YpG, and R′pG, as TLR9 agonists.13 In addition to synthetic motifs, IMO is composed of a novel DNA structure with 3′-3′ attached structure that has higher stability against 3′-exonuclease digestion and is a candidate for oral administration.13 The two 5′ ends facilitate increased TLR9 activation. IMO induces cytokine secretion profiles distinct from CpG dinucleotide. On the basis of immune profiles in various cell-based and in vivo studies, including in nonhuman primates, IMOs are broadly classified into 3 distinct groups.14 Phase I clinical trials in human subjects have shown increased levels of cytokine secretion (including IFN-γ/α). Subcutaneous trials in human subjects have used weekly dosing up to 3 to 4 weeks, with no dose-limiting toxicity observed and no evidence of adverse reactions. IMOs offer potential for innate immune responses, including localized delivery after exposure.
Hybridon has addressed Hackett's questions as follows: On the basis of animal models, TLR9 agonists provide broad protection against bacteria and viruses through TH1 immune responses. TLR9 agonists have produced strong antigen-specific IgG2a antibody responses lasting up to 26 weeks, with successful antigen challenge at week 24 in animals. IMO use could potentially address B anthracis, Yersinia species, variola, dengue, Ebola, Salmonella species, Listeria species, hepatitis A, West Nile Virus (WNV), influenza, rickettsia, rabies, and Severe Acute Respiratory Syndrome (SARS) infections. Humoral responses last up to 26 weeks. IMOs have been shown to be safe when given once a week for up to a year in patients with cancer and have been designed to induce a defined cytokine profile to avoid harmful inflammatory responses. On the basis of transgenic and knockout mouse models, autoimmune reactions might occur in susceptible individuals but are not observed in clinical trials with any CpG-containing oligonucleotides to date. There is no evidence that TLR9 single-nucleotide polymorphisms affect CpG responses or TLR9 signaling. One exception might be polymorphisms in IL-1 receptor–associated kinase 4, leading to decreased responsiveness to innate immunotherapy. CpG DNA and IMO share similar mechanisms of TLR9 activation. Lead IMO is in phase II trials for renal carcinoma and in phase I trials with Remune (HIV-1 whole killed virus) as an adjuvant. Several other IMOs with different immunostimulatory properties are at various stages of development and preclinical studies for a number of disease indications, including infectious diseases.
Thymalfasin
Thymalfasin (thymosin α1), manufactured by SciClone Pharmaceuticals, is a synthetic analog of a thymic peptide.15 It has been shown to have broad immunostimulatory activities both in vitro and in vivo, resulting in enhancement of adaptive and innate immune responses as evidenced by TH1 cell stimulation and increases in NK cell activity and DC activation. Current indications include hepatitis B virus (HBV) and HCV.15 Mouse models suggest innate stimulation in the setting of Aspergillus species infection, leading to increased survival.16
SciClone has addressed Hackett's questions as follows: Thymalfasin might be effective for a range of infections, including viruses (HCV, HBV, influenza, HIV, and herpes), fungi (Candida and Aspergillus species), and bacteria (Listeria species, Pseudomonas species, and Serratia marcescens). Biweekly treatment of human patients up to 1 year has been safe, with no cases of harmful inflammation or autoimmune activation seen in the thousands of human subjects treated with thymalfasin to date. The effect of genetic polymorphism is unknown. Thymalfasin has been approved in more than 30 countries and is in phase III testing in the United States (to be completed by year end 2005).
Ampligen
Recent evidence suggests that among TLRs in airway epithelial cells, TLR3 activation resulted in the greatest increase of innate immune response.17 Hemispherx Biopharma produces Ampligen (poly I: polyC12U, a synthetic double-stranded RNA), a TLR3 agonist that induces IFN cascade and activates critical enzymes (p68 kinase and 2′-5′ adenylate synthetase) normally induced by IFNs by mimicking double-stranded long cytoplasmic RNAs produced during viral infection. Synergistic immune activation is observed when Ampligen is given in conjunction with IFN-α. Ampligen has already been tested against more than 25 viruses and will be used in Advanced Biosystem's proposal for inhaled IFN (in conjunction with Alferon, see later). Ampligen has been demonstrated recently to mediate protective antiviral responses as long as 2 days after experimental infection with Coxsackie B3 virus in a murine model.18 Phase II/III trials of Ampligen have been completed successfully in human subjects with chronic fatigue syndrome and HIV infection. Ampligen therapy was generally well tolerated.
CRX-527
Corixa Corporation is developing TLR4 innate immunomodulators for airway delivery, including CRX-527 and CRX-527. A single-dose, dose-escalation, phase I clinical trial with intranasally delivered CRX-675 (a moderate-strength TLR4 agonist) is currently underway for the treatment of allergic rhinitis. CRX-527 is a highly active synthetic TLR4 agonist that induces innate resistance to airway challenge by viral and bacterial pathogens. Studies in mice have demonstrated that CRX-527 enhances protective immunity in the airways to L monocytogenes, influenza virus, respiratory syncytial virus, and Haemophilus influenzae, with resistance to infection persisting between 4 and 7 days accompanied by induction of cytokines, chemokines, defensins, and type 1 IFNs. Animal models suggest response within hours of administration and lasting up to a week. Weekly or biweekly administration can extend protection without inducing tolerance. Protective doses of CRX-527 are significantly lower than toxic doses, suggesting a safe therapeutic index.19, 20 A phase I, multidose, dose-escalation trial with CRX-527 (a high-activity TLR4 agonist) is scheduled for early 2006. It is expected that CRX-527's effects will last between 3 and 7 days, with biweekly dosing extending the duration for 6 to 12 weeks. Redosing might avoid prolonged innate stimulation and potential autoimmune reactions. Biweekly intranasal delivery has not been associated with systemic inflammation. Corixa does not anticipate that genetic polymorphism will significantly affect TLR4 agonist responses; however, a recent study suggests phylogenetic and individual diversity in TLR4 responses, suggesting possible variability in efficacy for TLR4 agonists. A phase I clinical trial with CRX675 was recently completed, with no serious or severe adverse events reported. Additional clinical trials with CRX527 (phase I safety study) and CRX675 (phase II) are planned for 2006.
IFNs
IFN-α plays a significant role in innate immunity to viral infections. There is also some evidence of viral decoy mechanisms, whereby some viruses have developed mechanisms to evade innate immunity.21 A multicomponent approach to IFN treatment could possibly overcome this potential form of innate resistance. Activating the innate immune system through IFNs raises safety issues regarding possible adverse reactions, including microbial reactions (as seen in sarcoid-like disease in mice)22 and those seen in current treatment of hepatitis infection with pegylated IFNs.23
Alferon
Hemispherx Biopharma also produces Alferon, a natural IFN-α. Alferon-N injection might soon be available in oral form and has a well-documented safety record. Animal studies suggest that oral IFN-α might be active at much lower doses than when administered parenterally. It is expected that oral doses will be 1000 times lower than those already FDA approved for parenteral administration. Alferon does not induce antibody formation (as opposed to recombinant forms of IFN-α) but does show broad-spectrum antiviral activity in chronic viral diseases. One of the benefits of Alferon might be that it is multispecies, offering various binding affinities yielding greater antiviral activity. Questions Hemispherx intends to address include dosing comparisons, route of delivery, in vivo versus in vitro peripheral blood responses, potential refractory period, synergy with Ampligen or other products, and whether Alferon renders effective prophylaxis.
Hemispherx is also developing Oragen, a class of 2′-5′ adenylate analogs that activate part of the IFN cascade, thus augmenting innate immunity. It is expected that Oragen, taken in combination with either Alferon or Ampligen will further increase innate protection against viral decoy devices. Oragen is a potential mediator of an end product of innate immunity cascade (RNAse-L activator). Hemispherx intends to address route of delivery (oral), effective prophylaxis, and synergy (with Ampligen or Alferon) to inactivate viral decoy mechanisms for Oragen.
Hemispherx Biopharma has addressed Hackett's questions regarding Ampligen, Alferon, and Oragen as follows: TLR3 agonists might provide the best innate immune response to respiratory exposure of microbiologic agents on the basis of the study by Sha et al.17 Coupling this with systemic IFNs should stimulate the broadest innate immune responses. Systemic innate antiviral immunity is expected by Alferon through buccal absorption. Ampligen lasts 3 to 4 days through TLR3 activation, with secondary activators (IFNs) lasting roughly the same length of time (as evidenced by animal and human experience). No evidence of inflammatory reactions has been observed in either Ampligen or Alferon. Administration of more than 50,000 doses of Ampligen has not resulted in a cell-mediated autoimmune response. Alferon should be superior to single recombinant IFN types with regard to binding to receptors that are structurally altered by genetic polymorphisms. Hemispherx doubts that there will be a significant role for genetic polymorphism in Ampligen responsiveness on the basis of the lack of clinically serious adverse reactions to date. A multivalent approach might optimize innate stimulation for pathogen protection while minimizing adverse reactions. FDA approval might be slow for agents proposed in combination. Hemispherx has initiated an Alferon LDO study in HIV-infected individuals to study the pattern of genes upregulated in the systemic circulation.
Nebulized IFNs
AFG Biosolutions (formerly Advanced Biosystems) suggests the use of nebulized IFNs (α and γ) in the form of a metered-dose inhaler. Data in mice demonstrate prevention of lethal respiratory vaccinia infections with IFN-α and IFN-γ.24 Proposed effectiveness might include early postexposure studies for bunyaviruses, arenaviruses, flaviviruses, coronaviruses, and orthomyxoviruses.
AFG Biosolutions has addressed the aforementioned questions as follows: IFNs ultimately provide the antiviral and immunoregulatory protection induced by TLR3, TLR7/TLR8, and TLR9 ligands for pre-exposure prophylaxis. Exogenous administration of IFNs will activate antiviral pathways faster than TLR ligands and might be an effective form of preventative therapy before and after exposure for certain viral infections. Anticipated duration is between days and weeks for a daily dose of IFNs given for 5 to 14 days (or biweekly doses given over 1 to 2 weeks if using pegylated IFNs). There are side effects to chronic IFN treatment seen thus far in HBV and HCV treatment, including flu-like symptoms, fatigue, depression, nausea, weight loss, musculoskeletal pain, and leucopenia. However, midterm to long-term protection can be administered for weeks to months at nontoxic doses. Prophylaxis through IFNs could trigger harmful inflammation because IFN therapy has resulted in autoimmune reactions in various case reports with large doses of IFNs. Smaller doses over a short period of time or autoantibody serial testing to monitor for exacerbations might limit possible autoimmune reactions. Genetic polymorphism is likely to play a role in IFN responsiveness. Race appears to affect responsiveness to IFN therapy in HCV (African American subjects have a reduced response relative to white subjects). Published data cited by AFG Biosolutions demonstrate the safety of aerosol IFNs; however, in-depth evaluation for treatment in infectious diseases in human subjects must be demonstrated. Literature from the mid-1980s using aerosolized IFNs for treatment of rhinovirus infections shows that aerosolized IFNs resulted in some reduction in virus replication but little relief from symptoms and increased blood and nasal secretions compared with placebo.25, 26 Inhaled IFN is likely to have similar side effects to current routes of administration, which might be acceptable in the setting of a biothreat agent (BTA). AFG Biosolutions, Inc, is conducting preclinical studies on IFN prophylaxis of viral infections: efficacy, toxicity, pharmacokinetics, optimal regimen, and delivery in rabbit models.
CEL-1000
NMRC/CEL-SCI suggests CEL-1000 as a strategy to enhance innate immune responses after pathogen exposure. This synthetic peptide contains an amino acid sequence derived from the β chain of MHC class II and stimulates TH1 responses to numerous infectious agents. Administration of CEL-1000 resulting in increased serum IFN-γ levels and number of CD4+/IFN-γ+ T cells and an antimalaria state.27 CEL-1000 is water soluble and can be administered through subcutaneous, intramuscular, or oral routes. Mouse models suggest prophylactic protection against Plasmodium yoelii, Leishmania major, HSV1, and an arboencephalitis virus.
NMRC/CEL-SCI/NEOUCOM have addressed the questions above as follows: CEL-1000 has been shown in mice to induce protection against diverse microbial infections (parasitic and viral) by CD4 molecules on T cells and CD4 T cells, leaving them in a state of readiness through TH1 responses on infectious challenge. Mouse studies suggest protection from lethal HSV1 between 2 weeks (intramuscularly and orally) and 2 months (subcutaneously) after administration of CEL-1000, which can be given up to 3 days after infection. Malaria models suggest a duration of several months, with protection present 7 days after a single CEL-1000 injection. Two injections of CEL-1000 induced complete protection within 2 to 4 weeks, decreasing after 4 months. No signs of harmful inflammation were observed in animal models with CEL-1000. No evidence suggests that CEL-1000 promotes autoimmune reactions or retroviral activation. No detectable antibodies to CEL-1000 or mouse cells (blood, spleen, and liver) were found after CEL-1000 administration. It is possible that genetic polymorphism might affect CEL-1000's responsiveness. Evidence was found in mice with a defect in the IL-12 receptor that were prone to TH2 responses that CEL-1000 was unable to completely protect against HSV1 or Plasmodium yoelii. CEL-1000 is in the early stages of development, and thus there are many unanswered questions regarding safety in human subjects and its therapeutic range.
SCV-07
SciClone proposes use of SCV-07, a dipeptide immunomodulator originally developed in Russia, to stimulate the innate immune system in the setting of BTA. SCV-07 appears to stimulate macrocytic phagocytosis and TH1 cytokines (IL-2), with use demonstrated against bacteria (tuberculosis)28 and viruses (Pichinde and HPV). Currently,it is administered subcutaneously, but oral dosage forms are bioavailable. Duration is expected to be between weeks and months. No cases of harmful inflammation, autoimmune reaction, or retroviral activation have been seen in the animals and hundreds of human subjects treated with SCV-07 to date. The effects of genetic polymorphism have not yet been evaluated with regard to SCV-07 responsiveness. SCV-07 is approved for use in Russia and is in phase I testing in the United States.
Cytokine blockade
TLR signal transduction might be essential to triggering the fatal sepsis cascade.29, 30 Kevin Tracey and colleagues propose 2 compounds, CAP68 and anti-high mobility group box 1 (anti-HMGB1) antibody, to counter septic shock.
HMGB1 is a late cytokine produced by activated macrophages that mediates lethal sepsis in animals.28 During lethal infection, HMGB1 can also be released by tissue, increasing organ damage. Anti-HMGB1 antibodies have been shown to prevent death in animal models by inhibiting the activity of HMGB1, which disrupts epithelial cells in organs.30 Tracey and colleagues are currently investigating this possibility for α7 expression and cholinergic agonist responsiveness. CAP68 (acetylcholine agonist) is a small molecule that prevents overproduction of cytokines like TNF and HMGB1. This is accomplished by targeting α7 receptors on macrophages, which in turn inhibit nuclear factor κB activity, the cytokine regulator responsible for the release of the lethal cytokines TNF and HMGB1. Blocking HMGB1 has already been shown to prevent death caused by peritonitis,30 supporting CAP68's possible role in lethal infection. It is likely that anti-HMGB1 antibodies will enter clinical trials within 12 to 18 months.
Treatment is expected to last 1 to 2 weeks, with suppression of cytokine overproduction lasting between days and weeks. Both compounds appear to be effective before exposure or after disease onset. Safety has not yet been determined. Human genetic polymorphisms could play a role in shock modulation because some patients might have a genetic predisposition to overexpress TNF, whereas others might have low levels of TNF but high levels of IFN-γ, which increases TNF activity.
Neuroactive agents
Webster and Sternberg (National Institute of Mental Health/National Institutes of Health)31 propose the use of drugs selected from the general category of agents that target the central nervous system as a potentially novel way to inhibit bacterial shock. Several arms of the central nervous system are known to regulate immune responses, including the hypothalamic-pituitary-adrenal axis, the sympathetic nervous system, the parasympathetic nervous system, and the peripheral nervous system. Many agents in this category are already available or have reached early clinical trials and are currently being developed for other purposes than BTA defense, suggesting a good possibility of approval in 5 years ( Fig 2).
Fig 2.
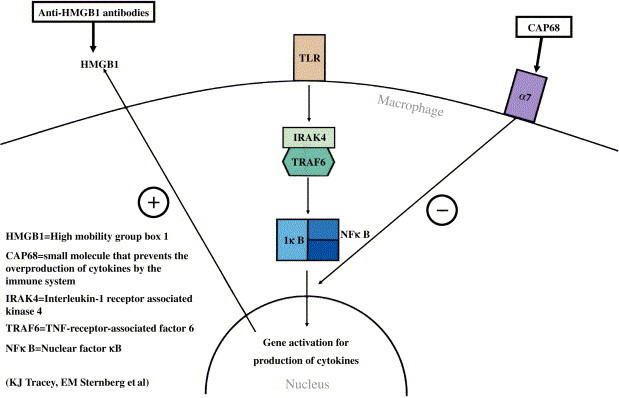
Small-molecule and antibody approach to cytokine overproduction.
Focused conclusions
After review of the literature and submitted proposals, we suggest the following responses to Hackett's questions:
-
1.
In theory, the most broadly active immunopotentiators should yield the broadest protective response. Candidates for innate immune receptor activation include TLR3, TLR4, TLR7, TLR8, TLR9, TH1, CD4, and the IFN-α and IFN-γ receptors. Concern exists for variable expression of targets between small animal models and human models (eg, human subjects express TLR9 and TLR8, whereas mice only express TLR9 but in a much greater variety of cell types). Thus a correlate between the efficacy and duration seen in animal models must take into account these species-specific differences to accurately represent human response to immunomodulation. The most likely route of attack (respiratory or gastrointestinal) is probably the ideal route of protection to minimize drug exposure.
-
2.
Innate immunity is relatively brief in duration, especially in the absence of an active threat or stimulus. Protection with immunomodulators usually lasts between 1 and 2 weeks, regardless of the mechanism targeted. The potential for readministration should extend the protective effects for greater than 2 weeks. The exact duration will vary on mechanism of action, delivery method, and any adverse effects of chronic use.
-
3.
Enhancing innate immunity carries an inherent risk of inflammatory response. Some immunomodulators, such as TLR4 agonists, raise concerns for the possibility of innate overactivity on the basis of their pivotal role in the development of septic shock.
-
4.
Some individuals are more prone to autoimmune reaction or retroviral activation with innate immunomodulation. This becomes increasingly more likely if the target yields polyclonal B-cell activation (ie, TLR9 and TLR7/TLR8 agonists) because of increased likelihood for loss of self-tolerance and development of autoantibodies. There is also some concern for the possible relationship between IFN and autoimmunity that remains to be established.
-
5.
Genetic polymorphism is an important potential variable, depending on the molecular target selected for therapy. Better comprehension of genetic variability could lead to improved treatment for nonresponders and discovery of novel targets for innate immunomodulation and could affect adverse events (inflammatory response) in predisposed individuals.
Acknowledgments
We thank the following: Charles J. Hackett, PhD, Deputy Director, Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Diseases; David Winter, PhD, Program Officer, Basic Immunology Branch, Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Disease; Esther M. Sternberg, MD, National Institute of Mental Health; Yupin Charoenvit, PhD, Naval Medical Research Center; Daniel Zimmerman, PhD, CEL-SCI Corporation; Kenneth Rosenthal, PhD, Northeastern Ohio Universities College of Medicine; Arthur M. Krieg, MD, Coley Pharmaceutical Group; David R. Strayer, MD, HemispheRx Biopharma; David H. Persing, MD, PhD, Corixa Corp; Cynthia W. Tuthill, PhD, SciClone Pharmaceuticals; Kevin J. Tracey, MD, Long Island Neurological Research Institute; Ekambar R. Kandimalla, PhD, Hybridon Inc; Claas Ulrich, PhD, Charite University; Mark A. Tomai, PhD, 3M Pharmaceuticals; Kenneth Alibek, MD, Ge Liu, PhD; Svetlana Hopkins, PhD and Alison Andrews, MD, AFG Biosolutions (formerly Advanced Biosystems); James A. Lederer, PhD, Harvard University; and Dara W. Frank, PhD and Peter G. Sohnle, MD, Medical College of Wisconsin for guidance. We also thank Deborah A. Dye for administrative support.
Milwaukee, Wis, Ann Arbor, Mich, and Washington, DC
Footnotes
Supported by a Distinguished Research Professorship (Dr. Whelan), the Center for Technology, National Security Policy National Defense University; the Chad Baumann Research Fund Endowment (Dr Whelan); the Bleser Foundation Endowed Professorship (Dr Whelan); and Defense Advanced Research Projects Agency contract DAAH01-03-C-R120 (Drs Whelan and Amlie-Lefond).
References
- 1.Hackett C.J. Innate immune activation as a broad spectrum biodefense strategy: prospects and research challenges. J Allergy Clin Immunol. 2003;112:686–694. doi: 10.1016/S0091-6749(03)02025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox J.T., Petry K.U., Rylander E., Roy M. Using imiquimod for genital warts in female patients. J Womens Health (Larchmt) 2004;133:265–271. doi: 10.1089/154099904323016428. [DOI] [PubMed] [Google Scholar]
- 3.Theos A.U., Cummins R., Silverberg N.B., Paller A.S. Effectiveness of imiquimod cream 5% for treating childhood molluscum contagiosum in a double-blind, randomized pilot trial. Cutis. 2004;74:134–138. 141-2. [PubMed] [Google Scholar]
- 4.Arevalo I., Ward B., Miller R., Meng T.C., Nagar E., Alvarez E. Successful treatment of drug-resistant cutaneous leishmaniasis in humans by use of imiquimod, an immunomodulator. Clin Infect Dis. 2001;33:1847–1851. doi: 10.1086/324161. [DOI] [PubMed] [Google Scholar]
- 5.Krug A., Rothenfusser S., Hornung V., Jahrsdorfer B., Blackwell S., Ballas Z.K. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur J Immunol. 2001;31:2154–2163. doi: 10.1002/1521-4141(200107)31:7<2154::aid-immu2154>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 6.Gursel M., Verthelyi D., Klinman D.M. CpG oligodeoxynucleotides induce human monocytes to mature into functional dendritic cells. Eur J Immunol. 2002;32:2617–2622. doi: 10.1002/1521-4141(200209)32:9<2617::AID-IMMU2617>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 7.Vollmer J., Weeratna R., Payette P., Jurk M., Schetter C., Laucht M. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur J Immunol. 2004;34:251–262. doi: 10.1002/eji.200324032. [DOI] [PubMed] [Google Scholar]
- 8.Obermeier F., Dunger N., Deml L., Herfarth H., Scholmerich J., Falk W. CpG motifs of bacterial DNA exacerbate colitis of dextran sulfate sodium-treated mice. Eur J Immunol. 2002;32:2084–2092. doi: 10.1002/1521-4141(200207)32:7<2084::AID-IMMU2084>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 9.Grajkowski A., Pedras-Vasconcelos J., Wang V., Ausin C., Hess S., Verthelyi D. Thermolytic CpG-containing DNA oligonucleotides as potential immunotherapeutic prodrugs. Nucleic Acids Res. 2005;33:3550–3560. doi: 10.1093/nar/gki657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klinman D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat Rev Immunol. 2004;4:249–259. doi: 10.1038/nri1329. [DOI] [PubMed] [Google Scholar]
- 11.Verthelyi D., Gursel M., Kenney R.T., Lifson J.D., Liu S., Mican J. CpG oligodeoxynucleotides protect normal and SIV-infected macaques from Leishmania infection. J Immunol. 2003;170:4717–4723. doi: 10.4049/jimmunol.170.9.4717. [DOI] [PubMed] [Google Scholar]
- 12.Leifer C.A., Verthelyi D., Klinman D.M. Heterogeneity in the human response to immunostimulatory CpG oligodeoxynucleotides. J Immunother. 2003;26:313–319. doi: 10.1097/00002371-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Wang D., Kandimalla E.R., Yu D., Tang J.X., Agrawal S. Oral administration of second generation immunomodulatory oligonucleotides induces mucosal Th1 immune responses and adjuvant activity. Vaccine. 2005;23:2614–2622. doi: 10.1016/j.vaccine.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 14.Kandimalla E.R., Bhagat B., Li Y., Yu D., Wang D., Cong Y.P. Immunomodulatory oligonucleotides containing a cytosine-phosphate-2′-deoxy-7-deazaguanosine motif as potent Toll-like receptor 9 agonists. Proc Natl Acad Sci U S A. 2005;102:6925–6930. doi: 10.1073/pnas.0501729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Billich A. Thymosin alpha1. SciClone Pharmaceuticals. Curr Opin Investig Drugs. 2002;3:698–707. [PubMed] [Google Scholar]
- 16.Romani L., Bistoni F., Gaziano R., Bozza S., Montagnoli C., Perruccio K. Thymosin alpha 1 activates dendritic cells for antifungal Th1 resistance through toll-like receptor signaling. Blood. 2004;103:4232–4239. doi: 10.1182/blood-2003-11-4036. [DOI] [PubMed] [Google Scholar]
- 17.Sha Q., Truong-Tran A.Q., Plitt J.R., Beck L.A., Schleimer R.P. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31:358–364. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- 18.Padalko E., Nuyens D., De Palma A., Verbeken E., Aerts J.L., De Clercq E. The interferon inducer ampligen [Poly(I)-Poly (C12U)] markedly protects mice against coxsackie B3 virus-induced myocarditis. Antimicrob Agents Chemother. 2004;48:267–274. doi: 10.1128/AAC.48.1.267-274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cluff C.W., Baldridge J.R., Stöver A.G., Evans J.T., Johnson D.A., Lacy M.J. Synthetic toll-like receptor 4 agonists stimulate innate resistance to infectious challenge. Infect Immun. 2005;73:3044–3052. doi: 10.1128/IAI.73.5.3044-3052.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stöver A.G., Da Silva Correia J., Evans J.T., Cluff C.W., Elliott M.W., Jeffery E.W. Structure-activity relationship of synthetic toll-like receptor 4 agonists. J Biol Chem. 2004;279:4440–4449. doi: 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]
- 21.Hahm B., Trifilo M.J., Zuniga E.I., Oldstone M.B. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22:247–257. doi: 10.1016/j.immuni.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 22.Nishiwaki T., Yoneyama H., Eishi Y., Matsuo N., Tatsumi K., Kimura H. Indigenous pulmonary propionibacterium acnes primes the host in the development of sarcoid-like pulmonary granulomatosis in mice. Am J Pathol. 2004;165:631–639. doi: 10.1016/S0002-9440(10)63327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matthews S.J., McCoy C. Peginterferon alfa-2a: a review of approved and investigational uses. Clin Ther. 2004;26:991–1025. doi: 10.1016/s0149-2918(04)90173-7. [DOI] [PubMed] [Google Scholar]
- 24.Liu G., Zhai Q., Schaffner D.J., Wu A., Yohannes A., Robinson T.M. Prevention of lethal respiratory vaccinia infections in mice with interferon-alpha and interferon-gamma. FEMS Immunol Med Microbiol. 2004;40:201–206. doi: 10.1016/S0928-8244(03)00358-4. [DOI] [PubMed] [Google Scholar]
- 25.Sperber S.J., Hayden F.G. Antiviral chemotherapy and prophylaxis of viral respiratory disease. Clin Lab Med. 1987;7:869–896. doi: 10.1016/S0272-2712(18)30721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayden F.G. Clinical applications of antiviral agents for chemophrophylaxis and therapy respiratory viral infections. Antiviral Res. 1985;(suppl 1):229–239. doi: 10.1016/s0166-3542(85)80033-4. [DOI] [PubMed] [Google Scholar]
- 27.Charoenvit Y., Brice G.T., Bacon D., Majam V., Williams J., Abot E. A small peptide (CEL-1000) derived from the beta-chain of the human major histocompatibility complex class II molecule induces complete protection against malaria in an antigen-independent manner. Antimicrob Agents Chemother. 2004;48:2455–2463. doi: 10.1128/AAC.48.7.2455-2463.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simbirtsev A., Kolobov A., Zabolotnych N., Pigareva N., Konusova V., Kotov A. Biological activity of peptide SCV-07 against murine tuberculosis. Russ J Immunol. 2003;8:11–22. [PubMed] [Google Scholar]
- 29.Erlandsson H., Andersson U. The nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol. 2004;34:103–112. doi: 10.1002/eji.200424916. [DOI] [PubMed] [Google Scholar]
- 30.Yang H., Ochani M., Li J., Quiang X., Tanovic M., Harris H.E. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webster J.I., Sternberg E.M. Role of the hypothalamic-pituitary-adrenal axis, glucocorticoids and glucocorticoid receptors in toxic sequelae of exposure to bacterial and viral products. J Endocrinol. 2004;181:207–221. doi: 10.1677/joe.0.1810207. [DOI] [PubMed] [Google Scholar]