

Abstract
Human rhinoviruses (HRV) frequently cause acute respiratory infections and chronic respiratory disease exacerbations. However, testing is not generally offered. We developed a modified HRV quantitative polymerase chain reaction (qPCR) assay to assess viral loads in the community and hospital patients. The assay had a lower limit of detection of 2 log10 viral copies/mL and displayed linearity over 5 log10 viral copies, with a lower limit of quantitation of 4 log10 viral copies/mL. Mean viral loads (95% confidence interval) for hospitalized children, university students, and institutionalized elderly, were 7.08 log10 viral copies/mL (6.7–7.5), 6.87 log10 viral copies/mL (6.5–7.2), and 7.09 log10 viral copies/mL (6.9–7.3), respectively (P = 0.67). Serial specimens of 14 university students showed a decrease of mean viral loads from 6.36 log10 viral copies/mL on day 1 to 2.32 log10 viral copies/mL 7 days past symptom onset (P < 0.001). Using an HRV qPCR, we showed that viral loads did not differ between the community and hospitalized populations and significantly decreased following symptoms onset in healthy individuals.
Keywords: Human rhinovirus, Quantitative, PCR, Viral load
1. Introduction
Human rhinoviruses (HRVs) cause 2 to 3 self-limited episodes of the common cold per person each year (Hershenson, 2010). It is a leading cause of respiratory virus infections causing hospitalizations in children: some 25–50% of children with respiratory symptoms test positive for HRV (Legg et al., 2005, van der Zalm et al., 2011). HRV exacerbate chronic respiratory diseases and increase morbidity rates in the elderly (Gerna et al., 2009, Jartti and Korppi, 2011, Longtin et al., 2010a). Viral infections cause 85% of asthma exacerbations in children, and HRV infections are responsible for 50–80% of these exacerbations (Kelly and Busse, 2008, Wark et al., 2005). Surveillance of long-term care facilities in Ontario found that, of 297 respiratory outbreaks, HRV was identified in 59% (Longtin et al., 2010b). Despite its increasing clinical importance, HRVs are not usually diagnosed by clinical laboratories. Traditional detection methods (i.e., antigen detection) are not feasible due to a large number of serotypes. Recently, molecular assays targeting the 5′UTR and commercial multiplex assays detecting human enterovirus (HEV) and HRV have been described (Gambarino et al., 2009, Jokela et al., 2005, Lu et al., 2008). Molecular tests targeting the conserved 5′UTR have improved sensitivity and increase the coverage of picornaviruses; however, they do not distinguish between HEV and HRV (Mahony, 2008). Not all resources, such as locked nucleotide analog primers, are available to everyone, as there are limited manufacturers limiting the possibilities of developing HRV-specific assays (Lu et al., 2008). Quantitative polymerase chain reaction (qPCR) assays have recently been developed for some respiratory viruses (Gianella et al., 2011, Hohaus et al., 2011, Ward et al., 2004), including HRV (Franz et al., 2010, Utokaparch et al., 2011), and used to assess responses to antiviral agents and predict patient outcomes.
The objectives of this study were to develop a modified qPCR assay for HRV from Lu et al. (2008), evaluate viral load in 3 symptomatic patient populations, and monitor viral loads in a student population for 7 days following symptom onset.
2. Methods
2.1. Clinical specimens
Nasopharyngeal swab (NP) and mid-turbinate nasal flocked swabs (NS) were collected by experienced healthcare professionals from 3 patient populations including hospitalized children, university students, and institutionalized elderly. This study was approved by McMaster University Research Ethics Board (Hamilton, ON, Canada).
One hundred NP specimens were submitted in universal transport medium (UTM) (Copan Italia, Brescia, Italy) to the Regional Virology Laboratory (Hamilton, ON, Canada) between September and December 2009 from children (0–16 years) representing the first population. Symptoms were recorded upon admission. The NP specimens were tested by xTAG™ RVP v. 1 (Luminex, Toronto, ON, Canada).
McMaster University undergraduate students (n = 422) participating in a cohort study between September and October 2009 and between September and October 2010 represented the second population. Mid-turbinate flocked nasal swabs in CyMol™ transport medium (Luinstra et al., 2011, Smieja et al., 2010) (Copan Italia, Brescia, Italy) were self-collected at symptom onset and for up to 7 days. Students submitted electronic questionnaires of upper respiratory tract infection (URTI) symptoms weekly. NS were tested with xTAG™ RVP v. 1 (Luminex).
The third group included 59 residents in a long-term care facility at Sunnybrook Health Sciences, Toronto, ON, Canada. NP specimens were collected in UTM (Copan Italia) from August 27 to October 13, 2010. Symptoms were recorded upon hospital admission. Samples were positive for HRV using the Resplex II v. 2 assay (Qiagen, Mississauga, ON, Canada).
2.2. Total nucleic acid extraction
Total nucleic acid was extracted from both NP and NS (200 μL) using the easyMAG™ automated extractor according to the manufacturer's instruction (bioMérieux, Montreal, QC, Canada). Twenty microlitres of MS2 bacteriophage (Luminex) was added to each sample.
2.3. Quantitative PCR
A 210-bp region of the 5′UTR of HRV was selected for amplification. Two modified forward primers (RAF and RBF), a reverse primer (LR), and a dual-labeled probe (Rhino-probe) were used in the qPCR (Table 1 ). Primers were synthesized at Mobix (Hamilton, ON, Canada), and the probe labeled with 6-carboxyfluorescein (5′) and Black Hole Quencher-1 (3′) was synthesized at Biosearch Technologies (Novato, CA, USA) (Table 1).
Table 1.
Primers and probe for HRV qPCR assay.
| Name | Sequence (5′ → 3′)a | Positionb |
|---|---|---|
| RAF, forwardc | GACAGTGTTCYAGCCTGCG | 342–360 |
| RBF, forwardc | RACHGTGTCYYAGCCTGCG | 342–360 |
| LR, reversed | GAAACACGGACACCCAAAGTA | 556–536 |
| Rhino-probed, e | TCCTCCGGCCCCTGAATGYGGC | 437–458 |
R = dA or dG; H = dA, dC, or dT; Y = dC or dT.
The nucleotide numbering is based on that of the HRV 60 (1473) sequence (accession no. FJ445143.1).
Primers modified from Lu et al. (2008).
Adapted from Lu et al. (2008).
The probe was 5′-end labeled with 6-carboxyfluorescein and 3′-end labeled with Black Hole Quencher-1 (Biosearch Technologies).
qPCR was performed using the QuantiTectProbe RT-PCR kit from Qiagen. The 20-μL reaction volume contained 1.5 μmol/L each of forward and reverse primers, 0.2 μmol/L of probe, and 5 μL of nucleic acid, and amplification was performed using the LightCycler 2.0 instrument (Roche, Laval, QC, Canada) as follows: reverse transcription, 30 min at 50 °C; polymerase activation, 15 min at 95 °C; and 45 cycles of 30 s at 95 °C and 60 s at 60 °C.
RNA transcripts were prepared with a representative HRV strain for analytical sensitivity and specificity studies. PCR amplicons were prepared from HRV 60 (American Type Culture Collection, Manassas, VA, USA) using primers that bracketed the qPCR region (Kiang et al., 2008). The transcripts were synthesized with the MEGAscript T7 In Vitro Transcription kit (Applied Biosystems, Carlsbad, CA, USA) and purified with PureLink PCR purification kit (Invitrogen, Burlington, ON, Canada). Transcripts were quantified using the NanoDrop 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Positive-sense transcripts were 400 bp in length with yields of 1.50 × 1013 viral copies/μL. A standard curve was generated with replicate serial 10-fold dilutions in easyMAG Extraction Buffer 3 (bioMérieux) and stored at −70 °C.
The specificity of the HRV qPCR assay was assessed using RSV, PIV 1–3, adenovirus, human metapneumovirus, human coronavirus 229E and OC43, influenza A H1N1 and H3N2, influenza B, herpes simplex virus 1, cytomegalovirus AD169, Chlamydophila pneumoniae, Legionella pneumophila, Streptococcus pneumoniae, and Mycoplasma pneumonia from clinical isolates. To assess the ability of the qPCR to detect a range of picornaviruses, 22 HEVs from clinical specimens were tested.
2.4. Sequencing
Sanger sequencing was used to distinguish between HEV and HRV targeting 400 bp of the 5′UTR of the HEV/HRV genome (Kiang et al., 2008). To identify species, a partial sequencing of the VP1 gene was amplified (Nix et al., 2006). Amplicons were purified using Purelink PCR purification (Invitrogen) and 5 μL was sequenced at Mobix on the ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
2.5. Statistical analysis
Statistical analyses were performed using GraphPad InStat 3 (GraphPad Software, La Jolla, CA, USA). A P < 0.05 was considered to be statistically significant.
3. Results
3.1. Development of qPCR assay for HRV
3.1.1. Sensitivity and specificity of qPCR
A qPCR assay amplifying 210 bp of the 5′UTR was developed to assess HRV viral loads. A panel of 18 different respiratory viruses and bacteria were tested to assess the specificity of the qPCR. All specimens tested negative by qPCR.
A panel of 22 recent HEV isolates were tested to assess cross-reactivity with HEV. Only 3 (13.6%) of 22 HEVs (mean threshold cycle [C T] 32.86; 95% confidence interval [CI], 31.7–34.0) were detected by the HRV qPCR assay.
3.1.2. Lower limit of detection and quantitation
HRV 60 RNA transcripts were used to assess amplification efficiency. Amplification was linear over a 5-log dynamic range (Fig. 1 ), an R 2 value of 0.996, and efficiency (Bustin et al., 2009) of 104.5%. The lower limit of detection (LLOD) was determined by testing 15 replicates of HRV 60 RNA transcripts (1.9 log10 to 3 log10 viral copies/mL each). Between 2 log10 and 3 log10 viral copies/mL, 15 of 15 replicates were positive, while, at 1.9 log10 viral copies/mL, only 3 of 15 replicates were positive. The LLOD was 2 log10 viral copies/mL. The lower limit of quantitation (LLOQ) was determined by plotting mean C T for dilutions 1.9 log10 to 3 log10 viral copies/mL as shown in Fig. 1. Linearity was lost below 4 log10 viral copies/mL, indicating an LLOQ of 104 copies of HRV RNA.
Fig. 1.
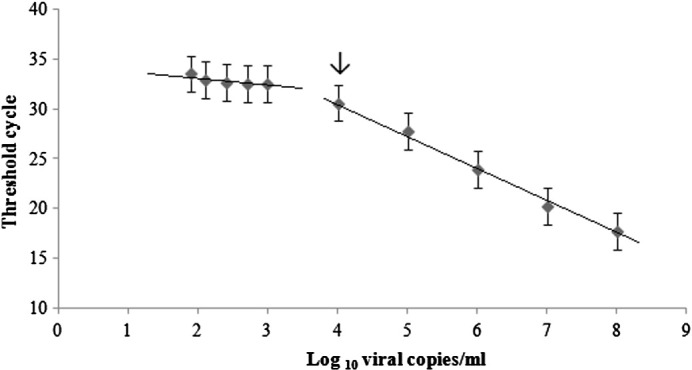
Dynamic range of the HRV qPCR assay and lower limit of quantitation. Mean threshold cycle (CT) values were plotted against the copy number. The lower limit of quantitation is 4 log10 viral copies/mL and is indicated by an arrow.
3.1.3. Reproducibility
Next, we assessed inter- and intra-assay reproducibility using serial dilutions of RNA transcripts ranging from 2 log10 to 8 log10 viral copies/mL by testing on 5 consecutive days. Mean C T (SD) from 2 log10 to 8 log10 ranged from 33.02 (1.14) to 17.52 (0.03) within runs, and 32.57 (0.71) to 16.48 (0.44) between runs, respectively. The assay was reproducible between 103 and 108 copies of RNA.
3.1.4. HRV viral loads of clinical specimens
A sampling of 187 HRV clinical isolates were tested using the HRV qPCR assay, consisting of 81 HRV A, 54 HRV B, and 52 HRV C confirmed by Sanger sequencing. All were detected and quantified by the assay.
3.2. Comparison of HRV viral loads in different populations
To determine HRV viral loads, we tested 36 NP from hospital-admitted children, 19 NS from university students, and 18 NP from institutionalized elderly on day 1 of illness. Mean viral loads from children, students, and elderly were 7.08 log10 viral copies/mL (95% CI 6.7–7.5), 6.87 log10 viral copies/mL (95% CI 6.5–7.2), and 6.86 log10 viral copies/mL (95% CI 6.5–7.3), respectively (P = 0.67). Twelve children reported symptoms of asthma exacerbations (33.3%), wheezing (8.3%), fever (50.0%), rhinitis (8.3%), and cough (41.7%). Eighteen students reported symptoms of rhinitis (77.8%), fever (22.2%), cough (66.7%), sore throat (38.9%), and wheezing (16.7%). Eighteen institutionalized elderly residents reported fever (25.0%), rhinitis (15.0%), and cough (90.0%). No individual in the 3 populations was co-infected with another respiratory virus as determined by xTAG™ RVP v. 1 (Luminex).
3.3. Serial daily sampling of HRV viral loads in university students
To determine the duration of HRV shedding, 14 undergraduate students with symptoms of an URTI self-collected NS for a period of 7 days from the onset of illness and then once weekly. Virus was detected on days 1–7, but none had detectable virus on day 14. Mean viral load decreased from 6.36 log10 viral copies/mL on day 1 to 2.32 log10 viral copies/mL on day 7 (P < 0.001) (Fig. 2 ). Sanger sequencing of HRV positives identified 5 HRV A, 5 HRV B, and 4 HRV C species. There was no significant difference in viral load found by species (P = 0.81). In addition, no significant difference in viral load decline was detected between species (P = 0.86).
Fig. 2.

Longitudinal surveillance of HRV viral loads in university students. Self-collected mid-turbinate nasal swabs (n = 98) were serially submitted over a 7-day period by undergraduate students (n = 14) following the onset of upper respiratory tract infection. Fourteen nasal swabs were tested per time point. Data are plotted as mean viral loads (log10 viral copies/mL) and SEM for various days following onset of symptoms (P < 0.001).
4. Discussion
In this study, we used an improved qPCR assay to evaluate viral loads in 3 patient populations and investigate HRV shedding in students. This assay detects a wider range of HRV genotypes due to the addition of a second forward primer. We found no significant difference in viral load between the 3 study populations (P = 0.67). Serial observations of students for 7 days post onset of symptoms demonstrated a significant decrease in viral load (P < 0.001).
The HRV qPCR assay targeted a 210-bp region in the 5′UTR. This is a highly conserved region with lower rates of recombination compared to HEV (Lu et al., 2008). The modified primers adapted from Lu et al. (2008) are effective at detecting all 3 HRV species, although not all genotypes were tested and future modifications to the primer and probe design may be necessary as novel genotypes emerge (Faux et al., 2011). Although the assay does not detect other respiratory pathogens, it detected some HEV (3/22 positives) at a low copy number (mean C T, 32.86). The relevance of low viral loads of HEV in clinical isolates is not known and will require further investigation. In addition, it is not known whether HRV viral loads below our LLOQ are clinically relevant and result in clinical presentations. We are currently investigating HRV viral loads in asymptomatic individuals.
We used the qPCR to test different symptomatic patient populations and expected to see different quantities between children, university students, and the elderly to correlate viral load with severity of clinical presentation. To our surprise, the mean titres of HRV were the same for these 3 populations. Sampling with NS and NPS is comparable and cannot account for differences in viral titres (Lambert et al., 2008, Larios et al., 2011). Timing may have affected our results as adult specimens were collected on day 1 following symptom onset, whereas the time of collection for children and symptom onset was not available. This is the first report, to our knowledge, of HRV viral loads in adult populations. Two previous reports measured HRV viral loads in hospitalized children (Franz et al., 2010, Utokaparch et al., 2011). Utokaparch et al. (2011) tested archived samples and reported lower viral loads (mean viral load, 1.07 log10 viral copies/mL) for children than we detected (Utokaparch et al., 2011). Lower viral loads in their study may have been due to prolonged specimen storage with potential RNA degradation, different assay methodology, or collection method. Future studies will be required to determine the range of viral loads in children as well as how viral load changes over time.
We evaluated the duration of infections in the community by testing otherwise healthy university students. There was no difference in the rate of decline for viral load of HRV A, B, or C (P = 0.86), although our sample size was small. Students were not infected with a distinct HRV genotype during the symptomatic episode nor did they experience a second episode. Additional studies with larger numbers of healthy individuals will be required to determine whether there are differences in viral loads across different HRV species.
The qPCR assay described here conducts rapid, sensitive, and precise quantitation of HRV. To date, there are no data on viral loads at various times following infection of hospitalized patients. Observations of hospitalized patients allowing comparisons to community infections may be warranted. qPCR tests provide accurate tools for investigation of HRV viral loads and could be used to measure novel antivirals currently under development.
References
- Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Franz A., Adams O., Willems R., Bonzel L., Neuhausen N., Schweizer-Krantz S. Correlation of viral load of respiratory pathogens and co-infections with disease severity in children hospitalized for lower respiratory tract infection. J Clin Virol. 2010;48:239–245. doi: 10.1016/j.jcv.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faux C.E., Arden K.E., Lambert S.B., Nissen M.D., Nolan T.M., Chang A.B. Usefulness of published PCR primers in detecting human rhinovirus infection. Emerg Infect Dis. 2011;17:296–298. doi: 10.3201/eid1702.101123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambarino S., Costa C., Elia M., Sidoti F., Mandtovani S., Gruosso V. Development of a RT real-time PCR for the detection and quantification of human rhinoviruses. Mol Biotechnol. 2009;42:350–357. doi: 10.1007/s12033-009-9164-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerna G., Piralla A., Rovida F., Rognoni V., Marchi A., Locatelli F. Correlation of rhinovirus load in the respiratory tract and clinical symptoms in hospitalized immunocompetent and immunocompromised patients. J Med Virol. 2009;81:1498–1507. doi: 10.1002/jmv.21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianella M., Alonso M., Viedma D.G., Roa P.L., Catalan P., Padilla B. Prolonged viral shedding in pandemic influenza A (H1N1): clinical significance and viral load analysis in hospitalized patients. Clin Microbiol Infect. 2011;17:1160–1165. doi: 10.1111/j.1469-0691.2010.03399.x. [DOI] [PubMed] [Google Scholar]
- Hershenson M.B. Rhinovirus and respiratory disease. In: Ehrenfeld E., Domingo E., Roos R.P., editors. The Picornaviruses. ASM Press; Washington, DC: 2010. pp. 369–382. [Google Scholar]
- Hohaus S., Giachelia M., Vannata B., Massini G., Cuccaro A., Martini M. The viral load of Epstein-Barr virus (EBV) DNA in peripheral blood predicts for biological and clinical characteristics in Hodgkin lymphoma. Clin Cancer Res. 2011;17:2885–2892. doi: 10.1158/1078-0432.CCR-10-3327. [DOI] [PubMed] [Google Scholar]
- Jartti T., Korppi M. Rhinovirus-induced bronchiolitis and asthma development. Pediatr Allergy Immunol. 2011;22:350–355. doi: 10.1111/j.1399-3038.2011.01170.x. [DOI] [PubMed] [Google Scholar]
- Jokela P., Joki-Korpela P., Maaronen M., Glumoff V., Hyypia T. Detection of human picornaviruses by multiplex reverse transcription-PCR and liquid hybridization. J Clin Microbiol. 2005;43:1239–1245. doi: 10.1128/JCM.43.3.1239-1245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J.T., Busse W.W. Host immune responses to rhinovirus: mechanisms in asthma. J Allergy Clin Immunol. 2008;122:671–682. doi: 10.1016/j.jaci.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiang D., Kalra I., Yagi S., Louie J.K., Boushey H., Boothby J. Assay for 5′ noncoding region analysis of all human rhinovirus prototype strains. J Clin Microbiol. 2008;46:3736–3745. doi: 10.1128/JCM.00674-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S.B., Whiley D.M., O'Neill N.T., Andrews E.C., Canavan F.M., Bletchly C. Comparing nose-throat swabs and nasopharyngeal aspirates collected from children with symptoms for respiratory virus identification using real-time polymerase chain reaction. Pediatrics. 2008;122:615–620. doi: 10.1542/peds.2008-0691. [DOI] [PubMed] [Google Scholar]
- Larios O.E., Coleman B.L., Drews S.J., Mazzulli T., Borgundvaaq B., Green K. Self-collected mid-turbinate swabs for the detection of respiratory viruses in adults with acute respiratory illnesses. PLoS ONE. 2011;6:1–4. doi: 10.1371/journal.pone.0021335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legg J.P., Warner J.A., Johnston S.L., Warner J.O. Frequency of detection of picornaviruses and seven other respiratory pathogens in infants. Pediatr Infect Dis J. 2005;24:611–616. doi: 10.1097/01.inf.0000168747.94999.aa. [DOI] [PubMed] [Google Scholar]
- Longtin J., Winter A., Heng D., Marchand-Austin A., Eshaghi A., Patel S. Severe human rhinovirus outbreak associated with fatalities in a long-term care facility, in Ontario, Canada. J Am Geriatr Soc. 2010;58:2036–2038. doi: 10.1111/j.1532-5415.2010.03091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtin J., Marchand-Austin A., Winter A., Patel S., Eshaghi A., Jamieson F. Rhinovirus outbreaks in long-term care facilities, Ontario, Canada. Emerg Infect Dis. 2010;16:1463–1465. doi: 10.3201/eid1609.100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Holloway B., Dare R.K., Kuypers J., Yagi S., Williams J.V. Real-time reverse transcription-PCR assay for comprehensive detection of human rhinoviruses. J Clin Microbiol. 2008;46:533–539. doi: 10.1128/JCM.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luinstra K., Petrich A., Castriciano S., Ackerman M., Chong S., Carruthers S. Evaluation and clinical validation of an alcohol-based transport medium for preservation and inactivation of respiratory viruses. J Clin Microbiol. 2011;49:2138–2142. doi: 10.1128/JCM.00327-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony J.B. Detection of respiratory viruses by molecular methods. Clin Microbiol Rev. 2008;21:716–747. doi: 10.1128/CMR.00037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix W.A., Oberste M.S., Pallansch M.A. Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J Clin Microbiol. 2006;44:2698–2704. doi: 10.1128/JCM.00542-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smieja M., Castriciano S., Carruthers S., So G., Chong S., Luinstra K. Development and evaluation of a flocked nasal mid-turbinate swab for self-collection in respiratory virus infection diagnostic testing. J Clin Microbiol. 2010;48:3340–3342. doi: 10.1128/JCM.02235-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utokaparch S., Marchant D., Gosselink J.V., McDonough J.E., Thomas E.E., Hogg J.C. The relationship between respiratory viral loads and diagnosis in children presenting to a pediatric hospital emergency department. Pediatr Infect Dis J. 2011;30:e18–e22. doi: 10.1097/INF.0b013e3181ff2fac. [DOI] [PubMed] [Google Scholar]
- van der Zalm M.M., Wilbrink B., van Ewijk B.E., Overduin P., Wolfs T.F.W., van der Ent C.K. Highly frequent infections with human rhinovirus in healthy young children: a longitudinal cohort study. J Clin Virol. 2011;52:317–320. doi: 10.1016/j.jcv.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Ward C.L., Dempsey M.H., Ring C.J.A., Kempson R.E., Zhang L., Gor D. Design and performance testing of quantitative real time PCR assays for influenza A and B viral load measurement. J Clin Virol. 2004;29:179–188. doi: 10.1016/S1386-6532(03)00122-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wark P.A.B., Johnston S.L., Bucchieri F., Powell R., Puddicombe S., Laza-Stanca V. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]