

Abstract
The generation of corroborative data has become a commonly used approach for ensuring the veracity of microarray data. Indeed, the need to conduct corroborative studies has now become official editorial policy for at least 2 journals, and several more are considering introducing such a policy. The issue of corroborating microarray data is a challenging one—there are good arguments for and against conducting such experiments. However, we believe that the introduction of a fixed requirement to corroborate microarray data, especially if adopted by more journals, is overly burdensome and may, in at least several applications of microarray technology, be inappropriate. We also believe that, in cases in which corroborative studies are deemed essential, a lack of clear guidance leaves researchers unclear as to what constitutes an acceptable corroborative study. Guidelines have already been outlined regarding the details of conducting microarray experiments. We propose that all stakeholders, including journal editorial boards, reviewers, and researchers, should undertake concerted and inclusive efforts to address properly and clarify the specific issue of corroborative data. In this article we highlight some of the thorny and vague areas for discussion surrounding this issue. We also report the results of a poll in which 76 life science journals were asked about their current or intended policies on the inclusion of corroborative studies in papers containing microarray data.
Introduction
Since the first papers describing their production and use were published in the mid-1990s [1], [2] microarrays have emerged as one of the most promising new tools in molecular biology since the polymerase chain reaction [3]. Part of the popularity of microarrays lies in the fact that they can be applied to research across a wide range of fields, a property that has driven an exponential increase in publications on their use over the past 6 years (Fig. 1) .
Fig. 1.
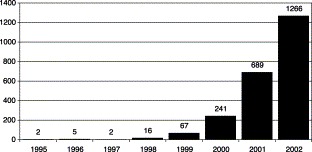
Number of articles in Entrez-PubMed containing keyword “microarray.” A search was conducted in Entrez-PubMed (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db = PubMed) using “microarray” as the sole search term. Searches were conducted for each year from 1995 through 2002.
In addition to their utility across many different life science fields, microarrays are also employed across a spectrum of experimental designs and objectives. For example, a relatively straightforward experimental objective may be a determination if a certain microbial species is present in a water or tissue sample. This approach was recently used to help pinpoint the nature of the virus causing severe acute respiratory syndrome [4]. Microarray experiments are more commonly used for either class comparison or class prediction [5]. In class comparison, genes that are expressed differentially in different types of samples are identified. A commonly reported approach has been to use gene expression patterns as a “fingerprint” to identify exposure of a tissue to a particular chemical class [6], [7], [8]. Such fingerprints have also been used to differentiate between different types and stages of neoplastic disease [5], [9] as well as nonneoplastic diseases and episodes such as stroke, seizures, hypoglycemia, and hypoxia [10]. Others have taken this approach a step further, into class prediction. By comparing expression profiles from treated and control samples with specific adverse outcomes, it has been possible to use microarray data to identify gene expression fingerprints that predict toxicity [11], response to drug treatment [12], or disease outcome [13].
Gene expression data from microarrays are not only useful in the discovery of new biomarkers. They can also supply information about the biological processes that are occurring in a particular model. By conducting sophisticated analysis of gene expression changes over dose and time, one can begin to identify cohorts of coregulated genes, which in turn facilitates both hypothesis generation and practical demonstration of the molecular mechanisms and pathways underlying a model of interest [14].
As a consequence of this broad applicability, the rapid and widespread increase in the use of microarrays has both revealed and introduced a number of problems related to specificity, sensitivity, reproducibility, and more. These issues are by no means unique to microarray technology. However, because the process of producing, using, and analyzing a microarray involves many discrete steps, there is ample opportunity for technical variability to swamp the biological variation that the tool intends to measure. Another significant concern is that each of the many steps of a microarray-based study can be, and often is, conducted differently between laboratories. Layer these methodological permutations on top of inherent biological and technological variation, and the result is a quality assurance/quality control challenge of utmost difficulty. Indeed, the current methods of conducting and reporting microarray data make it almost impossible for researchers to verify the veracity of data derived by another group.
In an attempt to introduce a degree of order to the microarray field, an international movement called the Microarray Gene Expression Data (MGED) Society was established in 1999 to facilitate the sharing of microarray data from functional genomics and proteomics experiments [15], [16]. MGED's initial goals include establishing standards for microarray data annotation and representation, facilitating the creation of microarray databases, and providing infrastructure for dissemination of experimental and data transformation protocols. Its longer term goal is to extend this same type of order into other functional genomic and proteomic high-throughput technologies. To fulfill its mission, MGED is pursuing four major standardization projects. These include:
-
(1)
Minimal Information about a Microarray Experiment (MIAME). “MIAME aims to outline the minimum information required to unambiguously interpret microarray data and to subsequently allow independent verification of this data at a later stage if required.”
-
(2)
MicroArray and Gene Expression (MAGE). The MAGE project is intended to provide a standard for the representation of microarray expression data that would facilitate the exchange of microarray information between different data systems.
-
(3)
Ontology—The primary purpose of the MGED Ontology project is to provide standard terms for the annotation of microarray experiments.
-
(4)
Data transformation—This goal is to develop a means by which microarray users can record how they normalized and transformed their data.
The primary benefit of the MGED projects is clearly the development of a set of standards that can be used to bring increased order to the diverse microarray field. Importantly, MGED's proposals are being supported by an increasing number of journals, including Cell, The EMBO Journal, The Lancet, The New England Journal of Medicine, and Nature. These journals have accepted the MIAME standards and consequently will no longer consider papers containing microarray data unless they conform to these standards. However, one of the most voiced concerns about MIAME standards and proposed offshoots such as MIAME-TOX [17] is that that public repositories of gene expression data such as Gene Expression Omnibus [18] or ArrayExpress [19] are not yet as user-friendly or as standardized as, say, the DDBJ/EMBL/GenBank trio of databases for DNA sequences [20], [21], [22]. Furthermore, many people consider the “minimal” part of MIAME to be a misnomer, since it can take a significant amount of time to learn how the systems work, input appropriate experimental details, and upload data.
Whatever the merits and drawbacks of the MIAME proposal, it is necessary and important that efforts are under way to bring some kind of order to the microarray field. However, what the MGED guidelines do not address are the so-called corroborative or validation studies that are often performed following analysis of microarray data. Indeed, there has been a wholesale lack of discussion on this issue across the microarray community, a situation that has led to exasperation when manuscripts that do not include such studies are rejected and confusion as to what exactly constitutes a corroborative study.
Confirmatory studies—the missing piece of the puzzle
Confirming the veracity of microarray data is deemed especially important at this early stage of development, since the technology is still rapidly evolving and still uses a wide variety of platform types, each with inherent limitations and biases. Furthermore, the technology appears set to be incorporated into a significant proportion of both mechanistic and applied life science research. Finally, and perhaps even more importantly, many experts believe that microarray data may eventually play an important role in the regulatory decision-making processes used to determine risk associated with approving the use of pharmaceuticals or other chemical compounds such as pesticides [23], [24], [25].
Corroborating microarray data usually takes the form of conducting some alternative means for quantitating mRNA abundance. Typically Northern blot or quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis of a limited number of genes on the microarray is performed to confirm that any observed changes are “real.” However, those working in the field will appreciate that there is an unspoken undercurrent of confusion and disagreement on the issue of whether gene expression data obtained from DNA arrays always needs to be validated in this way and to what degree.
The arguments for conducting corroborative studies for microarray data
In the early days of microarray development, calls to corroborate the data were virtually nonexistent, probably because the limited number of pioneers in the field had not then identified all potential pitfalls of the technique and its associated technologies. However, the collective experience of an enthusiastic and rapidly growing community of users has subsequently identified numerous such issues. For example, a whole group of problems that concern just the nature of the probes on the arrays have been identified. These include:
-
(1)
False positive signals generated by probes on the microarray cross-hybridizing with related but different genes in the mixed population of labeled cDNAs.
-
(2)
In the same vein, many genes have splice and tissue variants. Recent analyses of alternative splicing indicates that 40–60% of human genes may have alternative splice forms [26]. In extreme cases such as MDM2, more than 40 different splice variants have been identified [27]. It is probably important, therefore, to determine first if variants of a gene exist, what tissues they exist in, which variant of a gene is on one's array, and which, if any, of the other variants will hybridize to that particular probe.
-
(3)
Until recently, most “homemade” microarrays were printed with cDNAs, often amplified from IMAGE clones. Though these are now being replaced by long and short oligonucleotides [28], cDNA-based arrays have left these contemporary microarrays with a legacy of suspicion. This distrust is based on studies such as that of Halgren et al. [29], who reported that only 62% of IMAGE clones purchased from a commercial vendor were uncontaminated and contained cDNA inserts that had significant sequence identity to published data for the ordered clones. Even the use of sequence-verified clone libraries may not be enough to prevent the introduction of significant errors during the production of a microarray. One study found that by the time the PCR products from one such library were ready for printing, only 79% of the clones matched the original vendor database. Some of this error appeared to arise from mistakes in the vendor database itself, while the rest was introduced in the preparation of PCR products for printing [30].
-
(4)
Sequence problems have not been limited to cDNA clone stocks. Both commercial and noncommercial institutions are now producing oligonucleotide probes to use internally or sell in the form of premade arrays or print-ready oligonucleotide sets. Such institutions often rely in whole or in part on publicly available sequence data to design these probe sets. At least one high-profile problem resulted from the use of such data. In this case Affymetrix, an acknowledged innovator and leader in the microarray field, encountered problems when it used public sequence data to design probes for its mouse chips [31]. It was later discovered that the sequence data were inaccurate and that a significant proportion of probes on the affected chips were incorrect.
-
(5)
The general concern that the complex multiplex hybridization reaction on such a miniature scale can create unknown or unforeseen hybridization kinetics that can yield false positive or false negative results. It is indeed a tall order to design hundreds to thousands of probes that permit an equal number of discriminating hybridization reactions to occur under identical conditions.
A confusing message
Issues such as those enumerated above have worked together to introduce a general feeling of uncertainty about the veracity of data derived from microarrays. Over the past 5 years this uncertainty has translated into increasingly numerous calls for microarray data to be corroborated in some manner, increasing the burden for researchers who have chosen to invest in the technique. Where they have not been performed, corroborative studies to support microarray data are often called for by journal editors and reviewers before a paper will be accepted for publication. Unfortunately, these calls in many cases appear rather arbitrary. A combination of personal experience (as authors and reviewers), anecdotal comments from colleagues, and a close monitoring of the quality and content of microarray publications over the past few years has revealed mixed messages being sent by journals. For example, numerous microarray papers have in the past been published, even in high-impact journals, whose data are not supported by corroborative studies. A recent article by Goodman [32] reviewed the contents of 28 microarray papers published in 2002 by Science, Nature, and Cell. Of these 28, only 11 reported the results of confirmatory studies. Nine compared their results to previously published studies, while 10 reported no confirmation studies at all. On other occasions on which 2 or more microarray papers appear in the same or subsequent issues of a journal, one might provide corroborative data while the other does not. To readers and stakeholders there is often no clear reason behind such discrepancies. More often than not, the reason lies in which editor and reviewers appraise the manuscript, as it is apparent that even such highly knowledgeable professionals can have very different opinions on the need for corroborative data.
The arguments against conducting corroborative studies for microarray data
In light of the concerns mentioned earlier that would seem to dictate the inclusion of at least some corroborative data to accompany microarray experiments, what thought processes might occur in the minds of investigators, editors, and reviewers that ultimately persuade them that confirmatory studies are not needed? Clearly there are several possible factors that could contribute to such a decision:
-
(1)
If a large number of arrays are used on a particular set of control and treated samples, and relative changes in mRNA levels are identified by statistical analysis (as opposed to a simple “fold change”), this may be considered sufficient rigor such that corroborative studies are unnecessary for the most significantly changed genes (e.g., p < 0.001).
-
(2)
Where commonly used model systems are used, such as the effect of a particular hormone on a rodent reproductive organ or a toxicant on a particular cell line, at least a subset of the results has often been corroborated by previously published studies in the same system.
-
(3)
In studies in which the focus of the microarray data is not the response of a few novel genes, but rather the overall “pattern” of gene expression, there would seem to be limited utility in confirming individual gene expression differences. Take for example principal component analysis, an increasingly popular method of analyzing microarray data. In the high-dimensional expression space used in such analyses, a small number of nonsystematic errors in individual hybridizations would be unlikely to cause an artificial movement of points representing different time or dose groups. Indeed these types of “patterning” studies constitute a considerable percentage of all “array” papers and include identification of specific toxicant signatures, classification of tumor types, etc. Naturally, if such studies ventured into mechanistic discussions based on the behavior of specific genes, then corroborative data would be appropriate.
-
(4)
The vast majority of microarray publications have indicated that DNA arrays and Northern blot/RT-PCR analysis normally support each other qualitatively, i.e., the direction of the change is consistent. Of course, the fact that only concordant data are ever seen in the literature may be a bias in that only those that get confirmed get published.
-
(5)
Microarrays are still a relatively expensive research tool, and most researchers are reluctant to invest even more time and resources required to conduct RT-PCR or Northern blots.
-
(6)
Even today, not all laboratories have easy access to qRT-PCR resources. Though Northern blots may offer an accessible alternative, they require relatively large amounts of RNA, which are oftentimes not available.
-
(7)
Regardless of access to a qRT-PCR instrument, some researchers work with such small samples (e.g., embryonic rodent organs, laser capture microdissection tissue, human biopsies) that despite steadily improving RNA amplification techniques, there are often insufficient quantities of RNA for both microarray studies and corroborative experiments.
In the rare instances in which published microarray data are not in agreement with data generated by the corroborating method, it is difficult to judge which technique is “correct.” However, directly opposing results have rarely been published. Some as yet unpublished studies have found opposing results on different array platforms, but closer investigation revealed that either there was a problem with the probe sequences (they were incorrect) or the magnitude of change between experimental and control samples was so small that even minute variations in the method resulted in a directional change. In addition, in many cases in which a gene appears to have altered expression with one technique, but not the other, sensitivity of the assay may be the main cause.
This kind of difference can be attributed to the fact that different types of measurement techniques have certain inherent properties that contribute to their returning slightly different results, even if they are measuring changes in the same biological system. For example, it is well established that the dynamic range of microarrays tends to be lower than real-time PCR, thus giving, in essence, “compressed” results. Furthermore, this technical variability is apparent not only between the techniques themselves, but also among the methodological variations of each technique. Thus, when analyzing the same biological sample, differences can be expected between qRT-PCR and real-time PCR data and even between data generated by different types of real-time PCR (e.g., SYBR green vs Taqman). The situation is exacerbated with microarrays, of which there are multiple platform choices (e.g., two-color long oligonucleotide-based slide arrays versus single-color Affymetrix short oligonucleotide-based chips versus PCR-product-based radiolabeled membrane arrays). A recent study of cross-platform variability [33] has reported poor correlation between results obtained from spotted cDNA versus synthesized oligonucleotide arrays. However, as the authors point out, the experiments were performed independently by two different laboratories, making it difficult to affix all of the variability to intrinsic properties of the array platforms. It is not just technical differences that give rise to discrepancies. The computational tools and algorithms used to analyze the raw data from each of these techniques also use distinct criteria for extracting expression levels, thus introducing further sources of variation.
For these reasons, plus the fact that the vast majority of the manuscripts published to date indicate that corroborated DNA array data are representative, a compelling case can be made that data from microarrays are reliable and should be accepted at face value as long as the experimental design and statistical analysis is sound.
Which genes to corroborate?
It is also reasonable to suppose that some of the hesitance to corroborate microarray data stems in part from not knowing what constitutes an acceptable number or type of corroborative studies. In response to a brief “letter to the editor” submitted to raise the profile of this issue [34], a communication was received from another concerned scientist asking if there were any papers or standards already published that could help them select what genes should be validated by qRT-PCR. Other scientists have openly asked the same and related questions on the gene arrays listserv (gene-arrays@itssrv1.ucsf.edu), an online community dedicated to addressing issues and concerns of microarray users. In fact, two main questions need to be addressed: “How many genes are enough?” and “On what basis should I select the genes to be confirmed?” In reality, the corroboration of all microarray data is of course impractical because of the large number of genes they contain. Therefore, to avoid the “thou must confirm” kiss of rejection or major revision, most authors choose to provide corroborative Northern or RT-PCR data for a small number of genes found on their array. In many cases confirmatory studies have been carried out on only a handful of genes—in some cases as few as one or two [35].
The second issue is which genes to pick. The problem here of course is that the genes selected for corroboration can be cherry-picked to virtually ensure confirmation. For example, genes with a greater than fourfold change in expression on microarrays have consistently been validated by qRT-PCR [36]. So, by picking those genes that show the largest magnitude of change on the microarray, or those that have been confirmed to be changed in previous studies in the same or similar models, a researcher is virtually assured of attaining a passing grade for the corroborative studies section of their own study.
Furthermore, the actual process of corroborating expression of a few select genes with RT-PCR/Northern analysis does not mean that the same results would be obtained if all the genes were thus analyzed.
In addition, some journals have accepted corroborative studies in the form of protein expression analysis. This approach does not constitute a corroborate study per se, as it clearly addresses a different question (do mRNA and protein level changes correspond?). A change in expression of an mRNA species clearly cannot be confirmed by a method that measures protein expression.
Thus, a reasonable conclusion is that, in most circumstances, the true veracity of the microarray data is hardly more ensured than if the confirmatory studies had not been done at all.
Journal policies on providing corroborative data for microarray studies
In one of the most striking recent developments in the confirmatory studies issue, the journal Circulation Research made a seminal editorial decision to introduce a set of inclusion criteria for papers incorporating the use of microarray and other high-throughput genomics methods [37]. The first of these was “The salient results of the genomic screen must be confirmed by complementary methods (such as Northern blot analysis), for selected transcripts of greatest relevance or interest.” This decision was soon followed by the release of a similar set of criteria by Arthritis and Rheumatism, including one necessitating that “A non-array method must confirm changes in the expression of key genes” [38].
The problems with such well-intentioned measures include:
-
(1)
There is insufficient guidance on what statements such as “selected transcripts” mean.
-
(2)
As mentioned previously, some studies, such as those utilizing array data for class identification, rely on overall patterns of expression for hypothesis generation and testing and place minor emphasis on the behavior of any one or few specific genes.
-
(3)
As already alluded to, some studies simply do not yield sufficient quantities of RNA to conduct such studies.
Since we were interested in whether other journals were considering following the example set by Circulation Research, we wrote to the editor or editorial office of 76 life science journals that had published at least one article using microarray data and asked whether they were formulating policies regarding the corroborative studies issue. Of the 76 journals polled, responses covering 32 (42.1%) were received. The results of the poll are summarized in Table 1 .
Table 1.
Responses from solicited journals regarding guidelines for submission and corroboration of microarray data
| Journal title | Data submissiona | Corroborative studiesb |
|---|---|---|
| American Journal of Human Genetics | MIAME | CBC |
| Arthritis and Rheumatismc | CBC | COR |
| British Journal of Cancer | CBC | CBC |
| Cancer | CBC | CBC |
| Cell | MIAME | CBC |
| Circulation | CBC | CBC |
| Circulation Research | CBC | COR |
| Environmental Health Perspectives | CBC | CBC |
| Genome Biology | CBC | CBC |
| Genome Research | CBC | CBC |
| Human Reproduction/Molecular Human Reproduction | CBC | CBC |
| Journal of Biological Chemistry | CBC | CBC |
| Journal of Cellular Biochemistry | CBC | CBC |
| Journal of General Virology | CBC | CBC |
| Journal of Immunology | CBC | CBC |
| Journal of Molecular Biology | CBC | CBC |
| Lancet | MIAME | CBC |
| Molecular Cell | MIAME | CBC |
| Nature | MIAME | CBC |
| Nature Genetics | MIAME | CBC |
| Nature Medicine | MIAME | CBC |
| New England Journal of Medicine | MIAME | CBC |
| Oncogene | MIAME | CBC |
| Pharmacogenetics | CBC | CBC |
| Physiological Genomics | MIAME | CBC |
| Proceedings of the National Academy of Sciences of the USA | CBC | CBC |
| The Pharmacogenomics Journal | CBC | CBC |
| The Plant Cell | MIAME | CBC |
| The Plant Journal | MIAME | CBC |
| Toxicological Sciences | CBC | CBC |
| Toxicology | CBC | CBC |
| Toxicology In Vitro | CBC | CBC |
A letter was forwarded electronically to 76 journals that publish microarray data. Responses from the 31 journals covered by replies (in some cases, policies of multiple journals were included in one response) are reported.
Reported guidelines governing submission of microarray data. MIAME requires that submitted data conform to MIAME requirements; CBC requires completeness of data submission information evaluated by reviewers on a case-by-case basis.
Reported guidelines governing postarray corroboration of microarray data. COR requires that corroboration be performed on at least some array-identified genes; CBC requires that any corroborative data are evaluated by journal reviewers on a case-by-case basis.
Not e-polled (literature search).
Of the journals for which we received responses, only Circulation Research and Arthritis and Rheumatism have published guidelines indicating that the inclusion of some type of confirmatory data is mandatory. Even here, the guidelines are deliberately vague to permit reviewer latitude on whether the specific nature and number of “transcripts of greatest relevance or interest” have indeed been adequately selected for corroboration.
Responses from the other journals could be generally divided into two categories—one in which MIAME compliance is required as part of the data submission process, but inclusion of confirmatory experiments is left to the discretion of the authors and, of course, the reviewers (12 journals), and the other in which the acceptability of the chosen method for both microarray data presentation and confirmation is left to the discretion of the journal reviewers (18 journals). It must be noted that within the sum of these 30 responses, a number of journals indicated that depending on the type of study, corroboration of array data would be required. Furthermore, several editors noted that even though the need to conduct confirmatory studies was left to the discretion of the reviewers, the threshold for publishing such studies had clearly risen over the past few years as their journals had received more such papers and as the editors and reviewers had become aware of potential problems associated with microarray technology; indeed, two journals acknowledged that a policy of providing corroborative data had become an unwritten rule. A further five editors acknowledged that the requirement and/or guidelines for confirmatory studies was an increasingly thorny issue and one that was a part of ongoing journal editorial board discussions. For those rejecting the idea of imposing stipulations on the submission of microarray data, perhaps one editor put it best, saying that “Common sense must be the best guide …We have deliberately not defined rules, since each person is using the information in different ways. Flexibility is an asset in my opinion” (Dr Harry Klee, The Plant Journal, with permission).
What next for corroborative studies?
Since the results of corroborative studies can open or close the gate to publishing the results of a microarray study, it is surprising that this area has not yet been more closely scrutinized. Fortunately, this situation may not persist much longer. Recognition is growing among the scientific community that there are no clear standards in the confirmatory studies area and that while a certain degree of flexibility is called for, methods and guidelines developed to address this issue might be helpful to all interested parties.
Methods
Where methods are concerned, the development of RNA amplification techniques may enable “RNA-challenged” investigators to rest more easily. It has been shown that reproducible data can be obtained from a microarray starting with as little as 10 ng of RNA or less ([39], [40], G.M. Hellmann, unpublished observations). This provides a mechanism for those with limited amounts of total RNA (e.g., <1 μg) to conduct both microarray and confirmatory qRT-PCR studies. The caveat, however, is that while such amplification systems (e.g., Clontech's SMART system and Ambion's MessageAmp aRNA Kit) may facilitate such an approach, there still remains a degree of skepticism concerning the faithfulness of the final representation of amplified mRNA compared to the original sample. Additionally, this adds yet another step(s) to the overall process and therefore opportunity for the introduction of yet more measurement error. There is justifiable concern that amplification procedures may provide skewed representation of the original RNA population. In fact, these concerns constitute a valid argument for conducting mandatory corroborative studies when amplified samples are used.
Many researchers who use microarrays do not necessarily have expertise in designing probes for real-time PCR confirmatory studies. At least one company has seen that this is an underserved market segment just waiting to be exploited. In response to the burgeoning demand for real-time PCR primers, perhaps brought on in part by the need to corroborate microarray data, Applied Biosystems, Inc. (ABI; Foster City, CA, USA), has developed a product line called “assays-on-demand.” This is a collection of optimized gene-specific TaqMan reaction sets (primers and probes) for human and mouse genes. These currently consist of over 18,000 and 9000 reaction sets, respectively [41]. Whether intentional or not, it appears that this product line may facilitate the introduction of an element of standardization into the confirmatory studies process, assuming of course that ABI can persuade a high enough number of researchers to use it on a regular basis. Even if competitors develop similar products, as long as the number of companies offering them remains small it will surely serve to reduce the variability that can only ensue when multitudes of researchers design their own custom primer/probe sets.
A third and somewhat more provocative methodological consideration is whether it is in fact reasonable at all to validate oligomicroarrays using PCR-based methods. As alarming as this might sound to many microarray users for whom this has become the norm, there is in fact justifiable concern that this is not necessarily the best approach. As Chuaqui et al. pointed out [36], it appears that a subset of target cDNAs will hybridize strongly, not only to their intended probe on the array, but also to other probes that have no discernable sequence homology. Thus, it may be prudent to perform corroborative studies using techniques that are not hybridization-based (which PCR-based methods are). Chuaqui et al. suggest contrasting array-based results in silico with data from sequencing-based expression studies, e.g., serial analysis of gene expression, that do not rely on hybridization methods. Alternatively, Northern blotting enables hybridization specificity to be evaluated also on the basis of transcript size.
The call for these alternative RNA quantitation techniques is an acknowledgment that all techniques used to analyze biological systems suffer some kind of bias. For example, real-time PCR is rapidly becoming the “gold standard” for quantifying RNA in samples and is thus often used to corroborate microarray data. However, a gold standard technique usually earns this accolade not necessarily because it provides the most accurate representation of biological reality, but because it is widely used and accepted. In the case of real-time PCR, most researchers would be hard pressed to provide evidence that this technique accurately counts transcripts in a sample. Furthermore, unless and until complete sequence and annotation data of all genes are available, it is impossible to determine whether a particular real-time PCR is amplifying multiple splice forms and/or closely related gene sequences.
Standards and guidelines
As far as the imposition of standards for corroborating microarray data is concerned, it would seem that we are in a critical decision period. Two journals currently require corroborative data and several more are considering the introduction of a similar policy. However, the present consensus among the journals seems to be that each manuscript should be carefully judged on a case-by-case basis. In our minds, this should remain the standard. There are clearly times when corroborative studies may be impractical or provide no significant additional value to a study. All scientific journals have a responsibility to encourage and promote responsible scientific research, but in a way that does not set overly restrictive guidelines on how scientists should conduct their studies. In addition, one may argue that such rulings can alienate some authors who would otherwise have submitted their work to the journal. In fact, this same criticism has already been leveled at journals that have adopted the MIAME standards.
Where corroborative studies are deemed essential by reviewers, then both reviewers and editors have a responsibility to provide authors with guidance on what type of corroborative studies are acceptable and the appropriate amount of work required. Authors also need to know the value of electronic corroboration, a method that may become increasingly acceptable as more papers containing microarray data are published and more public gene expression databases, such as the Chemical Effects in Biological Systems [42], are populated with data. In many cases, arrays reveal changes in gene expression that have been confirmed one or more times previously in the same model, often with a different technique or microarray platform. If a scientist can identify 10 such genes, is this enough to justify that the array is working “correctly” and eliminate the need for confirmatory studies on other genes? While we are inclined to respond in the affirmative, it must be acknowledged that like other techniques, electronic corroboration of microarray data comes with its own set of problems. Whether in the published literature or a database, one can often find multiple studies supporting the changed expression of a particular gene. But clouding rather than clarifying the issue, there may be an equal number of studies that contradict this same finding. This may in part be due simply to biological variation or the fact that many models and methods that on the surface appear the same are in fact subtly different in some way. In addition, it can be difficult to confirm contradictory results. For example, since genes that do not change are not usually reported or discussed as extensively as those that do, it can require intensive review of original methods and data to determine whether the appropriate target for a gene one is trying to corroborate was present on the original array used in a published study and, if so, whether and how that gene's expression changed.
Finally, it is not just reviewers and editors who must shoulder responsibility for maintaining high and appropriate standards in studies utilizing microarrays. Authors also have a responsibility to assist editors, reviewers, and future readers by including in their manuscripts details on how and why corroborative studies were conducted and if they were not, why not. If we are truly to understand the power and limitations of microarray data, it should become a matter of routine for scientists to provide easy access to all raw data, as well as information on which of their corroborative experiments failed to support the RT-PCR data.
Summary
A lack of standards and guidance on corroborative or “confirmatory” studies for microarray data has been a source of frustration for many scientists. A conflict exists between the desirability of requiring a uniform set of procedures that apply to all microarray-containing journal submissions and the need for flexibility to accommodate the wide range of objectives, both current and in the future, that are being addressed by microarray technology. While most scientists are anxious to submit complete studies, the hard truth is that no one wants to do more work than necessary for an acceptable publication. In most cases, this is simply a matter of time and resources.
Chuaqui et al. [36] commented that the development of uniform validation methods will be important for the future development of microarray technology. We agree with this statement and are pleased to see that microarray standardization issues are now being addressed by stakeholders in this technology. On the other hand, while the editorial decisions of Circulation Research and Arthritis and Rheumatism not to accept microarray data papers without corroborative data are admirable in the sense that they are an attempt to bring some sort of order to the overgrown microarray field and weed out the ineffectual and incomplete studies, there are clearly circumstances in which it is not essential to conduct such studies to prove that the central message of a paper is correct. We are also concerned that this may set a potentially inappropriate precedent for other journals to follow. Guidelines such as MIAME are appropriate for the reporting of array design, experimental procedures, and other aspects of the mechanics of performing arrays, but such a rigid template is problematic when superimposed on postarray analyses such as the corroborative quantitative studies under discussion.
Finally, the question naturally arises as to whether and when enough microarray and corroborative studies will have been published to demonstrate that microarray data can stand alone on their own merit. We believe that this scenario is not too far away and that calls to corroborate microarray data with independent techniques will probably begin to subside within a few years. This will in part be the result of standardization of microarray methodology and in part be related to the maturation of the technology to a point at which data derived from microarray experiments is as trusted as PCR and DNA sequencing are trusted today. In the meantime, the approach to designing, conducting, reviewing, and publishing studies utilizing microarray data must be one of careful consideration from researchers, editors, and reviewers alike. Like any other component of experimental design, the need to corroborate and the methodology employed to do so must be carefully determined and justified to ensure that the microarray technique can flourish and yield in full the many valuable promises it offers.
Acknowledgements
We thank Drs. David Dix (U.S. EPA) and Wanda Fields (R.J. Reynolds) for critically reviewing the manuscript prior to submission. The information in this document has been funded in part by the U.S. Environmental Protection Agency. It has been subjected to review by the National Health and Environmental Effects Research Laboratory and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
References
- 1.Fodor S.P., Rava R.P., Huang X.C., Pease A.C., Holmes C.P., Adams C.L. Multiplexed biochemical assays with biological chips. Nature. 1993;364:555–556. doi: 10.1038/364555a0. [DOI] [PubMed] [Google Scholar]
- 2.Schena M., Shalon D., Davis R.W., Brown P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 3.Saiki R.K. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 4.Rota P.A. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 5.Golub T.R. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 6.Burczynski M.E. Toxicogenomics-based discrimination of toxic mechanism in HepG2 human hepatoma cells. Toxicol. Sci. 2000;58:399–415. doi: 10.1093/toxsci/58.2.399. [DOI] [PubMed] [Google Scholar]
- 7.Hamadeh H.K. Gene expression analysis reveals chemical-specific profiles. Toxicol. Sci. 2002;67:219–231. doi: 10.1093/toxsci/67.2.219. [DOI] [PubMed] [Google Scholar]
- 8.Thomas R.S. Identification of toxicologically predictive gene sets using cDNA microarrays. Mol. Pharmacol. 2001;60:1189–1194. doi: 10.1124/mol.60.6.1189. [DOI] [PubMed] [Google Scholar]
- 9.Stratowa C. cDNA microarray gene expression analysis of B-cell chronic lymphocytic leukemia proposes potential new prognostic markers involved in lymphocyte trafficking. Int. J. Cancer. 2001;91:474–480. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1078>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 10.Tang Y., Lu A., Aronow B.J., Sharp F.R. Blood genomic responses differ after stroke, seizures, hypoglycemia, and hypoxia: blood genomic fingerprints of disease. Ann. Neurol. 2001;50:699–707. doi: 10.1002/ana.10042. [DOI] [PubMed] [Google Scholar]
- 11.Kier L., Nolan T. Gene expression biomarkers that accurately predict kidney tubular necrosis. Toxicol. Sci. 2003;72(S-1):151. [Google Scholar]
- 12.Kaneta Y. Prediction of sensitivity to STI571 among chronic myeloid leukemia patients by genome-wide cDNA microarray analysis. Jpn. J. Cancer Res. 2002;93:849–856. doi: 10.1111/j.1349-7006.2002.tb01328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang E. Gene expression predictors of breast cancer outcomes. Lancet. 2003;361:1590–1596. doi: 10.1016/S0140-6736(03)13308-9. [DOI] [PubMed] [Google Scholar]
- 14.Kannan K., Kaminski N., Rechavi G., Jakob-Hirsch J., Amariglio N., Givol D. DNA microarray analysis of genes involved in p53 mediated apoptosis: activation of Apaf-1. Oncogene. 2001;20:3449–3455. doi: 10.1038/sj.onc.1204446. [DOI] [PubMed] [Google Scholar]
- 15.Brazma A., Robinson A., Cameron G., Ashburner M. One-stop shop for microarray data. Nature. 2000;403:699–700. doi: 10.1038/35001676. [DOI] [PubMed] [Google Scholar]
- 16.MGED, Microarray Gene Expression Data Society—MGED Society, accessed September 10, 2003, at http://www.mged.org/.
- 17.MIAME-TOX, Minimum Information About a Microarray Experiment: MIAME for toxicogenomics (MIAME/Tox), accessed June 18, 2003, at http://216.239.39.100/search?q = cache:cy23_6a5JNgJ: www.mged.org/MIAME1.1-DenverDraft.DOC+MIAME-TOX&hl = en&ie = UTF-8.
- 18.GEO, Gene Expression Omnibus, accessed June 9, 2003, at http://www.ncbi.nlm.nih.gov/geo/.
- 19.ArrayExpress, accessed June 9, 2003, at http://www.ebi.ac.uk/microarray/ArrayExpress/arrayexpress.html.
- 20.DDBJ, DNA Data Bank of Japan, accessed September 10, 2003, at http://www.ddbj.nig.ac.jp/.
- 21.EMBL, EMBL Nucleotide Sequence Database, accessed September 10, 2003, at http://www.ebi.ac.uk/embl/.
- 22.GenBank, accessed September 10, 2003, at http://www.ncbi.nlm.nih.gov/Genbank/index.html.
- 23.EPA 2002 Interim Genomics Policy, accessed June 24, 2003, at http://epa.gov/osp/spc/genomics.htm.
- 24.G.E. Marchant, Genomics and environmental regulation: scenarios and implications, accessed June 24, 2003, at http://www.epa.gov/osp/presentations/futures/marchant.pdf.
- 25.Robinson D.E., Petit S.D., Morgan D.G. Use of genomics in mechanism based risk assessment. In: Inoue T., Pennie W.D., editors. Toxicogenomics. Springer-Verlag; Tokyo: 2003. pp. 194–203. [Google Scholar]
- 26.Modrek B., Lee C. A genomic view of alternative splicing. Nat. Genet. 2002;30:13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 27.Bartel F., Taubert H., Harris L.C. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell. 2002;2:9–15. doi: 10.1016/s1535-6108(02)00091-0. [DOI] [PubMed] [Google Scholar]
- 28.Barrett J.C., Kawasaki E.S. Microarrays: the use of oligonucleotides and cDNA for the analysis of gene expression. Drug Discovery Today. 2003;8:134–141. doi: 10.1016/s1359-6446(02)02578-3. [DOI] [PubMed] [Google Scholar]
- 29.Halgren R.G., Fielden M.R., Fong C.J., Zacharewski T.R. Assessment of clone identity and sequence fidelity for 1189 IMAGE cDNA clones. Nucleic Acids Res. 2001;29:582–588. doi: 10.1093/nar/29.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor E. Sequence verification as quality-control step for production of cDNA microarrays. Biotechniques. 2001;31:62–65. doi: 10.2144/01311st01. [DOI] [PubMed] [Google Scholar]
- 31.Knight J. When the chips are down. Nature. 2001;410:460–461. doi: 10.1038/35073680. [DOI] [PubMed] [Google Scholar]
- 32.Goodman N. Microarrays: hazardous to your science. Genome Technol. April. 2003:42–45. [Google Scholar]
- 33.Kuo W.P., Jenssen T.K., Butte A.J., Ohno-Machado L., Kohane I.S. Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics. 2002;18:405–412. doi: 10.1093/bioinformatics/18.3.405. [DOI] [PubMed] [Google Scholar]
- 34.Rockett J.C. To confirm or not to confirm (microarray data)—that, is the question. Drug Discovery Today. 2003;8:343. doi: 10.1016/s1359-6446(03)02653-9. [DOI] [PubMed] [Google Scholar]
- 35.Carlisle A.J. Development of a prostate cDNA microarray and statistical gene expression analysis package. Mol. Carcinog. 2000;28:12–22. [PubMed] [Google Scholar]
- 36.Chuaqui R.F. Post-analysis follow-up and validation of microarray experiments. Nat. Genet. 2002;32:509–514. doi: 10.1038/ng1034. [DOI] [PubMed] [Google Scholar]
- 37.Circulation research: suitability criteria for manuscripts utilizing genomics array technology. Circ. Res. 2001;89:469. [Google Scholar]
- 38.Firestein G.S., Pisetsky D.S. DNA microarrays: boundless technology or bound by technology? Guidelines for studies using microarray technology. Arthritis Rheum. 2002;46:859–861. doi: 10.1002/art.10236. [DOI] [PubMed] [Google Scholar]
- 39.Aoyagi K. A faithful method for PCR-mediated global mRNA amplification and its integration into microarray analysis on laser-captured cells. Biochem. Biophys. Res. Commun. 2003;300:915–920. doi: 10.1016/s0006-291x(02)02967-4. [DOI] [PubMed] [Google Scholar]
- 40.Iscove N.N., Barbara M., Gu M., Gibson M., Modi C., Winegarden N. Representation is faithfully preserved in global cDNA amplified exponentially from sub-picogram quantities of mRNA. Nat. Biotechnol. 2002;20:940–943. doi: 10.1038/nbt729. [DOI] [PubMed] [Google Scholar]
- 41.Applied Biosystems, accessed June 18, 2003, at http://events-na.appliedbiosystems.com/mk/get/0905_ALLGENE_LANDING?isource = From www.allgenes.com.
- 42.Waters M. Systems toxicology and the Chemical Effects in Biological Systems (CEBS) knowledge base. EHP Toxicogenomics. 2003;111(1T):15–28. [PubMed] [Google Scholar]