

Abstract
Highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) was firstly characterized in 2006 in China. The virus has caused great economic loss to the Chinese swine production during the past years. Herein, we experimentally infected SPF pigs using two strains of PRRSV with different pathogenicity and observed the lung pathological changes looking for new sights on the possible pathogenesis associated with the virulence of HP-PRRSV. The results indicated that the HP-PRRSV-infected pigs died and exhibited severe pathological changes of lungs featuring increased neutrophils, mast cells and mononuclear macrophages, compared with the pigs inoculated with low pathogenic (LP-) PRRSV. Furthermore, the pigs infected with HP-PRRSV showed the higher levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, interleukin (IL)-8 and histamine, leukotriene B4 (LTB4), platelet activation factor (PAF) in sera than those inoculated with LP-PRRSV. Additionally, the fibrosis of lung was observed in the HP-PRRSV-infected pigs. At present, our findings suggest that the aberrant immune responses triggered by HP-PRRSV infection are closely related to acute lung injury (ALI), and especially the pathological changes in lung vascular system are of particular significance. These associated pathological changes of lung are in part responsible for the additional morbidity and mortality observed in HP-PRRSV infection.
Keywords: highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV), infection, pigs, lung, acute lung injury (ALI)
1. Introduction
Porcine reproductive and respiratory syndrome (PRRS) is generally considered as a severe viral disease in pigs, causing huge economic loss worldwide each year. The disease was first reported in the late 1980s in the United States (Keffaber, 1989), and similar clinical outbreak occurred in Germany in 1990 and was widespread throughout Europe by 1991 (Goyal, 1993). In the early 1990s, this viral disease was identified in Asia (Murakami et al., 1994, Shimizu et al., 1994). In 1995, a PRRS outbreak was firstly recognized in Beijing of China. Since then, PRRS has been verified in many provinces in China, proving to be one of main swine diseases in China (Gao et al., 2004, Guo et al., 1996). In China in 2006, an unparalleled large-scale, atypical PRRS outbreak caused by the highly pathogenic PRRSV (HP-PRRSV) dealt a heavy blow to the swine industry (Zhou and Yang, 2010, Tian et al., 2007).
Different pathogenic PRRSV strains in the field induced totally different consequences of infected pigs because of genetically extensive variation (Li et al., 2007, Meng, 2000, Nelsen et al., 1999). Our previous studies have shown that the low pathogenic PRRSV (LP-PRRSV) infection resulted in no fatality of pigs with light anatomical and histopathological changes in lungs of pigs, whereas the HP-PRRSV-infected pigs presented high mortality accompanying with high fever (40–42 °C), depression, anorexia, cough, asthma, severe dyspnea, disorder in the respiratory tract, lameness, shivering (Zhou et al., 2009). A recent study found that HP-PRRSV could exhibit extensive tissue tropism for pigs (Li et al., 2012). These observations of the pathological changes in tissues of HP-PRRSV-infected pigs suggested that HP-PRRSV infection induced severe pathological changes of lungs.
Various kinds of pathogenic factors can cause acute lung injury (ALI), such as severe infection, shock, thoracic trauma, disseminated intravascular coagulation, gastric acid, smog, and toxic gas aspiration clinically characterized by rapidly progressive dyspnea and intractable hypoxemia (Gattinoni et al., 1998, Atabai and Matthay, 2002). ALI may develop into acute respiratory distress syndrome (ARDS) characterized by damage to the alveolus-capillary interface, usually secondary to an intense inflammatory response of the host lung to infectious or noninfectious invasion (Levy et al., 2005, Bernard, 2005). It has been shown that severe acute respiratory syndrome (SARS) virus and avian influenza A H5N1 virus infection could result in high mortality due to the complication of ARDS. Therefore, infectious pathogens, in which that the majority are viruses, have become one of the most important causes of ARDS (Headley et al., 1997, Chen et al., 2005, Subbarao et al., 1998). Thus, it is intriguing to note that HP-PRRSV infection might be associated with ALI which may have the responsibility for the additional morbidity and mortality of the infected pigs.
Why two completely different consequences exist in the pigs infected with various PRRSV strains? To address this question, in this study, SPF pigs were infected with two strains of PRRSV, LP-PRRSV strain HB-1/3.9 and HP-PRRSV strain JXwn06, in an attempt to fully observe the distinct lung pathological changes and to better understand the lung pathogenesis induced by HP-PRRSV.
2. Materials and methods
2.1. Viruses, animals and experimental design
Two PRRSV strains, JXwn06 and HB-1/3.9 with different virulence, were used in the study. The virus JXwn06, a HP-PRRSV strain, was isolated from an intensive pig farm with atypical PRRS outbreak in 2006 (Zhou et al., 2009). The virus HB-1/3.9, a LP-PRRSV strain, was derived from HB-1(sh)/2002 adapted in MARC-145 cells (Gao et al., 2004). These viruses were propagated using highly permissive MARC-145 cells according to the method described previously (Zhou et al., 2009).
Twenty one 28-day-old SPF large white pigs (Beijing Center for SPF Swine Breeding & Management, Beijing, China) were divided randomly into three groups, the JXwn06-infected group (n = 9), the HB-1/3.9-infected group (n = 9) and control group (n = 3). Each group was then housed separately in different isolation rooms, with individual ventilation. Each pig was intranasally inoculated with 2 ml of 105 TCID50/ml virus (JXwn06 and HB-1/3.9), respectively. The pigs in the control group were mock-inoculated with the same dosages of MARC-145 cells culture supernatant, and then were euthanized and sampled. Clinical signs of virus-inoculated pigs were visually examined and simultaneously the rectal temperatures were measured daily.
Animal use and animal trials in this study have been approved by The Beijing Municipal Committee of Animal Management and The Ethics Committee of China Agricultural University.
2.2. Collection of samples, virus titration and ELISA
On days 3 and 5 post-inoculation (PI), 3 pigs in HB-1/3.9- and JXwn06-infected group were euthanized, and then once death of the rest 3 pigs in JXwn06-infected happened, the 3 pigs were sampled immediately. Meanwhile the rest 3 pigs in HB-1/3.9-infected group were euthanized accordingly. Two portions from each lobe of the left lung were collected immediately and fixed in 4% paraformaldehyde solution. The whole left lung was weighed before and after desiccation at 80 °C drying to constant weight, and then the lung wet:dry weight ratio was determined which was taken as one indicator of lung edema (Lang et al., 2005). Serum samples collected from the infected animals were detected for viral RNA using RT-PCR by amplifying a 312 bp ORF7 fragment of PRRSV. According to the method of Reed-Muench, virus titration in the lungs was determined with some modification. Briefly, three lobes of right lungs were sampled, frozen in liquid nitrogen and then ground into fine powder using a mortar and pestle, weighted for one milligram and homogenized in 1 ml cold phosphate-buffered saline. Clarified homogenates were titrated for viral infectivity in MARC-145 cells cultured in 96 well plates from initial dilutions of 1:10. Viral titers were expressed as mean TCID50 per milliliter. In addition, the concentrations of histamine, PAF, LTB4 and IL-8 in serum, and TNF-α in serum and homogenates of lungs were detected using R&D ELISA kit (R&D system, Inc. USA) according to the manufacture's procedure.
2.3. Lung histopathology
The fixed lung samples were dehydrated, embedded in paraffin, and serial sectioned (4 μm). Three sagittal sections from each lung, and six sections per animal, were stained with hematoxylin-eosin and Masson's trichrome. The severities of histopathological changes and pulmonary fibrosis of the lungs were assessed and scored. All the sections were numbered randomly and interpreted by three experimenters blinded to the experimental conditions. Histopathological changes were observed and scored under an Olympus microscope (Olympus Optical Co., Ltd.). Criteria for grading lung histopathological changes were as follows: Grade 0 = no obvious pathological changes; Grades 1–3 = light inflammatory cells infiltration, light hemorrhage, vasculitis or bronchiolitis; Grades 4–5 = inflammatory cells infiltration, hemorrhage, vasculitis or bronchiolitis, cell apoptosis and necrosis, microthrombus; Grades 6–10 = severe inflammatory cells infiltration, severe hemorrhage, vasculitis or bronchiolitis, obvious edema, cell apoptosis and necrosis, microthrombus, multinucleated giant cell.
Visual grading of pulmonary fibrosis was performed according to the Ashcroft's scoring with some modification (Ashcroft et al., 1988, Ouchi et al., 2008, Qiao et al., 2009). Briefly, the whole section was scanned for general qualitative observations under an Olympus microscope at ×100 magnifications, and each field was visually graded from 0 to 8. Criteria for grading lung fibrosis were as follows: Grade 0 = normal lung; Grade 1 = minimal fibrous thickening of alveolar or bronchiolar walls; Grades 2–3 = moderate thickening of walls without obvious damage to lung architecture; Grades 4–5 = increased fibrosis with definite damage to lung structure and formation of fibrous bands or small fibrous masses; Grades 6–7 = severe distortion of structure and large fibrous areas; Grade 8 = total fibrous obliteration of lung fields. After examination of the whole sections, the mean score of all the fields was taken as the fibrosis score for each animal.
2.4. Ultrastructural observation
To observe the ultrastructural changes of lungs, two portions (1–2 mm3) from each lobe of the right lungs were collected immediately and fixed in 2.5% (v/v) glutaraldehyde-polyoxymethylene solution for making semi-thin section and ultrathin section. Semi-thin section (2 μm) was stained with 0.1% crystal violet solution to determine specific tissue structures such as pulmonary alveoli, vessel and thicken alveolar septa. After determined the location, ultrathin section (0.5 μm) was made to observe the cell ultrastructural pathological changes of the lungs such as alveolar epithelial cells, vascular endothelial cells and immune cells.
2.5. Quantity of inflammatory cells in the lungs
Mast cells were examined by an improved toluidine blue staining according to the method described previously (Sun et al., 2008). Briefly, lung paraffin sections were dewaxed, rehydrated, and immersed in 0.8% toluidine blue (Sigma Co.) for 15 s. Sections were rinsed with distilled water, and then placed into 95% alcohol until the mast cells appeared deep reddish purple under the microscope, dehydrated and mounted. The number of mast cells was counted in 10 high-power fields under the microscope at ×400 magnifications and the means were calculated. Sampling of the sections was unbiased, with the samples coded and examinations performed by one investigator. The whole section was scanned for general qualitative observations, but detailed examination focused on mast cells. Mast cell density was expressed as cells per square millimeter.
The macrophages in the lungs were stained by immunohistochemistry using the antibody against CD86. The sections were incubated with the primary antibody to CD68 (Abcam Ltd. Hong Kong, China, 1:200) overnight at 4 °C, goat anti-mouse IgG conjugated with horseradish peroxidase (Zymed Laboratories Inc, Beijing, China) at 37 °C for 1 h, diaminobenzidine as chromogen (Zymed Laboratories Inc, Beijing, China) for 10 min at room temperature in the dark, and counterstained with haematoxylin. For negative control, the primary antibody was replaced with PBS. The number of positive cells was counted and the means calculated as described above in mast cell staining.
2.6. Statistical analysis
Data were expressed as means ± standard deviations (SD). The significance of the variability among different groups was determined by Two-way tests of variance using the GraphPad Prism software (version 5.0). P values < 0.05 were considered to be statistically significant.
3. Results
3.1. Clinical and gross pathologic observations of the infected pigs
The pigs in JXwn06-infected group showed severe clinical symptoms. From day 1 PI, all the pigs started to develop elevated body temperatures (>40 °C) with a peak of 41.5 °C. Signs of anorexia, depression and lethargy were first observed on day 3 PI. By day 7 PI, majority of the infected pigs abruptly presented severe clinical signs, including roughened hair, rubefaction, emaciation, lethargy, inappetence, respiratory distress, shiver, eyelid and limbs edema and posterior paresis. On day 8 PI, shiver and aspiration frequency were strengthened, and dyspnea, tachycardia, liquid diarrhea and cyanosis of ears, limbs or perineum concomitantly occurred. Two pigs died on day 9 PI and one pig died on day 16 PI. In contrast, the body temperature of HB-1/3.9-inoculated pigs was significantly lower than that of JXwn06-infected pigs, and no obvious clinical signs were observed except for occasional depression and anorexia. Gross observation showed slight edematous lesions in the lungs of HB-1/3.9-infected pigs were observed (Fig. 1A), while the lungs of JXwn06-infected pigs were highly edematous, emphysematous, with profuse areas of hemorrhage (Fig. 1B and C). No obvious gross lesions were observed in the lungs of control pigs (Fig. 1D).
Fig. 1.
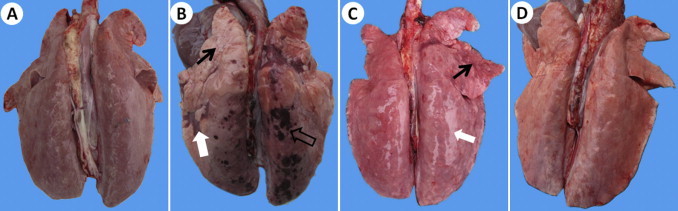
Gross pathological changes in the lungs of infected pigs. No obvious pathological changes with dark color in the lung of HB-1/3.9-infected pig (A). Emphysema (thin arrow), hemorrhage (thick-hollow arrow) and hepatization (thick-solid arrow) in the lung of JXwn06-infected pig on moribund (day 9 PI) (B). Consolidation (thin arrow) and severe edema with swelling (thick arrow) in the lung of JXwn06-infected pig on moribund (day 16 PI) (C). No obvious pathological changes in the lung of control pig (D).
By RT-PCR, PRRSV was detected to be positive from day 3 PI in the sera of JXwn06-infected pigs, but from day 5 PI in HB-1/3.9-infected pigs. The viral titers in the lungs of JXwn06- and HB-1/3.9-infected pigs were calculated to be 10−2.76 TCID50/ml and 10−1.76 TCID50/ml on moribund (including day 9 and 16 PI), respectively.
3.2. Histopathologic lesions in the lungs of infected pigs
The histopathological changes in lungs of the infected pigs were observed. As shown in Fig. 2, Fig. 3 , only predominantly peribronchiolitis and interstitial pneumonia with light haemorrhage in lungs of the HB-1/3.9-infected pigs were visible on days 3, 5, and 16 PI (Fig. 2A–C). On the contrary, in JXwn06-infected pigs, hyperaemia, initial peribronchiolar patchy pneumonia and interstitial pneumonia were observed on day 3 PI (Fig. 2D); by day 5 PI, lung histopathological lesions aggravated, including predominantly peribronchiolar lesions, fully developed bronchiolitis, and bronchopneumonia, and dropout of alveolar epithelial cells and narrowed alveolar space (Fig. 2E); severe histopathological lesions were observed in lungs of the died pigs, these lesions included destruction of lung structure with extensive hemorrhage and a large number of inflammatory cells infiltration, severe dropout of alveolar epithelial cells with exudation of inflammatory cells and erythrocytes into the alveolar spaces, vasculitis and thrombus, alveolar lumens flooded with edema fluid and pulmonary interstitial edema accompanied vascular trauma, and formation of multinucleated giant cells and epithelioid cells (Fig. 2F and G). The lungs of control pigs exhibited no obvious histopathological changes (Fig. 2H). Meanwhile, there was mild fibrosis with collagen fibers in HB-1/3.9-infected pigs on day 16 PI (Fig. 3A), while severe distortion of structure and diffuse fibrous areas were observed in the lungs of died pigs infected with JXwn06 (Fig. 3B). No obvious fibrosis was observed in the lungs of both HB-1/3.9- and JXwn06-infected pigs on days 3 and 5 PI. No fibrosis was in the lungs of control pigs (Fig. 3C). Severity scores for lung histopathological lesions are given in Fig. 4A. According to Ashcroft's method (Ashcroft et al., 1988), severity scores for lung fibrosis are given in Fig. 4B. These observations indicated that the lungs of JXwn06-infected pigs showed severer histopathological changes than those of HB-1/3.9-infected pigs.
Fig. 2.
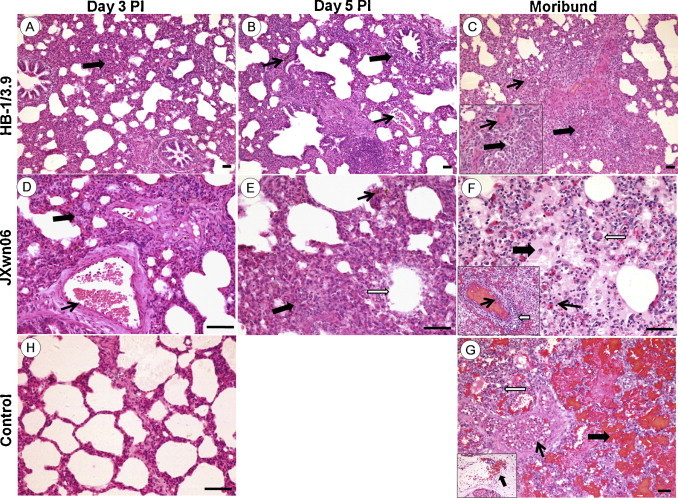
Histopathological changes in the lungs of the infected pigs. Inflammatory cells infiltration (solid and thick arrow) and hyperemia (solid and thin arrow) in the lung of HB-1/3.9- (A) and JXwn06- (D) infected pig on day 3 PI. Peribronchiolitis (solid and thick arrow), hemorrhage and hyperemia (solid and thin arrow) in the lung of HB-1/3.9-infected pig (B), and hemorrhage (solid and thin arrow), dropout of alveolar epithelial cells (open arrow), inflammatory cells infiltration and narrowed alveolar space (solid and thick arrow) in the lung of JXwn06-infected pig (E) on day 5 PI. Inflammatory cells infiltration and interstitial pneumonia (solid and thick arrow), and narrowed alveolar space (solid and thick arrow) in the lung of HB-1/3.9-infected pig (C), and microthrombus (solid and thin arrow, F), vasculitis (open arrow, F), alveolar lumens flooded with edema fluid mixed with erythrocytes and inflammatory cells (solid and thick arrow, F), vascular injury and severe hemorrhage (G), multinucleated giant cell (open arrow, F) and epithelioid cells (solid and thin arrow, F) in the lung of JXwn06-infected pig on day 16 PI and moribund. No significant changes in the lung of control pig (H).
Fig. 3.
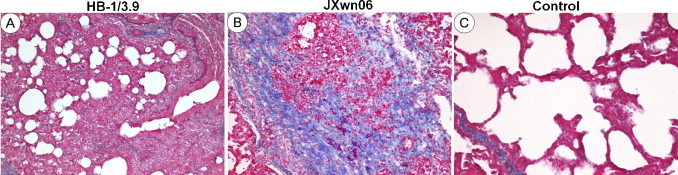
Fibrosis in the lungs of infected pigs. Mild fibrosis with collagen fibers (solid arrow) in the lung of HB-1/3.9-infected pig on day 16 PI (A). Severe distortion of structure and diffuse fibrous areas (solid arrow) in the lung of JXwn06-infected pig (B). No fibrosis in the lung of control pig (C). Bar = 50 μm.
Fig. 4.
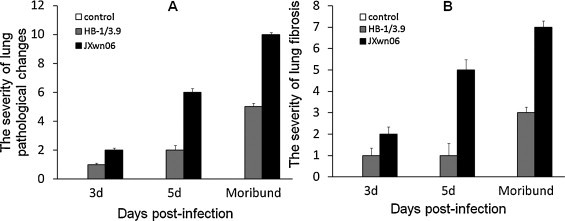
The pathological scores of the lungs of infected pigs. The severity of lung pathological lesions (A). The severity of lung fibrosis (B). The pathological lesions were scored according to the methods described in Materials and methods.
3.3. HP-PRRSV infection could induce severe lung edema in pigs
Severe lung edema in some cases could result in ALI which was associated with death (Mannam et al., 2013, Meers et al., 2011, Dulu et al., 2006). In our study, the lungs of JXwn06-infected pigs exhibited severe edema which was significantly different from HB-1/3.9-infected and control groups. With regard to edema, the lung wet:dry weight ratio and the concentrations of histamine, PAF and LTB4 in serum were measured. As shown in Fig. 5A, the lung wet:dry weight ratio of JXwn06-infected pigs was higher than that of BH-1/3.9-infected pigs, with a significant difference on moribund (p < 0.01). The levels of histamine in JXwn06-infected pigs significantly increased from day 3 PI to moribund compared with those of HB-1/3.6-infected and control groups (p < 0.01), and on day 16 PI, the levels of histamine in the lungs of HB-1/3.9-infected pigs had significant difference compared with those of control group (Fig. 5B, p < 0.05). PAF in JXwn06-infected pigs also elevated significantly from day 5 PI to moribund compared to control group (p < 0.05), whereas no obvious changes of PAF were observed in HB-1/3.9-infected pigs (Fig. 5C). Dramatic increase of LTB4 in JXwn06-infected pigs was detected with highly significant difference compared to those of control group on day 5 PI to moribund (p < 0.01), and was significantly different from those of HB-1/3.9-infected group (p < 0.05). While no difference of LTB4 was found between HB-1/3.9-infected and control groups (Fig. 5D).
Fig. 5.
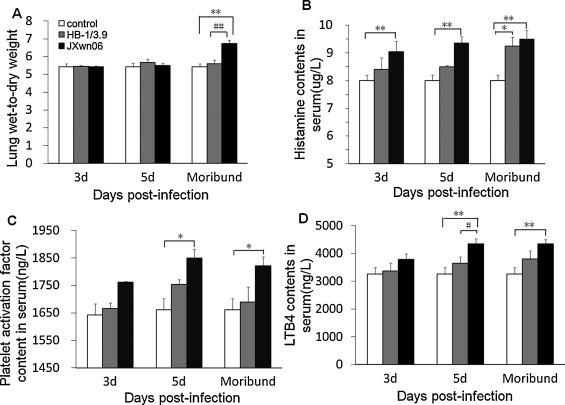
Changes of indicators for lung edema. Lung and serum samples were collected on days 0, 3, 5 PI and moribund. Lung wet-to-dry weight ratios (A), histamine (B), platelet activation factor (C), and leukotriene B4 (D) were measured by the methods described in Materials and methods. Data were shown as means ± SD from 3 pigs each group. *p < 0.05 and **p < 0.01 between control and virus infection groups. #p < 0.05 and ##p < 0.01 between HB-1/3.6- and JXwn06-infected groups.
3.4. PRRSV infection resulted in severe inflammation in the lungs of infected pigs
Compared with control group, the lungs of HB-1/3.9- and JXwn06-infected pigs showed an increased inflammatory cell population as early as day 3 PI. To quantify the immune cell sub-populations responding to viral infection, we determined the cell numbers of specific inflammatory cell populations in the lungs of infected pigs using specific stainings. Mast cells, mononuclear-macrophages and polymorphonuclear neutrophils accounted for the highest percentage of the total leukocytes following infection (Fig. 6 ). On days 3 to 5 PI, mast cell numbers in the lungs of JXwn06- and HB-1/3.9-infected pigs were significantly higher than those of control group (p < 0.05), while on day 5 PI, more mast cells were in the lungs of JXwn06-infected pigs compared to those of HB-1/3.9-infected group. On days 9 and 16 PI (moribund), no difference in mast cells in mast cells was showed between JXwn06-infected and control groups, but mast cell numbers in the lungs of HB-1/3.9-infected pigs were dramatically higher than those of control group (Fig. 7A). There were extremely significant differences of mononuclear-macrophages numbers in the lungs of pigs between control and virus-infected group on day 3 PI to moribund (p < 0.01), while on day 5 PI to moribund, the number of mononuclear-macrophages in lungs of JXwn06-infected group was highly significantly different from those of HB-1/3.9-infected group (p < 0.01) (Fig. 7B). Compared with control and HB-1/3.9-infected group, the number of neutrophils of JXwn06-infected pigs increased significantly from day 3 PI to moribund (p < 0.01), while from day 5 PI to moribund there was highly significant difference in nutrophils number between HB-1/3.9-infected and control groups (p < 0.01) (Fig. 7C).
Fig. 6.
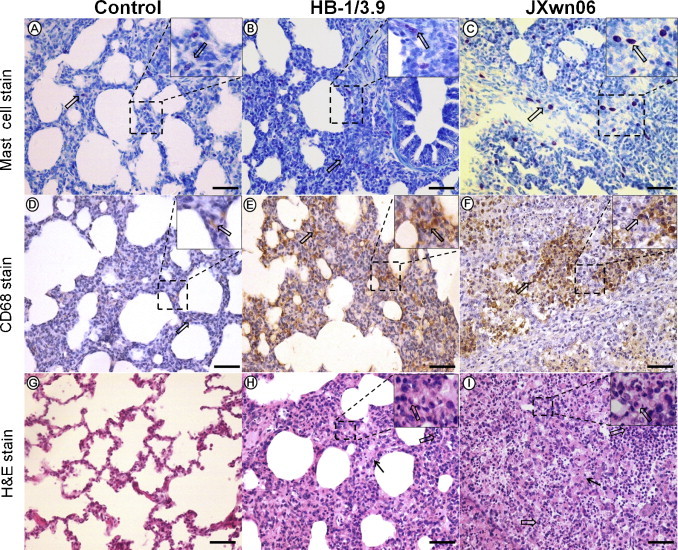
Changes of mast cells, mononuclear-macrophages and neutrophils in the lungs of infected pigs. Mast cells with reddish purple (arrows) in the lung of control pig (A), HB-1/3.9-infected pig (B) and JXwn06-infected pig (C). Mononuclear-macrophages with brown orange (arrows) in the lung of control pig (D), HB-1/3.9-infected pig (E) and JXwn06-infected pig (F). Neutrophils in the lung of control pig (G), HB-1/3.9-infected (H) and JXwn06-infected pig (I).
Fig. 7.
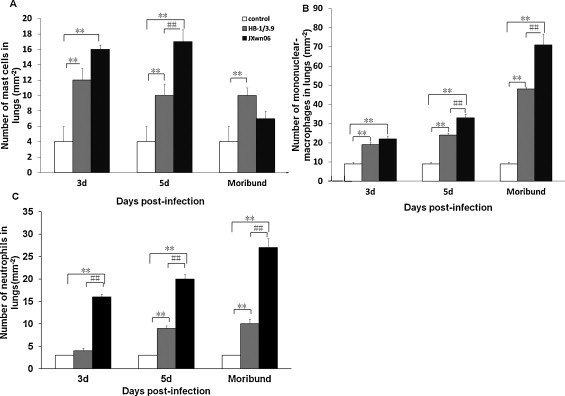
Numbers of mast cells, mononuclear-macrophages and neutrophils in the lungs of infected pigs. Numbers of mast cells, mononuclear-macrophages and neutrophils were counted. Data were shown as means ± SD mm−2 from 3 pigs each group. *p < 0.05 and **p < 0.01 between control and virus infection groups. #p < 0.05 and ##p < 0.01 between HB-1/3.6- and JXwn06-infected groups.
As for pro-inflammatory cytokines and chemokines, the concentrations of TNF-α, IL-1β and IL-8 in sera and TNF-α in the lungs were measured. As shown in Fig. 8 , the levels of TNF-α in the lungs of JXwn06-infected group significantly increased on day 3 PI to moribund (p < 0.01), and on day 5 PI there was highly significant difference between JXwn06- and HB-1/3.9-infected group (p < 0.01). On day moribund, significantly different levels of TNF-α in HB-1/3.9-infected group were detected compared to those of control group (p < 0.05). However, TNF-α level in sera also elevated from day 3 PI to moribund, but there was no significant alteration between control and virus infection group. Compared with control group, IL-8 level was dramatically increased in the sera of JXwn06-infected group from day 3 PI to moribund (p < 0.01), and there were remarkable and great differences between JXwn06- and HB-1/3.9-infected group on day 5 PI and moribund. Significant difference of IL-8 level between control and HB-1/3.9-infected group was detected on day moribund (p < 0.05). The concentration of IL-1β in sera of infected pigs increased from day 3 PI to moribund, but only on moribund, significant difference between control group and JXwn06-infected group could be observed (<0.01).
Fig. 8.
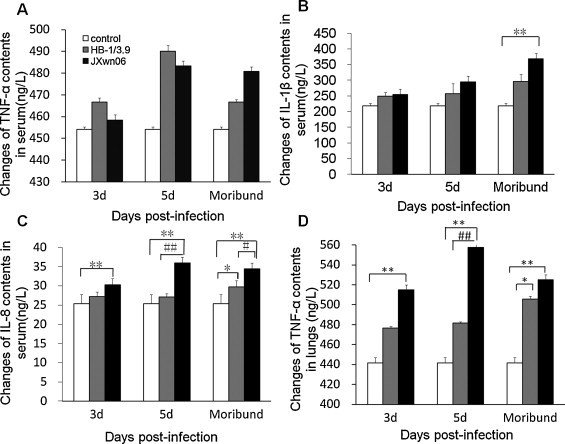
Concentrations of IL-1β, IL-8 and TNF-α in the sera and lungs of infected pigs. Lung and serum samples were collected on days 0, 3, 5 PI and moribund. TNF-α in sera (A) and in lungs (D), IL-1β (B) and IL-8 in sera (C) were detected by the methods described in Materials and methods. Data were shown as means ± SD from 3 pigs each group. *p < 0.05 and **p < 0.01 between control and virus infection groups. #p < 0.05 and ##p < 0.01 between HB-1/3.6- and JXwn06-infected groups.
3.5. Severe ultrastructural pathological changes in the lungs of infected pigs
Compared with control group, cell ultrastructural pathological changes in the lungs of HB-1/3.9- and JXwn06-infected pigs were observed (Fig. 9 ). For HB-1/3.9-infected pigs, the inflammatory cells were recruited into the alveolar septa and a few alveolar epithelial cells with ultrastructural pathological changes were observed, including fragmentation of cell membrane, mitochondrial swelling, and a few death cells shown as karorrhexis and karyolysis. However, for JXwn06-infected pigs, severe ultrastructural pathological changes including vascular wall injury, haemorrhage, alveolar epithelial cells necrosis and apoptosis could be seen, showing as fragmentation of cell membrane, nucleolemma introcession and karyopycnosis, cell vacuolization, a large number of cells death with severe mitochondria swelling and crista fragmentation, nuclear pyrosis, nuclear fragmentation and the formation apoptotic body. In addition, obvious fibroplasias could be observed under transmission electron microscope.
Fig. 9.
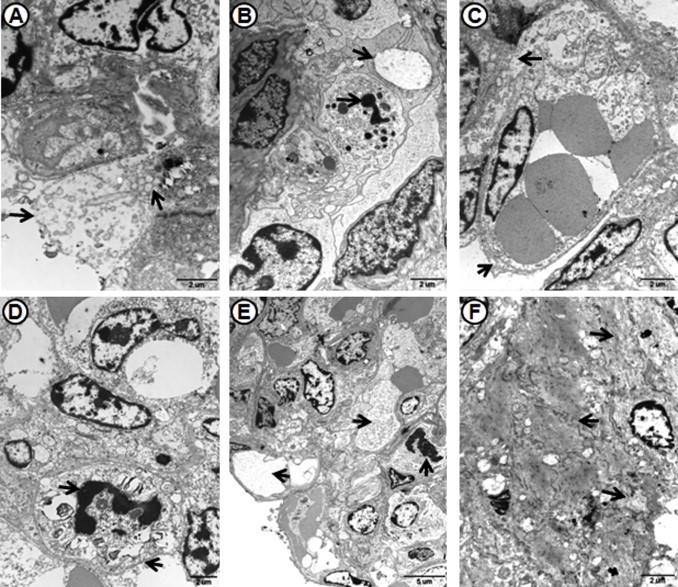
Ultrastructural pathological changes in the lungs of infected pigs. Alveolar epithelial cells with fragmentation of cell membrane and mitochondrial swelling (arrow) in the lung of HB-1/3.9-infected pig on day 5 PI (A). A few cells undergone apoptosis with karorrhexis, karyolysis, and organelles dissolution in the lung of HB-1/3.9-infected pig on day 16 PI (B). Obvious injury of vascular wall (arrow) in the lung of JXwn06-infected pig on day 5 PI (C). Alveolar epithelial cells occurred apoptosis with fragmentation of cell membrane, nucleolemma introcession and karyopycnosis in the lung of JXwn06-infected pig on day 5 PI (D). Cell vacuolization (arrows) in the lung of JXwn06-infected pig (E). Obvious fibroplasias and cell necrosis (arrow) in the lung of JXwn06-infected pig on moribund (F).
4. Discussion
Highly pathogenic PRRSV emerging in China could give rise to high mortality and severe lung pathological lesions (Zhou and Yang, 2010, Zhou et al., 2009, Xiao et al., 2010). In this report, differences in pathological changes, as well as a possible lung pathogenesis following HB-1/3.9 and JXwn06 infections were investigated. The results of pathological changes showed that unlike HB-1/3.9-infected group with mild lung pathological changes, more severe pathological lesions were observed in the lungs of JXwn06-infected pigs due to ALI. Firstly, almost the infected pigs exhibited clinical signs of respiratory disease, including visually prominent signs of respiratory distress in this study. Secondly, gross pathological changes and the dramatically increased lung wet: dry weight ratios showed that the infected pigs had highly edematous lungs. Thirdly, the infected pigs displayed a similar histopathologic pattern, including an initial peribronchiolar patchy pneumonia and interstitial pneumonia on day 3 PI and predominantly peribronchiolar lesions and light haemorrhage, and fully developed bronchiolitis, interstitial pneumonia and bronchopneumonia by day 5 PI. Pathological lesions in the lungs were characterized by inflammatory cellular infiltration, interstitial and alveolar edema, and hemorrhage. Lastly, on day 7 PI, the infected pigs suddenly presented prominent clinical signs of respiratory distress, especially on day 9 PI; the infected pigs showed severe dyspnea accompanying with tachycardia and cyanosis. These changes demonstrated that the infected pigs may develop progressive and severe hypoxemia in accordance with the time course of clinical signs and pulmonary lesions of ALI. Moreover, the ultramicroscopical changes of lungs suggested that after HB-1/3.9 infection, increased inflammatory cells in alveolar septa and a small number of death cells did not change the gas exchange function. On the contrary, blood cells deposition in vessel and alveolar space, severe alveolar epithelial cells death and serious pathological changes of vascular endothelial cells resulted in the ventilation and gas exchange dysfunction of lungs after JXwn06 infection. Furthermore, the death of type II alveolar epithelial cells serving as latent stem cells could not protect the normal lung structure through proliferation, leading to further damage of gas exchange function. The lung pathological changes observed in our report are consistent with those of previous studies (Guo et al., 2013, Li et al., 2012, Tian et al., 2007, Zhou et al., 2009, Li et al., 2007) which strongly support our contention that HP-PRRSV contributes substantially to the ALI. Moreover, ALI induced by HP-PRRSV infection in this study showed similar clinical and histopathological changes to those induced by other pathogens, such as avian influenza virus and SARS virus (Gattinoni et al., 1998, Atabai and Matthay, 2002, Levy et al., 2005, Bernard, 2005, Headley et al., 1997).
Previous studies have demonstrated that PRRSV replicates extensively in pulmonary alveolar macrophages (Van Gorp et al., 2008, Lee and Lee, 2010, Van Breedam et al., 2010a, Van Breedam et al., 2010b). In our study, we found that compared with HB-1/3.9-infected pigs, earlier viremia and more infected cells in the lungs occurred in JXwn06-infected pigs which were similar to the results of IHC and virus titers in Guo's report (Guo et al., 2013). Our previous study also found that HP-PRRSV displayed an expanded tissue tropism (Li et al., 2012). After HB-1/3.9 infection, mild interstitial pneumonia and cytokines reaction were detected, but JXwn06 could induce more immune cells infiltration and higher levels of pro-inflammatory cytokines with regard to severe vascular injury, exudation and hemorrhage which flooded the alveolar space and increased the thickness of alveolar-capillary membranes and decreased the number of ventilated alveoli associated with hypoxia. These data suggest that extreme inflammatory injury rather than uncontrolled infection might be a determinant of the fatal outcome after HP-PRRSV infection.
As reported that an intense, neutrophil-predominant host inflammatory response could result in ALI. After stimulation by pro-inflammatory cytokines, the activated neutrophils could release free radicals, inflammatory cytokines and proteases which had contributions to lung lesions (Ware and Matthay, 2000, Avasarala et al., 2013). In our study, we found that increased immune cells also included mononuclear phagocytes and mast cells besides neutrophils. On day 3 to 5 PI, the number of immune cells in the lungs of JXwn06-infected pigs increased by nearly 2-3 times compared with HB-1/3.9-infected and control pigs, suggesting that mononuclear phagocytes and mast cells also play an important role in the process of ALI caused by HP-PRRSV infection. Previous studies indicated that the cytokines such as TNF-α were believed to be one of important factors involved in ALI (Ni et al., 2011, Grommes and Soehnlein, 2011, Goodman et al., 2003). Xu and co-workers found that Chicken/HB/108 H5N1 infection could induce ALI and high levels of TNF-α in the lungs of mice, but TNF-α in serum showed significantly no change (Xu et al., 2006). Several studies have indicated that PRRSV infection also could induce high levels of pro-inflammatory cytokines (Gomez-Laguna et al., 2010, Gimeno et al., 2011, Borghetti et al., 2011). Our data demonstrated that there was no obvious difference of TNF-α level in serum between control and infection group, but TNF-α expression in the lungs significantly increased in JXwn06-infected pigs. Meanwhile, the levels of IL-1β which played an important role in inflammation only increased significantly in serum of JXwn06-infected pigs on day moribund. In addition, the levels of chemokine IL-8 in serum increased significantly in JXwn06-infected group from day 3 PI to moribund compared to HB-1/3.9-infected and control groups. The results of cytokines changes in our study were consistent with those of Guo's experiment (Guo et al., 2013), except TNF-alpha level was higher in the control group. These results indicated that IL-8, released into the blood in the early stage of PRRSV infection, gave a hand in immune cells infiltration into the sites of lung damage involving in inflammation. Then, TNF-α and IL-1β mainly produced by these activated inflammatory cells highly accumulated in the lung, and then aggravated the inflammation, inducing irreversible damage to the lung. Simultaneously, these results suggested the inflammatory cells such as mononuclear phagocytes were mostly recruited, sequestrated and activated in the lungs but not returned to circulation, which might be helpful for illustrating the phenomenon that no difference of TNF-α in serum existed between the groups. In addition, in JXwn06-infected group, the levels of histamine, LTB4 and PAF significantly increased in serum primarily in response to increased permeability of the vessel and inflammatory cells accumulation. Owing to the fact that histamine and LTB4 are mainly produced by mast cells, and PAF is principally produced by neutrophils and endothelial cells, we propose that mast cell mostly located around blood vessels might be activated directly by PRRSV or the cytokines from activated macrophages, and then released inflammatory factors resulting in increased vessel permeability and inflammatory cells recruitment. Taken together, these results suggested that in the early stage of PRRSV infection, the immune cells in the lung were activated and then highly expressed proinflammatory cytokines causing a serious effect on the permeability of the vessels and recruitment of inflammatory cells into the lung leading to severe haemorrhage and exudation.
As one of tissue repair mechanisms, fibrosis not only plays an important role in the late stage of inflammation, but also acts an adverse effect on the process of some tissue repair especially in the lung. Many studies found that fibrosis occurred in ARDS during convalescence leading to respiratory failure in a few years and an increased risk of death (Martin et al., 1995, Jeon et al., 2006). Qiao and co-workers found that in mouse, ALI model following H5N1 virus infection, most mice developed fibrosis gradually at late stage; especially the survived mice underwent severe fibrosis on day 30 PI (Qiao et al., 2009). In our study, on day 16 PI, severe lung fibrosis of one pig in JXwn06-infected group were observed, while no fibrosis or light fibrosis was observed in the lungs of other pigs as well as in those of HB-1/3.9-infected and control groups. We first observed the fibrosis involved in lung injury occurred at the late stage of inflammation after JXwn06 infection, suggesting a close relation with fatal outcome of the infected pig. It is worthy to be noted that on moribund some pathological changes induced by secondary bacterial infection were also observed in JXwn06-infected pigs. Therefore, it is necessary for further investigating the role of bacterial co-infection in the pathogenesis of HP-PRRSV.
In summary, we demonstrated that HP-PRRSV—JXwn06 could induce ALI concomitantly occurred with haemorrhage, edema, cell death and fibrosis, which may be in part responsible for the pigs’ death. As well known, the vascular system is widely distributed in the lung, thus it may become the direct or indirect invasion target by PRRSV, especially HP-PRRSV, which belongs to the family Ateriviridae. Moreover, mast cell may play a major role in activating endothelial cells and increasing vascular permeability after HP-PRRSV infection. Our findings will be helpful for further investigation on the pathogenesis of ALI induced by HP-PRRSV and will provide valuable clues better clinical treatment for this disease.
Acknowledgments
This work was supported by National Key Basic Research Plan Grant (2014CB542700) from the Chinese Ministry of Science and Technology and Key project of National Natural Science Funds from National Natural Science Foundation of China (31330077), and the earmarked fund for Modern Agro-industry Technology Research System of China (CARS-36).
Contributor Information
Hanchun Yang, Email: yanghanchun1@cau.edu.cn.
Kedao Teng, Email: kedao@cau.edu.cn.
References
- Ashcroft T., Simpson J.M., Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atabai K., Matthay M.A. The pulmonary physician in critical care. 5: acute lung injury and the acute respiratory distress syndrome: definitions and epidemiology. Thorax. 2002;57:452–458. doi: 10.1136/thorax.57.5.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avasarala S., Zhang F., Liu G., Wang R., London S.D., London L. Curcumin modulates the inflammatory response and inhibits subsequent fibrosis in a mouse model of viral-induced acute respiratory distress syndrome. PLoS ONE. 2013;8:e57285. doi: 10.1371/journal.pone.0057285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghetti P., Saleri R., Ferrari L., Morganti M., De Angelis E., Franceschi V., Bottarelli E., Martelli P. Cytokine expression, glucocorticoid and growth hormone changes after porcine reproductive and respiratory syndrome virus (PRRSV-1) infection in vaccinated and unvaccinated naturally exposed pigs. Comp. Immunol. Microbiol. Infect. Dis. 2011;34:143–155. doi: 10.1016/j.cimid.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Bernard G.R. Acute respiratory distress syndrome: a historical perspective. Am. J. Respir. Crit. Care Med. 2005;172:798–806. doi: 10.1164/rccm.200504-663OE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Lee C.H., Liu C.Y., Wang J.H., Wang L.M., Perng R.P. Clinical features and outcomes of severe acute respiratory syndrome and predictive factors for acute respiratory distress syndrome. J. Chin. Med. Assoc. 2005;68:4–10. doi: 10.1016/S1726-4901(09)70124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulu A., Pastores S.M., Park B., Riedel E., Rusch V., Halpern N.A. Prevalence and mortality of acute lung injury and ARDS after lung resection. Chest. 2006;130:73–78. doi: 10.1378/chest.130.1.73. [DOI] [PubMed] [Google Scholar]
- Gao Z.Q., Guo X., Yang H.C. Genomic characterization of two Chinese isolates of porcine respiratory and reproductive syndrome virus. Arch. Virol. 2004;149:1341–1351. doi: 10.1007/s00705-004-0292-0. [DOI] [PubMed] [Google Scholar]
- Gattinoni L., Pelosi P., Suter P.M., Pedoto A., Vercesi P., Lissoni A. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? Am. J. Respir. Crit. Care Med. 1998;158:3–11. doi: 10.1164/ajrccm.158.1.9708031. [DOI] [PubMed] [Google Scholar]
- Gimeno M., Darwich L., Diaz I., de la Torre E., Pujols J., Martin M., Inumaru S., Cano E., Domingo M., Montoya M., Mateu E. Cytokine profiles and phenotype regulation of antigen presenting cells by genotype-I porcine reproductive and respiratory syndrome virus isolates. Vet. Res. 2011;42:9–18. doi: 10.1186/1297-9716-42-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Laguna J., Salguero F.J., Barranco I., Pallares F.J., Rodriguez-Gomez I.M., Bernabe A., Carrasco L. Cytokine expression by macrophages in the lung of pigs infected with the porcine reproductive and respiratory syndrome virus. J. Comp. Pathol. 2010;142:51–60. doi: 10.1016/j.jcpa.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman R.B., Pugin J., Lee J.S., Matthay M.A. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14:523–535. doi: 10.1016/s1359-6101(03)00059-5. [DOI] [PubMed] [Google Scholar]
- Goyal S.M. Porcine reproductive and respiratory syndrome. J. Vet. Diagn. Invest. 1993;5:656–664. doi: 10.1177/104063879300500435. [DOI] [PubMed] [Google Scholar]
- Grommes J., Soehnlein O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B.Q., Chen Z.S., Liu W.X. Isolation of PRRSV from aborted fetus with typical clinical PRRS manifestations. Chin. J. Prevent. Vet. Med. 1996;2:1–5. [Google Scholar]
- Guo B., Lager K.M., Henningson J.N., Miller L.C., Schlink S.N., Kappes M.A., Kehrli M.E., Jr., Brockmeier S.L., Nicholson T.L., Yang H.C., Faaberg K.S. Experimental infection of United States swine with a Chinese highly pathogenic strain of porcine reproductive and respiratory syndrome virus. Virology. 2013;435:372–384. doi: 10.1016/j.virol.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headley A.S., Tolley E., Meduri G.U. Infections and the inflammatory response in acute respiratory distress syndrome. Chest. 1997;111:1306–1321. doi: 10.1378/chest.111.5.1306. [DOI] [PubMed] [Google Scholar]
- Jeon K., Chung M.P., Lee K.S., Chung M.J., Han J., Koh W.J., Suh G.Y., Kim H., Kwon O.J. Prognostic factors and causes of death in Korean patients with idiopathic pulmonary fibrosis. Respir. Med. 2006;100:451–457. doi: 10.1016/j.rmed.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Keffaber K.K. Reproductive failure of unknown etiology. Am. Assoc. Swine Prac. News. 1989;1:1–9. [Google Scholar]
- Lang J.D., Figueroa M., Sanders K.D., Aslan M., Liu Y., Chumley P., Freeman B.A. Hypercapnia via reduced rate and tidal volume contributes to lipopolysaccharide-induced lung injury. Am. J. Respir. Crit. Care Med. 2005;171:147–157. doi: 10.1164/rccm.200302-305OC. [DOI] [PubMed] [Google Scholar]
- Lee Y.J., Lee C. Porcine reproductive and respiratory syndrome virus replication is suppressed by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. Virus Res. 2010;152:50–58. doi: 10.1016/j.virusres.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Levy M.M., Baylor M.S., Bernard G.R., Fowler R., Franks T.J., Hayden F.G., Helfand R., Lapinsky S.E., Martin T.R., Niederman M.S., Rubenfeld G.D., Slutsky A.S., Stewart T.E., Styrt B.A., Thompson B.T., Harabin A.L. Clinical issues and research in respiratory failure from severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 2005;171:518–526. doi: 10.1164/rccm.200405-621WS. [DOI] [PubMed] [Google Scholar]
- Li L.M., Zhao Q., Ge X.N., Teng K.D., Kuang Y., Chen Y.H., Guo X., Yang H.C. Chinese highly pathogenic porcine reproductive and respiratory syndrome virus exhibits more extensive tissue tropism for pigs. Virol. J. 2012;9:203–208. doi: 10.1186/1743-422X-9-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Wang X., Bo K., Tang B., Yang B., Jiang W., Jiang P. Emergence of a highly pathogenic porcine reproductive and respiratory syndrome virus in the Mid-Eastern region of China. Vet. J. 2007;174:577–584. doi: 10.1016/j.tvjl.2007.07.032. [DOI] [PubMed] [Google Scholar]
- Mannam P., Zhang X., Shan P., Zhang Y., Shinn A.S., Lee P.J. Endothelial MKK3 is a critical mediator of lethal murine endotoxemia and acute lung injury. J. Immunol. 2013;190:1264–1275. doi: 10.4049/jimmunol.1202012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C., Papazian L., Payan M.J., Saux P., Gouin F. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. A study in mechanically ventilated patients. Chest. 1995;107:196–200. doi: 10.1378/chest.107.1.196. [DOI] [PubMed] [Google Scholar]
- Meers C.M., De Wever W., Verbeken E., Mertens V., Wauters S., De Vleeschauwer S.I., Vos R., Vanaudenaerde B.M., Verleden G.M., Van Raemdonck D.E. A porcine model of acute lung injury by instillation of gastric fluid. J. Surg. Res. 2011;166:e195–e204. doi: 10.1016/j.jss.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Meng X.J. Heterogeneity of porcine reproductive and respiratory syndrome virus: implications for current vaccine efficacy and future vaccine development. Vet. Microbiol. 2000;74:309–329. doi: 10.1016/S0378-1135(00)00196-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y., Kato A., Tsuda T., Morozumi T., Miura Y., Sugimura T. Isolation and serological characterization of porcine reproductive and respiratory syndrome (PRRS) viruses from pigs with reproductive and respiratory disorders in Japan. J. Vet. Med. Sci. 1994;56:891–894. doi: 10.1292/jvms.56.891. [DOI] [PubMed] [Google Scholar]
- Nelsen C.J., Murtaugh M.P., Faaberg K.S. Porcine reproductive and respiratory syndrome virus comparison: divergent evolution on two continents. J. Virol. 1999;73:270–280. doi: 10.1128/jvi.73.1.270-280.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y.F., Kuai J.K., Lu Z.F., Yang G.D., Fu H.Y., Wang J., Tian F., Yan X.L., Zhao Y.C., Wang Y.J., Jiang T. Glycyrrhizin treatment is associated with attenuation of lipopolysaccharide-induced acute lung injury by inhibiting cyclooxygenase-2 and inducible nitric oxide synthase expression. J. Surg. Res. 2011;165:e29–e35. doi: 10.1016/j.jss.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Ouchi H., Fujita M., Ikegame S., Ye Q., Inoshima I., Harada E., Kuwano K., Nakanishi Y. The role of collagenases in experimental pulmonary fibrosis. Pulm. Pharmacol. Ther. 2008;21:401–408. doi: 10.1016/j.pupt.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Qiao J., Zhang M., Bi J., Wang X., Deng G., He G., Luan Z., Lv N., Xu T., Zhao L. Pulmonary fibrosis induced by H5N1 viral infection in mice. Respir. Res. 2009;10:107–115. doi: 10.1186/1465-9921-10-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M., Yamada S., Murakami Y., Morozumi T., Kobayashi H., Mitani K., Ito N., Kubo M., Kimura K., Kobayashi M. Isolation of porcine reproductive and respiratory syndrome (PRRS) virus from Heko-Heko disease of pigs. J. Vet. Med. Sci. 1994;56:389–391. doi: 10.1292/jvms.56.389. [DOI] [PubMed] [Google Scholar]
- Subbarao K., Klimov A., Katz J., Regnery H., Lim W., Hall H., Perdue M., Swayne D., Bender C., Huang J., Hemphill M., Rowe T., Shaw M., Xu X., Fukuda K., Cox N. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science. 1998;279:393–396. doi: 10.1126/science.279.5349.393. [DOI] [PubMed] [Google Scholar]
- Sun Q., Wang D., She R., Li W., Liu S., Han D., Wang Y., Ding Y. Increased mast cell density during the infection with velogenic Newcastle disease virus in chickens. Avian Pathol. 2008;37:579–585. doi: 10.1080/03079450802499092. [DOI] [PubMed] [Google Scholar]
- Tian K., Yu X., Zhao T., Feng Y., Cao Z., Wang C., Hu Y., Chen X., Hu D., Tian X., Liu D., Zhang S., Deng X., Ding Y., Yang L., Zhang Y., Xiao H., Qiao M., Wang B., Hou L., Wang X., Yang X., Kang L., Sun M., Jin P., Wang S., Kitamura Y., Yan J., Gao G.F. Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS ONE. 2007;2:e526. doi: 10.1371/journal.pone.0000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gorp H., Van Breedam W., Delputte P.L., Nauwynck H.J. Sialoadhesin and CD163 join forces during entry of the porcine reproductive and respiratory syndrome virus. J. Gen. Virol. 2008;89:2943–2953. doi: 10.1099/vir.0.2008/005009-0. [DOI] [PubMed] [Google Scholar]
- Van Breedam W., Delputte P.L., Van Gorp H., Misinzo G., Vanderheijden N., Duan X., Nauwynck H.J. Porcine reproductive and respiratory syndrome virus entry into the porcine macrophage. J. Gen. Virol. 2010;91:1659–1667. doi: 10.1099/vir.0.020503-0. [DOI] [PubMed] [Google Scholar]
- Van Breedam W., Van Gorp H., Zhang J.Q., Crocker P.R., Delputte P.L., Nauwynck H.J. The M/GP(5) glycoprotein complex of porcine reproductive and respiratory syndrome virus binds the sialoadhesin receptor in a sialic acid-dependent manner. PLoS Pathog. 2010;6:e1000730. doi: 10.1371/journal.ppat.1000730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware L.B., Matthay M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- Xiao S., Mo D., Wang Q., Jia J., Qin L., Yu X., Niu Y., Zhao X., Liu X., Chen Y. Aberrant host immune response induced by highly virulent PRRSV identified by digital gene expression tag profiling. BMC Genomics. 2010;11:544–562. doi: 10.1186/1471-2164-11-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T., Qiao J., Zhao L., Wang G., He G., Li K., Tian Y., Gao M., Wang J., Wang H., Dong C. Acute respiratory distress syndrome induced by avian influenza A (H5N1) virus in mice. Am. J. Respir. Crit. Care Med. 2006;174:1011–1017. doi: 10.1164/rccm.200511-1751OC. [DOI] [PubMed] [Google Scholar]
- Zhou L., Zhang J., Zeng J., Yin S., Li Y., Zheng L., Guo X., Ge X., Yang H. The 30-amino-acid deletion in the Nsp2 of highly pathogenic porcine reproductive and respiratory syndrome virus emerging in China is not related to its virulence. J. Virol. 2009;83:5156–5167. doi: 10.1128/JVI.02678-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Yang H. Porcine reproductive and respiratory syndrome in China. Virus Res. 2010;154:31–37. doi: 10.1016/j.virusres.2010.07.016. [DOI] [PubMed] [Google Scholar]