

Graphical abstract
Abbreviations: SI, selectivity index; IC50, the concentrations that inhibit HBV DNA replication, and HBsAg and HBeAg secretions to 50%; CC50, the concentration that achieved 50% cytotoxicity against cultured cells; ADV, adefovir dipivoxil
Keywords: Polyoxometalates, Cs2K4Na[SiW9Nb3O40]·H2O, Antiviral agent, Hepatitis B virus, HepG2.2.15
Abstract
To synthesise and characterize the polyoxometalate Cs2K4Na[SiW9Nb3O40]·H2O 1 for its anti-hepatitis B virus (HBV) properties by using the HepG2.2.15 cell. The methylthiazol tetrazolium assay was used to evaluate the growth inhibitory effect of Compound 1 on HepG2.2.15 cell. By using ELISA and real-time PCR, respectively, the presence of extracellular hepatitis B surface antigen (HBsAg), e antigen (HBeAg), and HBV DNA were measured. The levels of intracellular HBV DNA and mRNA were determined by using Southern blot or reverse-transcription-PCR, respectively. Intracellular distribution of antigen were measured by Western blot. A 1995 μmol/L concentration of the commercially-available hepatitis B drug, adefovir dipivoxil (ADV), was required to achieve 50% cytotoxicity against cultured cells (CC50) by day nine; in contrast, only 1747 μmol/L of Compound 1 was required for the same result. Treatment of HepG2.2.15 cells with Compound 1 effectively suppress the secretion of HBV antigens and HBV DNA in a dose-dependent and time-dependent manner. IC50 values were determined to be 80 μmol/L for HBsAg, 75 μmol/L for HBeAg and 3.72 μmol/L for supernatant HBV DNA at day nine post-exposure, as opposed to 266, 296, 30.09 μmol/L, respectively, for ADV. Intracellular HBV DNA, mRNA and antigen were also found to be decreased by Compound 1. The same dose of ADV yielded a significantly less robust inhibitory effect. Compound 1 can clear HBV from hepatic cells and may represent a therapeutic agent to treat HBV infection.
Approximately 2 billion people worldwide have been infected with hepatitis B and about 350 million live with chronic infection. HBV belongs to the Hepadnaviridae family and is a non-cytopathic DNA virus that can replicate via reverse-transcription of an RNA intermediate.1 The pregenomic RNA (pgRNA) intermediate is produced inside the cellular nucleus. Next, the pgRNA is exported to the cytoplasm, where a virally-encoded polymerase can then convert the pgRNA into minus-strand DNA; ultimately, a double-stranded, relaxed circular DNA molecule is generated.2, 3 This life cycle places HBV into the larger family of retroviruses, which are characterized by the reverse-transcription activity involving an RNA intermediate.
Chronic hepatitis B patients are commonly treated with either interferon alpha (INF-α) or the nucleoside analog lamivudine (3TC), adefovir, entecavir, or telbivudine, which are all synthetic reverse-transcriptase inhibitors.4, 5 INF-α inhibits viral replication and acts as an immuno-modulator, but is limited in effectiveness (40% response rate),6 is costly, and elicits serious, undesirable side effects. 3TC mediates a reversible inhibition event, but continuous treatment has been associated with development of drug-resistant HBV variants in up to 70% of patients after 4 years of treatment.7 Adefovir, entecavir, and telbivudine are used when 3TC-resistant strains are detected; however, resistances to these drugs have also been reported.8 Combination therapy, such as a nucleoside analog plus INF-α, is a promising approach that may yield additive or synergistic effects and may reduce the emergence of resistant strains. Unfortunately, the serious side effects associated with each of the agents used remain and may even be compounded. Therefore, it is critical to develop therapeutic agents with improved efficacy and minimal to no side effects.
Polyoxometalates (POMs) are metal-oxide cluster anions which have applicable potential in electronics, magnetics, homogeneous and heterogeneous photo-catalysis, and in medicine where they can act as anti-tumor, -viral, and -bacterial agents.9 Many studies have found evidence that POMs can inhibit the replication of both RNA viruses and DNA viruses.9, 10, 11, 12 In particular, decreased activities of the human immunodeficiency virus, severe acute respiratory syndrome coronavirus, influenza virus, and herpes simplex virus have been observed.10 The mechanism of antiviral action remains to be fully elucidated, but may occur at any of the life cycle stages, including viral adsorption, penetration, or reverse-transcription.10, 11, 13
The niobium-substituted-heteropolytungstates were shown to have potent anti-HIV-1 activity while being minimally cytotoxic.14 Therefore, we sought to investigate the antiviral effect of a POM Cs2K4Na[SiW9Nb3O40]·H2O 1, by using HepG2.2.15 cells, which secrete HBsAg, HBeAg, and complete DNA particles. The results indicate that Compound 1 exhibits strong antiviral activities against the HBV viruses with low cytotoxicity, indicating that it is a potential medicinal candidate against HBV viruses. To date, very few studies of hepatitis B virus treatment by POMs have been reported, yet the potent antiviral activity of these molecules is promising for clinical application.
X-ray crystallography has shown that Compound 1 consists of a polyoxoanion [SiW9Nb3O40]7−, alkali cations, and an isolated water molecule (Table 1 ). In Compound 1, the polyoxoanion [SiW9Nb3O40]6− exhibits a well-known Keggin structure, which is quite similar to that found in other previously described POMs14, 15 (Fig. 1 ). The central Si atom is surrounded by a tetrahedron with oxygen vertices that are each linked to one of the M3O13 (M = W, Nb) groups. Each M3O13 consists of three MO6 octahedra linked in a triangular arrangement with shared edges, and the four M3O13 units are linked together by their corners. In the polyoxoanion, there exist three sites with occupancy disorder in the W4, W5, W6 centers; that is, Nb1 and W4, Nb2 and W5, and W6 and Nb3 atoms share the same site with 50% occupancy, respectively, forming the polyoxoanion [SiW9Nb3O40]6−. The Si–O bond distance is in the range of 1.57(9) to 1.60(6) Å. The W(Nb)–O distances can be grouped into two sets: M–Ot (terminal; 1.67(4)–1.71(5) Å), M–Ob (bridge; 1.70(7)–2.29(6) Å).
Table 1.
Crystal data and structure refinements for Compound 1
| Compound | 1 |
|---|---|
| Formula | H2Cs2K4NaNb3O41SiW9 |
| fw | 3064.70 |
| T[K] | 293(2) |
| Crystal system | Trigonal |
| Space group | P-3c1 |
| a [Å] | 22.6866(10) |
| b [Å] | 22.6866(10) |
| c [Å] | 16.9770(16) |
| α [°] | 90 |
| β [°] | 90 |
| γ [°] | 120 |
| V [Å3] | 7567.1(9) |
| Z | 6 |
| ρcalcd [Mg m−3] | 4.035 |
| μ [mm−1] | 22.955 |
| Rint | 0.0807 |
| Data/restraints/parameters | 4482/30/200 |
| Goodness-of-fit on F2 | 1.106 |
| R1a [I>2σ(I)] | 0.1216 |
| wR2b [I>2σ(I)] | 0.3175 |
| Largest residuals [e.Å−3] | 9.874/−5.713 |
Figure 1.
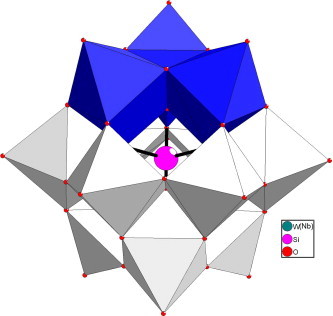
Polyhedral representation of the [SiW9Nb3O40]7− polyoxoanion. The M centers are in blue, and the grey spheres represent the tungsten atoms.
To determine whether Compound 1 suppressed HBV production by inducing cytotoxicity, HepG2.2.15 cells were exposed to the drug and cellular viability was analyzed by MTT assay. No apparent cytotoxicity was observed for up to 9 days after treatment with Compound 1 at lower concentrations (156.25 and 312.5 μmol/L) (Fig. 2 ). This finding suggested that the suppressive activities of Compound 1 on supernatant viral DNA and antigen levels was not a result of induced cytotoxicity. The CC50 for Compound 1 was determined to be 1747 μmol/L at day nine (Fig. 2). The corresponding CC50 for ADV was 1995 μmol/L.
Figure 2.
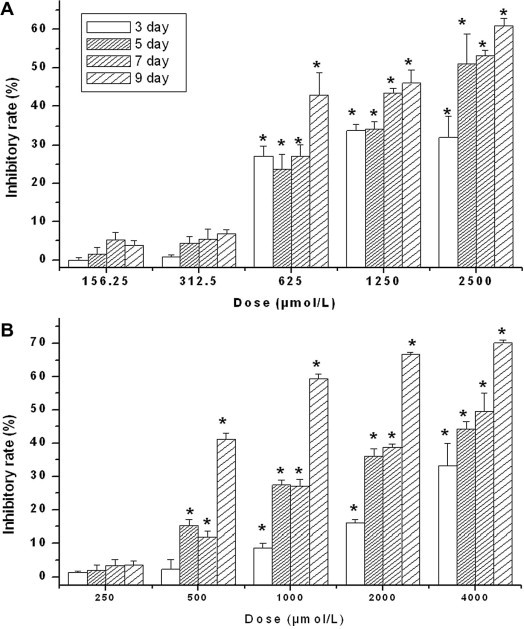
Cytotoxicity of Compound 1 (A) and ADV (B) in HepG2.2.15 cells. The data represent mean ± SD (n = 4). ∗P <0.05 versus the corresponding non-treated control (Student’s t-test).
Anti-HBV activities of Compound 1 and ADV were investigated and compared. HepG2.2.15 cells were treated with various concentrations of Compound 1 or ADV. Secretion of HBeAg, HBsAg, and HBV DNA were found to be significantly less for each compared to levels detected in supernatants from non-treated controls (Fig. 3 ).
Figure 3.
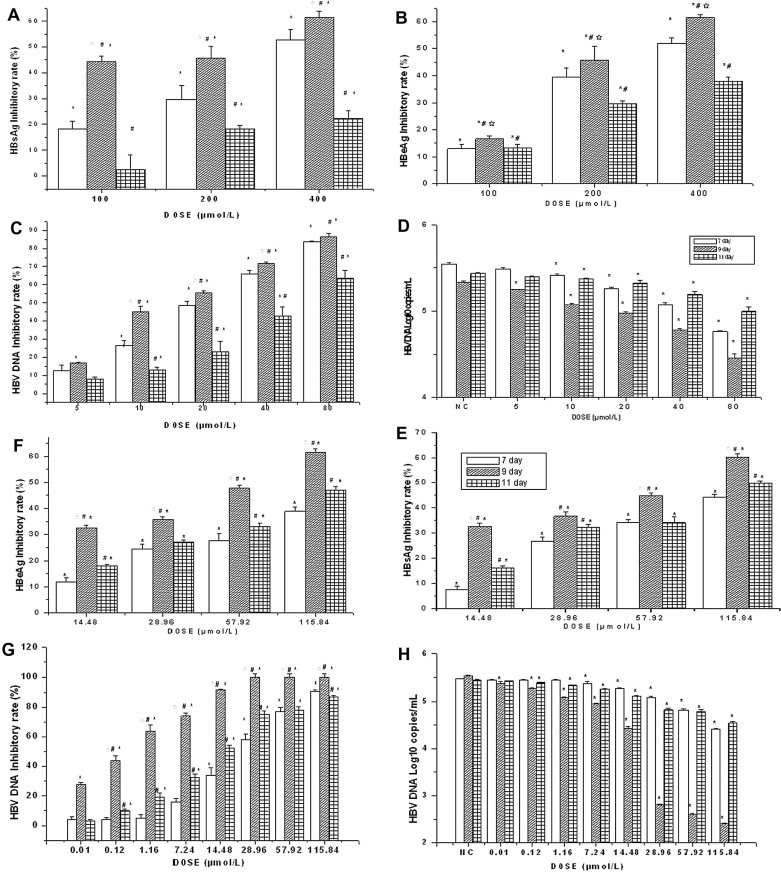
Effects of ADV (left panels) or Compound 1 (right panels) on secreted HBsAg (A, E), HBeAg (B, F), and HBV DNA (C, D, G, H) from HepG2.2.15 cells. Inhibition rate of secreted antigen levels are presented as % of that detected in non-treated controls. Viral DNA levels are presented as% (C, G) and copies/mL (D, H). Data are expressed as mean ± SD (n = 4). NC: negative control group. ∗P <0.05 versus the corresponding non-treated control; #P <0.05 versus the corresponding day 7 outcome of the same dose; ∗P <0.05 versus the corresponding day 11 outcome of the same dose.
As shown in Figure 3, treatment with 14.48 μmol/L Compound 1 significantly reduced HBsAg and HBeAg in a dose-dependent manner, as compared with non-treated controls (P <0.01). The dose of 0.01 μmol/L was able to reduce HBV DNA at 9 days after treatment.
The data clearly showed that the inhibitory activity of Compound 1 on HBsAg, HBeAg, and HBV DNA was promoted over time (vs non-treated controls, P <0.01). The inhibitory activity of Compound 1 on the ninth day of treatment was higher than on day seven. Notably, these results indicated that the production of HBsAg and HBeAg was only slightly suppressed by low doses of Compound 1, but supernatant HBV DNA levels were dramatically suppressed by the same dosage.
The IC50 for Compound 1 and ADV at 9 days is shown in Table 2 . The Compound 1’s SI was greater than that for ADV at day nine, especially when HBV DNA was considered.
Table 2.
Anti-HBV activity and cytotoxicity of Compound 1 in vitro at day 9 of treatment
| Compound (μmol/L) | Cytotoxicity | HBV DNA |
HBeAg |
HBsAg |
|||
|---|---|---|---|---|---|---|---|
| CC50a | IC50b | SI | IC50b | SI | IC50b | SI | |
| Compound 1 | 1747 | 3.72 | 469.0 | 75 | 23.0 | 80.00 | 21.0 |
| ADV | 1995 | 30.09 | 66.3 | 296 | 6.7 | 266.00 | 7.5 |
Selectivity index was determined as the CC50/IC50 value.
The cytotoxicity concentration of Compound 1 or ADV that reduced cell viability to 50% (CC50).
The concentrations of Compound 1 or ADV needed to inhibit.
Interestingly, at day 11 (4 days after the end of exposure to the drug), the inhibition rates of Compound 1 on serum HBsAg, HBeAg, and HBV DNA were lower than those observed on day nine. However, they were not lower than those on day seven, indicating that Compound 1 may have a persistent effect on suppressing HBV. In contrast, the inhibition rate of ADV on serum HBsAg, HBeAg, and HBV DNA declined steadily and were lower than on day seven (Fig. 3).
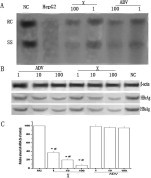
To characterize the intracellular effect by which Cs2K4Na[SiW9Nb3O40]·H2O inhibits HBV in HepG2.2.15 cells, we analyzed the amounts of HBV DNA (intermediates), mRNA present after 4 days of exposure to Cs2K4Na[SiW9Nb3O40]·H2O. Intracellular HBV intermediates were analyzed by Southern blotting. As shown in Figure 4 , Cs2K4Na[SiW9Nb3O40]·H2O and ADV can reduce the level of HBV intermediates in a dose-dependent manner. qRT-PCR results revealed that the levels of HBV mRNA was decreased significantly compared to those detected from the negative control group (P <0.05). The peak inhibition rate of mRNA (93%) was achieved with a dose of 100 μmol/L, as determined when compared with levels detected in the non-treated control group. Moreover, the levels of mRNA was lower than those detected in cells treated with a similar dose of ADV (P <0.05). These results suggested that Compound 1 acted by detrimentally impacting viral DNA replication and viral mRNA transcription in HepG2.2.15 cells and that the anti-HBV effect of Compound 1 seemed to be at least partially similar to that of ADV and the suppression effect was more robust.
Figure 4.
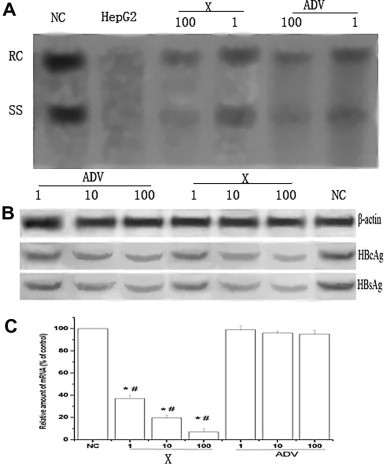
Effects of Cs2K4Na[SiW9Nb3O40]·H2O or adefovir dipivoxil on intracellular HBV DNA replication (A), intracellular HBcAg and HBsAg; (B) and intracellular mRNA; (c) in HepG2.2.15 cells. Cells were treated with compound for 4 days. Data are expressed as mean ± SD (n = 4). 1:1 μmol/L, 10:10 μmol/L, 100:100 μmol/L, NC: negative control, ADV: adefovir dipivoxil, X: Cs2K4Na[SiW9Nb3O40]·H2O (A) Intracellular HBV DNA replication was determined by Southern blot analysis. RC: Relaxed circular DNA; SS: Single stranded DNA. HepG2: HepG2 cell was used as control, which replaced HepG2.2.15 cell; (B) Intracellular HBcAg and HBsAg was determined by Western blot analysis; (C) Intracellular mRNA was determined by RT-PCR. Relative amounts of intracellular mRNA are presented as%, as compared to levels detected in non-treated controls (NC). ∗P <0.05 versus the corresponding non-treated controls; #P <0.05 versus the corresponding positive controls at the same dose.
We next investigated whether Compound 1 was able to reduce the HBsAg and HBcAg proteins. HepG2.2.15 cells were treated with Compound 1 for 4 days. Western blot analysis with anti-HBsAg, anti-HBcAg and β-actin indicated that the drug caused a dose-dependent decrease in the levels of intracellular production of HBsAg and HBcAg when compared with the negative control (Fig. 4). At the same dose, ADV treatment was able to inhibit the antigens but not to the same extent.
POMs are negatively charged, inorganic substances that are principally comprised of oxide anions and early transition-metal cations.16 Several of the POMs have been characterized as having a broad range of antiviral activities. In particular, these complexes have shown excellent inhibitory activity against human immunodeficiency viruses HIV-1 and HIV-2, herpes simplex virus, and influenza virus.17 As the POMs are non-toxic to mammalian cells and do not appear to readily induce drug-resistant strains of their target pathogens, they are very promising candidates for development as human therapeutic agents. Research into POMs as small molecule drugs has become so heavily pursued that during our short study period a number of POMs had been synthesized and evaluated for their potential antiviral activity.12, 18
Our study examined the anti-HBV activities of Compound 1 in vitro. We employed the HepG2.2.15 cell line, a widely used model for the evaluation of anti-HBV drugs, which contains two copies of the HBV genome.19 Compound 1 elicited a dose-dependent and time-dependent inhibitory effect on the secretion of HBV DNA and the HBsAg and HBeAg antigens after 9 days of treatment. A lower dose of the compound was required to effectively inhibit secreted HBV DNA than to inhibit secreted antigens. The effect of Compound 1 on inhibiting HBV activity in HepG2.2.15 cells was determined to not be a cytotoxic event. It is possible that the compound acts on the exported virions outer protein coats. Though the mechanisms mediating the antiviral effects by Compound 1 remain unclear, we have deduced that Compound 1 might block the secretion of HBV containing nucleocapsids or destabilize HBV DNA-containing nucleocapsids.20
We observed that the inhibition rates on HBsAg, HBeAg and HBV DNA were dependent on the concentration of and time of exposure to Compound 1 and ADV. Interestingly, the in vitro anti-HBV properties of Compound 1 against rebound of serum HBV DNA, HBsAg, and HBeAg were more robust than those of ADV, as indicated by evaluation of treated cells 4 days after termination of treatment. This character of sustained activity may have significant clinical implications for Compound 1 and supports further efforts to develop Compound 1 as an anti-HBV drug.
Treatment of patients with chronic HBV infection remains an important clinical challenge. As persistent HBV viraemia is strongly associated with development of liver cirrhosis and hepatocellular carcinoma, complete resolution of the infection is critical. Most of the currently available treatment options, however, can only suppress viral replication.21 Fortunately, the nucleoside/tide analog-based therapeutics are associated with lower rates of antiviral drug resistance in the long-term and they induce a rapid virologic response.22 However, there is still a demand for new and improved therapies that will completely clear the HBV infection. The ADV drug competitively inhibits HBV polymerase, which is structurally similar to dATP. It is also known to reduce HBV DNA and HBsAg both in vitro and in vivo by a phosphorylation event that facilitates its physical incorporation into nascent viral DNA by the activity of HBV polymerase during replication.23
In our study, we observed an apparent reduction in the levels of intracellular HBV intermediates and mRNA in response to Compound 1 treatment. Therefore, it was not surprising that the levels of antigen translated from mRNA, were reduced.24 The underlying molecular mechanism(s) by which Compound 1 acts on the viral polymerase and processes of protein expression remain unknown and will be elucidated in further investigations.
When we performed the experiments using non-HepB gene expressing HepG2 cells, instead of HepB gene expressing HepG2.2.15 cells, no cytotoxicity was observed up to 9 days after exposure to Compound 1 or ADV for any of the dosages used with HepG2.2.15 (data not shown). As anticipated, no HBsAg, HBeAg, or viral DNA was detected in the supernatants of the HepG2 negative control cells. Intracellular HBV DNA, mRNA, and antigen were not detected either. The results from our negative control cells confirmed that the results from the HepG2.2.15 were specific for HepB, which is uniquely expressed in those cells.
The most attractive feature of POMs for their development as therapeutics is that nearly every molecular property that mediates the recognition and reactivity of POMs with target biological macromolecules can be manipulated. For example, fine tuned alterations can be made to the redox potentials, polarity, shape, surface charge distribution, and acidity. Both organic derivatization and metal substitution have extended the number of POMs available. Moreover, POMs-based raw materials are relatively inexpensive and technically simple to generate and amplify; hence, they promise significant economic and technical advantages over the currently available pharmaceuticals.15, 17, 25
Since the regulatory mechanisms involved in viral replication are quite different from those for the protein expression of HBV antigen, our observations that Compound 1 can inhibit not only the HBV DNA replication and RNA transcription but also the HBV antigen secretion, indicates that there might be at least three targets of Compound 1 antiviral activity. However, the exact mechanism of the anti-HBV activity of Compound 1 remains to be completely understood and will require further investigation. In conclusion, the present study demonstrated that the Compound 1 effectively inhibits HBV antigen secretion and HBV replication in a dose-dependent and time-dependent manner in vitro. Compound 1 is, therefore, worthy of further investigation and is an excellent candidate for potential clinical application.26, 27, 28, 29, 30
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation (NO. 30972611, NO. 30872174 and NO. 81172726) and the National S T Major Project of China (No. 2009ZX09103 and NO. 2008ZX10002-004).
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2011.12.115.
Contributor Information
Juan Li, Email: ChunYWag@126.com.
Junqi Niu, Email: junqinu@126.com.
Supplementary data
Experimental procedure.
References and notes
- 1.Ganem D., Prince A.M.N. Engl. J. Med. 2004;350:1118. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 2.Dixon J.S., Boehme R.E. Acta Gastroenterol. Belg. 2000;63:348. [PubMed] [Google Scholar]
- 3.Doo E.C., Ghany M.G. Clin. Liver Dis. 2010;14:397. doi: 10.1016/j.cld.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crosby I.T., Bourke D.G., Jones E.D., Jeynes T.P., Cox S., Coates J.A., Robertson A.D. Bioorg. Med. Chem. Lett. 2011;21:1644. doi: 10.1016/j.bmcl.2011.01.109. [DOI] [PubMed] [Google Scholar]
- 5.Buster E.H., Janssen H.L.Neth. J. Med. 2006;64:175. [PubMed] [Google Scholar]
- 6.Jones J., Shepherd J., Baxter L., Gospodarevskaya E., Hartwell D., Harris P., Price A. Health Technol. Assess. 2009;13:1. doi: 10.3310/hta13350. [DOI] [PubMed] [Google Scholar]
- 7.Fischer K.P., Gutfreund K.S., Tyrrell D.L. Drug Resist. Updat. 2001;4:118. doi: 10.1054/drup.2001.0190. [DOI] [PubMed] [Google Scholar]
- 8.Almeida A.M., Ribeiro A.Q., Pádua C.A., Brandão C.M., Andrade E.I., Cherchiglia M.L., Carmo R.A., Acurcio F.A. Rev. Soc. Bras. Med. Trop. 2010;43:440. doi: 10.1590/s0037-86822010000400021. [DOI] [PubMed] [Google Scholar]
- 9.Ogata A., Mitsui S., Yanagie H., Kasano H., Hisa T., Yamase T., Eriguchi M. Biomed. Pharmacother. 2005;59:240. doi: 10.1016/j.biopha.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 10.Dan K., Miyashita K., Seto Y., Fujita H., Yamase T. Pharm. Res. 2002;46:357. doi: 10.1016/s1043661802001706. [DOI] [PubMed] [Google Scholar]
- 11.Shigeta S., Mori S., Kodama E., Kodama J., Takahashi K., Yamase T. Antiviral Res. 2003;58:265. doi: 10.1016/s0166-3542(03)00009-3. [DOI] [PubMed] [Google Scholar]
- 12.Juan L., Yan F.Q., Wang E.B. J. Coord. Chem. 2004;9:715. [Google Scholar]
- 13.Yamase T. J. Mater. Chem. 2005;15:4773. [Google Scholar]
- 14.Richard G.Finke, Michael W.Droege. J. Am. Chem. Soc. 1984;106:7274. [Google Scholar]
- 15.Kim Z. Inorg. Chem. 2003;42:5537. doi: 10.1021/ic0341845. [DOI] [PubMed] [Google Scholar]
- 16.Shigeta S., Mori S., Yamase T., Yamamoto N., Yamamoto N. Biomed. Pharmacother. 2006;60:211. doi: 10.1016/j.biopha.2006.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhule J.T., Hill C.L., Judd D.A., Schinazi R.F. Chem. Rev. 1998;98:327. doi: 10.1021/cr960396q. [DOI] [PubMed] [Google Scholar]
- 18.Xi H.Y., Di W., Zheng B.H., Zhi W.Sun, Juan L. Chin. J. Lab Diagn. 2008;2:199. [Google Scholar]
- 19.Sells M.A., Chen M.L., Acs G. Proc. Natl. Acad. Sci. U.S.A. 1987;84:1005. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang K.L., Lai Y.K., Lin C.C., Chang J.M. World J. Gastroenterol. 2006;12:5721. doi: 10.3748/wjg.v12.i35.5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C.J., Yang H.I., Su J., Jen C.L., You S.L., Lu S.N., Huang G.T., Iloeje U.H. JAMA. 2006;295:65. doi: 10.1001/jama.295.1.65. [DOI] [PubMed] [Google Scholar]
- 22.Locarnini S., Qi X., Arterburn S., Snow A., Brosgart C., Currie G. J. Hepatol. 2005;42:17. [Google Scholar]
- 23.Marcellin P., Chang T.T., Lim S.G., Sievert W., Tong M., Arterburn S., Borroto-Esoda K., Frederick D., Rousseau F. Hepatology. 2008;48:750. doi: 10.1002/hep.22414. [DOI] [PubMed] [Google Scholar]
- 24.He Y.W., Guo C.X., Pan Y.F., Peng C., Weng Z.H. World J. Gastroenterol. 2008;14:1592. doi: 10.3748/wjg.14.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strasfeld L., Chou S. Infect. Dis. Clin. North Am. 2010;24:809. doi: 10.1016/j.idc.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Sheldrick,; G. M. SHELXS97, Program for Crystal Structure Solution, University of Göttingen: Göttingen, Germany 1997.
- 27.Sheldrick,; G. M. SHELXL97, Program for Crystal Structure Refinement, University of Göttingen: Göttingen, Germany 1997.
- 28.Han Y.Q., Huang Z.M., Yang X.B., Liu H.Z., Wu G.X. J. Ethnopharmacol. 2008;118:148. doi: 10.1016/j.jep.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 29.Xu W.S., Zhao K.K., Miao X.H., Ni W., Cai X., Zhang R.Q., Wang J.X. World J. Gastroenterol. 2010;16:2028. doi: 10.3748/wjg.v16.i16.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livak K.J., Schmittgen T.D. Methods. 2001;25:402. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedure.