

Graphical abstract
Two predicted binding modes of the inhibitors in active site of SARS-CoV Mpro, with the 3-chloropyridine moieties located in the S1 pocket.

Keywords: SARS coronavirus, 3C-like proteinase, Small molecule docking, Non-peptidyl proteinase inhibitor, Drug design, Chloropyridine
Abstract
The 3C-like main proteinase of the severe acute respiratory syndrome (SARS) coronavirus, SARS-CoV Mpro, is widely considered to be a major drug target for the development of anti-SARS treatment. Based on the chemical structure of a lead compound from a previous screening, we have designed and synthesized a number of non-peptidyl inhibitors, some of which have shown significantly improved inhibitory activity against SARS-CoV Mpro with IC50 values of ∼60 nM. In the absence of SARS-CoV Mpro crystal structures in complex with these synthetic inhibitors, molecular docking tools have been employed to study possible interactions between these inhibitors and SARS-CoV Mpro. The docking results suggest two major modes for the initial binding of these inhibitors to the active site of SARS-CoV Mpro. They also establish a structural basis for the ‘core design’ of these inhibitors by showing that the 3-chloropyridine functions common to all of the present inhibitors tend to cluster in the S1 specificity pocket. In addition, intrinsic flexibility in the S4 pocket allows for the accommodation of bulky groups such as benzene rings, suggesting that this structural plasticity can be further exploited for optimizing inhibitor–enzyme interactions that should promote a tighter binding mode. Most importantly, our results provide the structural basis for rational design of wide-spectrum antiviral drugs targeting the chymotrypsin-like cysteine proteinases from coronaviruses and picornaviruses.
1. Introduction
In the winter of 2002, a form of atypical pneumonia, later termed the severe acute respiratory syndrome (SARS), broke out in southern China and rapidly spread to 32 countries and regions on five continents. Approximately 8098 people were infected and of these, 774 deaths were reported in the ensuing months.1
The causative agent of SARS is a novel human coronavirus (SARS-CoV).2, 3, 4, 5 Similar to other coronaviruses, the RNA genome of SARS-CoV encodes at least two viral proteinases, one of which is the main proteinase (Mpro). Mpro, a highly conserved proteinase among coronaviruses, is responsible for the majority of the proteolytic processing of two large viral polyproteins, replicases pp1a and pp1ab. All of the cleavage products from these replicases are required for the assembly of membranous complexes in infected cells where the viral RNA genome is replicated. Interruption of either viral RNA translation or polyprotein proteolytic processing abolishes viral RNA replication.6, 7 Because the indispensable involvement in polyprotein processing of SARS-CoV Mpro is upstream of many replicative events in the viral life cycle, reducing or blocking the peptidase function of Mpro could have a profound impact on lowering progeny virus yield and enhancing cell survival. Therefore, SARS-CoV Mpro is an attractive target for the development of anti-SARS drugs.8
The functional quaternary assembly of SARS-CoV Mpro is believed to be a dimer.9 The two protomers are oriented almost perpendicular to one another and each protomer consists of three domains: I (residues 1–101), II (residues 102–184), and III (residues 201–306). Domains I and II are made of β-barrel motifs similar to the domains in the chymotrypsin-type serine proteases and in the 3C proteinases of picornaviruses. The substrate binding sites, and the nearby catalytic dyad consisting of residues Cys145 and His41, are located in a cleft between domains I and II. Domain III contains five α-helices connected to domain II via an extended loop (residues 185–200); it plays an essential role for the proteolytic activity of SARS-CoV Mpro by maintaining the proper conformation of the dimer.9, 10, 11
The substrate binding sites of SARS-CoV Mpro have been probed using several classes of inhibitors.9, 12, 13 Chloromethyl ketones, epoxides, and Michael acceptors are among the functional groups known to form a covalent bond with the nucleophilic sulfur atom of Cys145, thereby leading to the inactivation of SARS-CoV Mpro’s enzymatic activity. Similar to the main proteinases of transmissible gastroenteritis virus (TGEV) and human coronavirus strain 229E, SARS-CoV Mpro exhibits well-defined S1, S2, and S4 pockets that seem to be highly structurally conserved among coronaviruses.8, 12, 13 Both in vitro and in vivo studies show that the residues at the P1, P2, and P4 positions of the substrate are involved in substrate binding, recognition, and cleavage.8, 14, 15 Consequently, the S pockets of SARS-CoV Mpro have been the principal targeting sites for anti-SARS (or wide-spectrum anti-coronavirus) drugs.
Various experimental techniques and modeling methods, such as structure-based discovery,16, 17, 18, 19, 20 experimental screening,21, 22, 23, 24, 25 and virtual screening,26, 27, 28, 29, 30, 31, 32, 33 have been employed in the search for effective anti-SARS inhibitors. Although the efficacies of some compounds were verified and characterized by biochemical and/or cell culture-based assays, the inhibition mechanism remains unclear for some of them primarily due to the lack of structural information.
Recently, a large-scale high-throughput screening of anti-SARS compounds identified 2-(5-chloropyridin-3-yl)-1-(thiophen-2-yl)ethanone (MAC-5576) as a potent inhibitor for SARS-CoV Mpro with an IC50 value of 0.5 ± 0.3 μM.22 In order to derive even better anti-SARS-CoV Mpro compounds, we then synthesized a number of non-peptidic compounds based on the chemical structure of MAC-5576. Some of these compounds showed significant improvement over MAC-5576 in their inhibitory activity.34 In this paper, we report the in vitro inhibition of SARS-CoV Mpro by a series of non-peptidic compounds designed on the basis of MAC-5576 (Table 1, Table 2 ) and analyze the possible modes of interaction between this new class of non-peptidyl inhibitors and SARS-CoV Mpro using molecular docking tools. Our docking results also allow us to put forward constructive suggestions for the future design of anti-SARS Mpro inhibitors.
Table 1.
Structures and inhibitory activity of a set of non-peptidyl compounds (group I) based on the chemical structure of MAC-5576 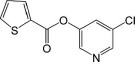
| No. | Compound | IC50 (nM) | ΔG 1sta (kcal/mol) | ΔG 2ndb (kcal/mol) | c (2A5K) (Å) | d (1UK4) (Å) | e (2A5K) (Å) | f (1UK4) (Å) |
|---|---|---|---|---|---|---|---|---|
| 1 | 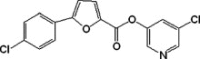 |
63 | −9.33 | −9.88 | 4.41 | 3.70 | 6.88 | 5.62 |
| 2 |  |
60 | −9.34 | −9.80 | 4.51 | 3.82 | 6.65 | 5.48 |
| 3 | 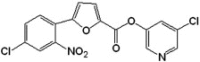 |
122 | −9.82 | −9.84 | 3.80 | 3.10 | 6.54 | 5.30 |
| 4 | 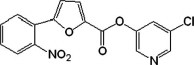 |
208 | −8.73 | −9.47 | 3.56 | 2.80 | 7.12 | 5.88 |
| 5 | 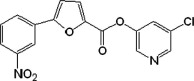 |
500 | −9.33 | −10.19 | 4.66 | 3.80 | 6.92 | 5.68 |
The binding free energies predicted in the S2–S1 binding mode.
The binding free energies predicted in the S4–S1 binding mode.
Distances between the carbonyl carbon of the compounds and the sulfur atom of Cys145 (PDB 2A5K) in the S2–S1 binding mode.
Distances between the carbonyl carbon of the compounds and the sulfur atom of Cys145 (PDB 1UK4) in the S2–S1 binding mode.
Distances between the carbonyl carbon of the compounds and the sulfur atom of Cys145 (PDB 2A5K) in the S4–S1 binding mode.
Distances between the carbonyl carbon of the compounds and the sulfur atom of Cys145(PDB 1UK4) in the S4–S1 binding mode.
Table 2.
Structures and inhibitory activity of a set of non-peptidyl compounds (group II) based on the chemical structure of MAC-5576
| No. | Compound | IC50 (nM) | ΔGa (kcal/mol) | Dc–sb (2A5K) (Å) | Dc–sc (1UK4) (Å) |
|---|---|---|---|---|---|
| 6 | 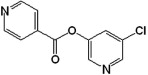 |
164 | −6.92 | 4.50 | 3.03 |
| 7 | 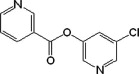 |
697 | −6.93 | 4.49 | 3.02 |
| 8 | 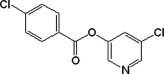 |
434 | −7.49 | 4.45 | 3.01 |
| 9 |  |
333 | −6.79 | 4.43 | 3.10 |
| 10 | 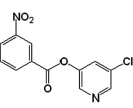 |
684 | −7.23 | 4.60 | 3.15 |
| 11 | 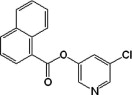 |
124 | −8.41 | 4.14 | 2.85 |
| 12 | 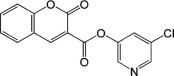 |
108 | −8.58 | 3.74 | 3.38 |
The binding free energies predicted in the Cys–S1 binding mode.
Distances between the carbonyl carbon of the acyclic ester bond and the sulfur atom of Cys145 (PDB 2A5K) in the Cys–S1 binding mode.
Distances between the carbonyl carbon of the acyclic ester bond and the sulfur atom of Cys145 (PDB 1UK4) in the Cys–S1 binding mode.
2. Results
Prior to the virtual docking, the compounds were sorted into two groups based on their chemical characteristics. Both groups contain the ester function and the chloropyridine moiety inherited from the parent compound. In addition, group I compounds have a furan ring connected to a derivatized benzene moiety (Table 1); whereas the group II compounds have a derivatized, six-membered cyclic aromatic system that is linked to the central ester function (Table 2).
2.1. In vitro inhibition of SARS-CoV Mpro by non-peptidyl inhibitors.
In general, group I compounds are better inhibitors of SARS-CoV Mpro than group II compounds as shown by their lower IC50 values (Table 1, Table 2). Interestingly, the positions of the electron-withdrawing substituents on their terminal aromatic rings seem to influence the inhibitory efficacy of these compounds dramatically. For example, the difference in the position of the nitrogen atom of the pyridine moiety relative to the carbonyl function in compounds 6 and 7 is associated with a fourfold disparity in their IC50 values. The best group I inhibitors, compounds 1–3, contain either a chloride or a nitro group at the para position relative to the furan function. Moving the nitro group of compound 2 to either the ortho (compound 4) or the meta (compound 5) position led to a threefold and eightfold increase in their IC50 values, respectively. In a similar manner to compounds 2, 4, and 5, compounds 8–10 of group II bear a chloro- or a nitro-substituent on the benzene ring directly connected to the central ester function at the para, ortho, and meta position relative to the ester bond, respectively. The position of the substituents in the latter three compounds has much less of an impact on the inhibitory activity against SARS-CoV Mpro, although the meta-substituted compound 10 is a markedly weaker inhibitor than either of compounds 8 and 9. Compounds 11 and 12, having terminal conjugated rings, showed inhibitory activities roughly equivalent to those of the best group I compounds.
2.2. Docked conformations of group I compounds
Two possible binding modes for compounds 1–5, which have similar chemical structures except for the different substituents on the benzene ring, were clearly identified. The first binding mode (the S2–S1 mode) of the group I compounds occupies primarily the volume extending from the S2 to the S1 substrate binding sites (Fig. 1 a). Figure 1b shows the predicted interactions between the enzyme and compound 1 in the active site as an example. The chloropyridine moieties of these inhibitors bind inside the S1 pocket in an orientation wherein the pyridine plane is parallel to the two ‘walls’ of the S1 sites formed by residues Phe140, Leu141, Asn142, Ser144, and residues Glu166, His172, respectively. In the docked complexes of compounds 1, 2, and 5, the common nitrogen atom of the chloropyridine moieties receives a hydrogen bond from Nε2 of His163, which is the P1 specificity-determining residue (Fig. 1b). Similarly, the furan moieties of group I inhibitors occupy the same volume between the S1 and S2 pockets and form a few van der Waals interactions with Met165 (not shown). In contrast, the benzene rings of the group I inhibitors in the S2–S1 mode show relatively larger conformational variations. Compounds 1 and 2, which contain para-substituted benzene rings, can fit into the S2 pocket well. Compounds 3–5 all contain ortho- or meta-orienting groups on their benzene rings. This causes the benzene moieties in these compounds to be pushed outward from S2 site so as to avoid steric hindrance between the benzyl substituents and the side chains of His41 and Met49. The docking models suggest that hydrophobic interactions are formed between residues His41, Met49, and Gln189 and the benzene ring moieties, whereas the main chain NH of Gln189 forms a hydrogen bond with the ortho-orienting nitro substituent on the benzene rings.
Figure 1.

(a) The S2–S1 binding mode of group I compounds. The side chain of Met49, which forms the outer lid of the S2 pocket, was removed to allow clearer visualization of the S2 pocket. (b) Interactions between SARS-CoV Mpro (white ribbon) and compound 1 (yellow carbon sticks) in the S2–S1 binding mode. Hydrogen bonds are shown as green dashed lines.
In the S2–S1 binding mode, the ester functions of the inhibitors are approximately 3.5–4.7 Å from Sγ of the catalytic residue Cys145 (Table 1 column 6); the carbonyl oxygen atoms of the inhibitors point into the oxyanion hole of SARS-CoV Mpro such that compounds 1, 3, and 4 receive hydrogen bonds from the main chain NH of Gly143. Esters are known substrates for chymotrypsin-type serine proteases, enzymes that are structurally similar to the catalytic domains of SARS-CoV Mpro. This suggests that the ester bonds of group I inhibitors bound in this conformation could be attacked by the nucleophilic sulfur of Cys145, leading to the formation of an acylated enzyme.
The second binding mode of group I compounds extends from the S4 to the S1 specificity pockets (the S4–S1 mode) (Fig. 2 a). In a similar fashion to the S2–S1 binding mode, the chloropyridine moieties of group I inhibitors are also located in the S1 pocket. However, the orientations of the chloropyridine rings are roughly perpendicular to those of the chloropyridine rings in the S2–S1 binding mode. The planes of the rings are parallel to the ‘floor’ of the S1 site formed by the side chains of His163 and His172. The m-chlorine on the pyridine ring points into the solvent channel at the back side of the S1 pocket, occupying a volume very close to that of a chloride ion observed in the crystal structure of an unliganded SARS-CoV Mpro 35 (PDB code 2A5A). In the docked complexes containing compounds 1, 4, and 5, this chlorine is hydrogen bonded to Oγ of Ser144 (Fig. 2b). Residues Cys145 and Met165 form hydrophobic interactions with the pyridine rings of these inhibitors. The furan rings in the S4–S1 binding mode are situated on top of Met165 and Glu166, making a few van der Waals interactions with these residues. The S4 pocket is large enough to accommodate the benzene rings of the group I inhibitors despite the presence of various substituents at different positions on the rings. Residues Met165, Glu166, Leu167, and Gln189 contribute mostly hydrophobic interactions with the benzene rings. The main chain NH of Glu166 forms two hydrogen bonds with the oxygen atom on the furan and the carbonyl oxygen at the β-position of the central ester bond of group I compounds. In addition, the nitro function of compound 5 forms a hydrogen bond with Nε2 of Gln192 (Fig. 2b). In the S4–S1 binding mode, the distances between the carbonyl carbons of the ester functions of the inhibitors and Sγ of Cys145 range from 6.5 to 7.2 Å (Table 1 column 8). The inhibitors bound in this mode are very unlikely to be attacked by the sulfur nucleophile of Cys145 due to this long distance constraint. Therefore, the S4–S1 binding mode likely represents a mechanism of competitive inhibition in which the inhibitors block the activity of SARS-CoV Mpro by denying entry of substrates into the active site. Most significantly, this binding hypothesis suggests a way to improve the design of more potent non-covalent SARS-CoV Mpro inhibitors that promote tighter S4–S1 binding (discussed in later section).
Figure 2.

(a) The S4–S1 binding mode of group I compounds. (b) Interactions between SARS-CoV Mpro (white ribbon) and compound 5 (yellow carton sticks) in the S4–S1 binding mode. Hydrogen bonds are shown as green dashed lines.
2.3. Docked conformations of group II compounds
The extended end-to-end lengths of group II compounds are shorter than those of the group I compounds. Consequently, they do not span more than one substrate binding sites. The most commonly observed binding mode has an orientation that extended from the catalytic dyad to the S1 pocket (Cys–S1 mode). As with the S2–S1 binding mode of group I compounds, the m-chloropyridine rings of the group II inhibitors are reliably docked inside the S1 site with the m-chlorine atom pointing to solvent (Fig. 3 a). With the derivatized benzene functions of group II compounds docked near the catalytic residues, the Cys–S1 mode can be viewed as a partial S2–S1 binding mode (akin to fragment binding). The aromatic systems on the carbonyl side of the ester bond form mostly van der Waals contacts with residues Phe140, Leu141, Cys145, Met165, and Glu166. However, their orientations with regard to the rest of the compound vary substantially (Fig. 3a).
Figure 3.
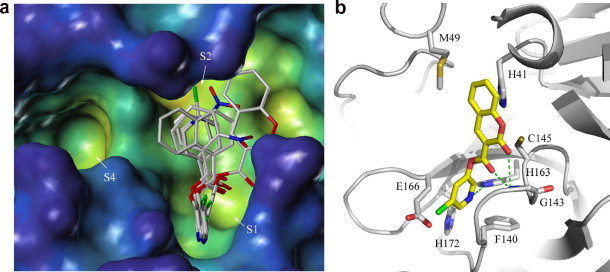
(a) The Cys–S1 binding mode of group II compounds. (b) Interactions between SARS-CoV Mpro (white ribbon) and compound 12 (yellow carbon sticks) in the Cys–S1 binding mode. Hydrogen bonds are shown as green dashed lines.
The hydrogen bonds formed between group II compounds and SARS-CoV Mpro also show polarized patterns. The nitrogen atom of the pyridine ring of compound 12 is hydrogen bonded to Nε2 of His163, which also receives a hydrogen bond from the β-carbonyl oxygen atoms of other group II compounds. Furthermore, in compound 12, the main chain N of Gly143 forms a bifurcated hydrogen bond to the carbonyl oxygen of the ester bond and to the carbonyl oxygen of the coumarin moiety, respectively (Fig. 3b).
In the Cys–S1 binding mode of the group II inhibitors, the average distance between the carbonyl carbon atoms of the central ester bonds and the sulfur atom of Cys145 is about 3.7–4.6 Å. Compared to those inhibitors of group I in the S4–S1 binding mode, the ester bonds in the group II inhibitors are placed much closer to the catalytic dyad; the carbonyl oxygen atoms of the central ester point into the oxyanion hole. Therefore, the group II compounds bound in the Cys–S1 mode are potentially exposed to higher risk of enzymatic hydrolysis.
2.4. Binding free energy and inhibitor efficacy
The estimated free energies of binding (ΔG) between the enzyme and the 12 inhibitors were predicted by AutoDock based on the new scoring function, which was developed using the AMBER force field and a set of coefficients.36 Although AutoDock cannot provide an absolute binding free energy, these predicted ΔGs can be used for approximate ranking of the inhibitors according to their predicted binding affinities. For group I compounds, there are no significant differences in the binding free energies between the S2–S1 mode (ΔG 1st) and the S4–S1 binding mode (ΔG 2nd) (Table 1). This suggests that group I compounds have comparable tendencies to adopt either the S4–S1 or the S2–S1 conformation prior to their possible covalent modification of SARS-CoV Mpro enzyme. The binding free energies for group II compounds are significantly higher than those of group I compounds, with the exception of compounds 11 and 12. This is in agreement with the general trend that SARS-CoV Mpro is more strongly inhibited by group I than group II compounds. Compounds 11 and 12 show binding free energies closer to those exhibited by the group I compounds, probably because the larger aromatic stabilization effects of the naphthalene moiety of compound 11 and the coumarin moiety of compound 12 make their central ester bonds less susceptible to nucleophilic attack by Sγ of Cys145. Correspondingly, these two compounds exhibit better anti-SARS-CoV Mpro activity than other group II inhibitors.
3. Discussion
The inhibitors used in this study bind to the active site of SARS-CoV Mpro primarily through hydrophobic contacts. Our docking results clearly show that the 3-chloropyridine moieties of the ester-based non-peptidyl inhibitors have a strong propensity to enter the S1 specificity pocket of SARS-CoV Mpro. Accordingly, the residues forming the S1 pocket play a major part in the interactions between the inhibitors and SARS-CoV Mpro. This is significant, as the chloropyridine function does not resemble the cognate P1-Gln residue in terms of chemical properties. Consequently, some interactions between SARS-CoV Mpro and chloropyridine moiety likely differ from those between P1-Gln and SARS-CoV Mpro. Further derivatization of the chloropyridine group has yielded only marginal improvement on the efficacy of the resultant inhibitors, indicating that our design may have its maximal potential concerning the S1 pocket of SARS-CoV Mpro. Since the S1 pockets of all coronaviral Mpro are structurally conserved and are similar to those of the picornaviral 3Cpro, the inhibitors described in this study or at least their basic designs should prove useful in developing wide-spectrum antiviral compounds. An early indication of that came from the observation that the parent compound MAC-5576 showed very good inhibitory activity against both the SARS-CoV Mpro and the HAV 3Cpro with corresponding IC50 values in the high nanomolar range.22
A second ‘hotspot’ that could be targeted by anti-SARS-CoV Mpro compounds is residue His41. His41 plays the dual role of activating Sγ of Cys145 during the catalytic cycle as a general base as well as forming part of the S2 specificity pocket. In the S2–S1 binding mode, His41, together with Met165 and Glu166, forms more than half of the total hydrophobic interactions with the group I inhibitors (Table 3 ). Met165 and Glu166 form the ‘wall’ of the S2 pocket opposite to that of His41; these residues are also major contributors of hydrophobic interactions in the S4–S1 and the Cys-S1 binding modes (Table 4, Table 5 ).
Table 3.
Numbers of hydrophobic contacts between residues of SARS-CoV Mpro and group I compounds in the S2–S1 binding mode
| Residues | Number of hydrophobic contacts |
||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| His41 | 10 | 9 | 8 | 4 | 5 |
| Met49 | 5 | 6 | 5 | 4 | 1 |
| Phe140 | 2 | 3 | 3 | 3 | 3 |
| Leu141 | 2 | 4 | 3 | 3 | 4 |
| Cys145 | 3 | 2 | 3 | 4 | 1 |
| His163 | 2 | 1 | 2 | 2 | 1 |
| Met165 | 4 | 4 | 3 | 3 | 3 |
| Glu166 | 7 | 7 | 3 | 3 | 7 |
| His172 | 1 | ||||
| Arg188 | 2 | ||||
| Gln189 | 2 | 6 | |||
| Total | 36 | 29 | 30 | 28 | 33 |
Table 4.
Numbers of hydrophobic contacts between residues of SARS-CoV Mpro and group I compounds in the S4–S1 binding mode
| Residues | Number of hydrophobic contacts |
||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| Leu141 | 1 | ||||
| Asn142 | 1 | ||||
| Cys145 | 1 | 2 | 1 | 1 | |
| Met165 | 12 | 10 | 11 | 10 | 13 |
| Glu166 | 5 | 2 | 4 | 2 | 3 |
| Leu167 | 5 | 7 | 5 | 4 | 5 |
| Pro168 | 1 | 1 | 1 | 1 | |
| Gln189 | 3 | 3 | 4 | 3 | 2 |
| Gln192 | 2 | 1 | |||
| Total | 27 | 26 | 28 | 21 | 25 |
Table 5.
Numbers of hydrophobic contacts between residues of SARS-CoV Mpro and group II compounds in the Cys–S1 binding mode
| Residues | Number of hydrophobic contacts |
||||||
|---|---|---|---|---|---|---|---|
| 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| His41 | 2 | 2 | 2 | 2 | 6 | ||
| Met49 | 3 | ||||||
| Phe140 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Leu141 | 3 | 2 | 2 | 2 | 2 | 3 | 3 |
| Asn142 | 1 | ||||||
| Cys145 | 3 | 3 | 3 | 3 | 3 | ||
| His163 | 1 | 1 | 1 | 2 | 1 | 1 | |
| His164 | 2 | 2 | 1 | 1 | |||
| Met165 | 3 | 3 | 3 | 4 | 7 | 8 | |
| Glu166 | 6 | 6 | 6 | 7 | 6 | 6 | 3 |
| His172 | 1 | 1 | 1 | 1 | |||
| Total | 23 | 22 | 21 | 17 | 18 | 26 | 22 |
The distances between Sγ of Cys145 and the carbonyl carbon atoms in the ester functions of group II inhibitors are significantly shorter than those between the nucleophilic sulfur and the corresponding atoms of group I inhibitors in the S4–S1 binding mode (Table 1 column 8 and Table 2 column 5). It is worth mentioning that the model structure used in docking is that of a SARS-CoV Mpro covalently modified at Sγ of Cys145. The position of the nucleophilic sulfur atom, relative to the other active site residues, has shifted significantly from that of Sγ in the unliganded enzyme structures (PDB codes, e.g., 1UK4 or 2A5A) (Fig. 4 a). Our recent X-ray crystallographic analyses of SARS-CoV Mpro in complex with some peptidyl inhibitors indicate that the formation of tetrahedral intermediates during substrate hydrolysis can take place without the requirement of the previously observed conformational changes in Cys145.37 Consequently, we aligned the 1UK4 structure onto the docking molecule 2A5K to obtain the alternate coordinates of the nucleophilic sulfur, which should represent the position of Sγ in its resting state and when the substrate/inhibitor first approaches. The distances involving Sγ of Cys145 were recalculated. It is clear that the carbonyl carbon atoms of the group II compounds are closer to Sγ of Cys145 than those of the group I compounds (Table 2 column 6). Note that some of the calculated distances (for group II compounds and for compounds 3 and 4 in the S2–S1 binding mode) are shorter than the sum of van der Waals radii of carbonyl oxygen and thiol sulfur atoms, suggesting a strong propensity for the covalent attachment of these inhibitors to Sγ of Cys145.
Figure 4.

(a) Alignment of SARS-CoV Mpro crystal structures: 1UK4 (pink) and 2A5K (white). APE and the catalytic residues of His41 and Cys145 are shown in the stick mode. Residues Met49 and Gln192-Gly195 are shown in lines to illustrate the structural plasticity in the S2 and S4 pockets. The structural changes in these pockets involve both the side chain and main chain atoms in the residues involved. (b) The binding conformation of APE in the active site of SARS-CoV Mpro (2A5K). APE is shown in the ball and stick mode. Oxygen, nitrogen and carbon atoms are shown in red, blue, and gray, respectively. Color of surface shows the cavity depth from the exterior (blue) to the interior (yellow) of protein.
In summary, our docking study provides a reasonable structural interpretation for the in vitro activities of a new class of non-peptidyl anti-SARS-CoV Mpro inhibitors. It suggests that a chloropyridine function in the context of an ester compound specifically binds to the S1 substrate binding site of SARS-CoV Mpro. The interactions between the S1 site of SARS-CoV Mpro and the chloropyridine functions likely play an important role in the initial binding of our inhibitors to the enzyme in vitro, whereas the rate of subsequent water-mediated hydrolysis of the resultant acyl enzyme depends on the chemical properties of the leaving groups on the carbonyl side of the ester bond. It is worth mentioning that additional docking studies indicate that the chloropyridine function of these compounds likely targets the S1 site of HAV 3Cpro as well (data not shown). This correlates well with the high structural similarity in the active sites of SARS-CoV Mpro and HAV 3Cpro.
The information obtained through our structural analyses may be incorporated in the future design of more potent anti-SARS-CoV Mpro inhibitors. Due to the hydrolyzability of the ester bonds in these compounds, their inhibitory effect on SARS-CoV Mpro is likely to involve a covalent attachment of the inhibitors to the nucleophilic cysteine Cys145. Further modifications (individually or in combination) on the basis of compound 1 may improve its efficacy of inhibition against SARS-CoV Mpro and/or other structurally similar viral proteinases.
-
1.
The ester bond could be replaced by other functional groups that react with Cys145. This should enhance the inhibitory effect of inhibitors more prone to take on the S2–S1 or Cys–S1 conformations. Possible candidates for the reactive function are: epoxides, Michael acceptors, and azides.
-
2.
Side chain improvement aimed at increasing the interactions between the S2 or S4 sites and the inhibitor would create a mode of inhibition by virtually denying the entry of peptidyl substrates. A bifurcated modification diverging at the furan ring could, in principle, involve both the S2 and S4 sites in the binding of the inhibitor to the enzyme, thereby maximizing their interactions.
4. Materials and methods
4.1. Syntheses of compounds and in vitro characterization of their inhibitory effects on SARS-CoV Mpro
The chemical syntheses and in vitro characterization of the compounds described in this study were done essentially as reported previously.34 In summary, the pyridinyl esters were prepared through parallel synthesis, by the coupling reactions between 5-chloro-3-pyridinol and commercially available carboxylic acids using method A or B. Briefly, in method A, EDCI (0.5 mmol, 1.0 equiv), HOBt (0.5 mmol, 1.0 equiv), DIPEA (0.5 mmol, 1.0 equiv), and 5-chloro-3-pyridinol (0.5 mmol, 1.0 equiv) were added to a solution of carboxylic acid (0.5 mmol, 1.0 equiv) in DMF (2 mL) at RT. After 24 h of stirring, the solvent was removed in vacuo to afford the crude mixture. In method B, to a solution of carboxylic acid (1 mmol, 1.0 equiv) in DCM (5 mL) at RT was added thionyl chloride (2.6 mmol, 2.6 equiv) dropwise and a catalytic amount of DMF (2 drops). After overnight stirring, the solvent was removed in vacuo to afford the acyl chloride product. A solution of the acyl chloride in DCM (5 mL) was then added dropwise to a solution of 5-chloro-3-pyridinol (1 mmol, 1.0 equiv) and pyridine (0.09 mL, 1.1 equiv) in DCM (5 mL) at 0 °C. After 3 h of stirring at RT, the solvent was removed in vacuo to afford the crude mixture. The crude mixtures obtained from both methods A and B were purified by HPLC–MS automated instrument.
4.2. Choice and preparation of protein structures
To choose rationally a conformation of the SARS-CoV Mpro enzyme for the docking experiments, several crystal structures of SARS-CoV Mpro deposited in Protein Data Bank (PDB)38 were first aligned by a least-square fit algorithm in the Sybyl 7.1 package (Tripos Inc., St. Louis, USA). The conformations of the S2 and S4 pockets clearly showed structural variations among these structures (Fig. 4a, for clarity, only two models were overlaid). The depths of the S2 and S4 pockets could affect docking results dramatically simply because larger inhibitors will not be docked into the smaller pockets. The crystal structure of SARS-CoV Mpro in complex with an aza-peptide epoxide (APE)35 (PDB code: 2A5K, chain A) was chosen to construct the predictive model for this series of inhibitors. The benzene rings in the P2 and P4 positions of APE were accommodated cozily in the S2 and S4 pockets as a result of the relatively larger size of these pockets in available crystal structures of SARS-CoV Mpro (Fig. 4a and b). Therefore, using this structure of the enzyme is, in principle, to allow the inhibitors to adopt conformations that would maximize the interactions between the inhibitors and the S2 and S4 sites of the enzyme. The coordinates of the inhibitor, APE and the solvent molecules were first removed from the corresponding PDB file. Then essential hydrogen atoms and Kollman united atom charges were added to the protein structure using the molecular modeling software, Sybyl version 7.1 (Tripos Inc., St. Louis, USA). Fragmental volumes and atomic solvation parameters that are important for computing the binding free energies were assigned using the ADDSOL module of the AutoDock36 program.
4.3. Preparation of inhibitor structures
The 3D structures of the studied compounds were constructed using Sybyl 7.1 (Tripos Inc., St. Louis, USA). Hydrogen atoms were added to the inhibitor structures and partial atomic charges were calculated using the Gasteiger–Marsili method.39 A geometry optimization was performed by applying the Tripos force field in Sybyl 7.1 (Tripos Inc., St. Louis, USA). All possible rotatable bonds in the compounds were assigned using the program AutoTors in AutoDock.36 This allowed searching for flexible conformations of the compounds during the docking process.
4.4. Molecular docking
The docking program AutoDock 3.0.536 was used to perform the automated molecular docking. The Lamarckian genetic algorithm (LGA) was applied to deal with the inhibitor–enzyme interactions. The grid map with 60 × 60 × 60 points spaced equally at 0.375 Å was generated using the AUTOGRID program to evaluate the binding energies between the compounds and the protein. Docking parameters were set to default values except for the step size for translation (0.2 Å) for the orientation and torsion angles (5°), the number of generations (37,000), and the energy evaluations (1,500,000). The docked inhibitor–enzyme complexes were ranked according to the predicted binding energies and to the conformity to ideal geometry of the docked structures.
Acknowledgments
We thank Ting-Wai Lee (University of Alberta) for helpful discussions. This work has been supported by the Alberta Heritage Foundation for Medical Research (AHFMR), the Canadian Institutes of Health Research (CIHR) and the Natural Sciences and Engineering Research Council of Canada and the Canada Research Chairs Program. J.Y. is a recipient of the postdoctoral fellowships from Izaak Walton Killam Foundation and AHFMR.
References and notes
- 1.World Health Organization. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003 (based on data as of the 31 December 2003). http://www.who.int/csr/sars/country/table2004_04_21/en/.
- 2.Fouchier R.A., Kuiken T., Schutten M., van Amerongen G., van Doornum G.J., van den Hoogen B.G., Peiris M., Lim W., Stohr K., Osterhaus A.D. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 5.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 6.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 7.Thiel V., Herold J., Schelle B., Siddell S.G. J. Virol. 2001;75:6676–6681. doi: 10.1128/JVI.75.14.6676-6681.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 9.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi J., Song J. FEBS J. 2006;273:1035–1045. doi: 10.1111/j.1742-4658.2006.05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan J., Verschueren K.H., Anand K., Shen J., Yang M., Xu Y., Rao Z., Bigalke J., Heisen B., Mesters J.R., Chen K., Shen X., Jiang H., Hilgenfeld R. J. Mol. Biol. 2005;354:25–40. doi: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shan Y.F., Xu G.J. Acta Biochim. Biophys. Sin. (Shanghai) 2005;37:807–813. doi: 10.1111/j.1745-7270.2005.00114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh A.K., Xi K., Ratia K., Santarsiero B.D., Fu W., Harcourt B.H., Rota P.A., Baker S.C., Johnson M.E., Mesecar A.D. J. Med. Chem. 2005;48:6767–6771. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 18.Shie J.J., Fang J.M., Kuo C.J., Kuo T.H., Liang P.H., Huang H.J., Yang W.B., Lin C.H., Chen J.L., Wu Y.T., Wong C.H. J. Med. Chem. 2005;48:4469–4473. doi: 10.1021/jm050184y. [DOI] [PubMed] [Google Scholar]
- 19.Jain R.P., Pettersson H.I., Zhang J., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James M.N., Wishart D.S., Vederas J.C. J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H.Z., Zhang H., Kemnitzer W., Tseng B., Cinatl J., Jr., Michaelis M., Doerr H.W., Cai S.X. J. Med. Chem. 2006;49:1198–1201. doi: 10.1021/jm0507678. [DOI] [PubMed] [Google Scholar]
- 21.Wu C.Y., Jan J.T., Ma S.H., Kuo C.J., Juan H.F., Cheng Y.S., Hsu H.H., Huang H.C., Wu D., Brik A., Liang F.S., Liu R.S., Fang J.M., Chen S.T., Liang P.H., Wong C.H. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kao R.Y., Tsui W.H., Lee T.S., Tanner J.A., Watt R.M., Huang J.D., Hu L., Chen G., Chen Z., Zhang L., He T., Chan K.H., Tse H., To A.P., Ng L.W., Wong B.C., Tsoi H.W., Yang D., Ho D.D., Yuen K.Y. Chem. Biol. 2004;11:1293–1299. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martina E., Stiefl N., Degel B., Schulz F., Breuning A., Schiller M., Vicik R., Baumann K., Ziebuhr J., Schirmeister T. Bioorg. Med. Chem. Lett. 2005;15:5365–5369. doi: 10.1016/j.bmcl.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaeppler U., Stiefl N., Schiller M., Vicik R., Breuning A., Schmitz W., Rupprecht D., Schmuck C., Baumann K., Ziebuhr J., Schirmeister T. J. Med. Chem. 2005;48:6832–6842. doi: 10.1021/jm0501782. [DOI] [PubMed] [Google Scholar]
- 26.Xiong B., Gui C.S., Xu X.Y., Luo C., Chen J., Luo H.B., Chen L.L., Li G.W., Sun T., Yu C.Y., Yue L.D., Duan W.H., Shen J.K., Qin L., Shi T.L., Li Y.X., Chen K.X., Luo X.M., Shen X., Shen J.H., Jiang H.L. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 27.Rajnarayanan R.V., Dakshanamurthy S., Pattabiraman N. Biochem. Biophys. Res. Commun. 2004;321:370–378. doi: 10.1016/j.bbrc.2004.06.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei D.Q., Zhang R., Du Q.S., Gao W.N., Li Y., Gao H., Wang S.Q., Zhang X., Li A.X., Sirois S., Chou K.C. Amino Acids. 2006 doi: 10.1007/s00726-006-0361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai K.C., Chen S.Y., Liang P.H., Lu I.L., Mahindroo N., Hsieh H.P., Chao Y.S., Liu L., Liu D., Lien W., Lin T.H., Wu S.Y. J. Med. Chem. 2006;49:3485–3495. doi: 10.1021/jm050852f. [DOI] [PubMed] [Google Scholar]
- 30.Dooley A.J., Shindo N., Taggart B., Park J.G., Pang Y.P. Bioorg. Med. Chem. Lett. 2006;16:830–833. doi: 10.1016/j.bmcl.2005.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X.W., Yap Y.L., Altmeyer R.M. Eur. J. Med. Chem. 2005;40:57–62. doi: 10.1016/j.ejmech.2004.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z., Huang C., Fan K., Wei P., Chen H., Liu S., Pei J., Shi L., Li B., Yang K., Liu Y., Lai L. J. Chem. Inf. Model. 2005;45:10–17. doi: 10.1021/ci049809b. [DOI] [PubMed] [Google Scholar]
- 33.Lu I.L., Mahindroo N., Liang P.H., Peng Y.H., Kuo C.J., Tsai K.C., Hsieh H.P., Chao Y.S., Wu S.Y. J. Med. Chem. 2006;49:5154–5161. doi: 10.1021/jm060207o. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J., Pettersson H.I., Huitema C., Niu C., Yin J., James M.N., Eltis L.D., Vederas J.C. J. Med. Chem. 2007;50:1850–1864. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- 35.Lee T.W., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N. J. Mol. Biol. 2005;353:1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 37.Yin J., Niu C., Cherney M.M., Zhang J., Huitema C., Eltis L.D., Vederas J.C., James M.N. J. Mol. Biol. 2007;371:1060–1074. doi: 10.1016/j.jmb.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gasteiger J., Marsili M. Tetrahedron. 1980;36:3219–3228. [Google Scholar]