

Abstract
Genetic studies of host susceptibility to infection contribute to our understanding of an organism's response to pathogens at the immunological, cellular, and molecular levels. In this review we describe how the study of host genetics in mouse models has helped our understanding of host defense mechanisms against viral infection, and how this knowledge can be extended to human infections. We focus especially on the innate mechanisms that function as the host's first line of defense against infection. We also discuss the main issues that confront this field, as well as its future.
Viral infections in humans are notable for the diversity of host responses, rates of progression, and disease outcomes. A large body of evidence indicates that these differential responses depend not only on viral factors but also on inherited components affecting host susceptibility. Significant advances in our understanding of the host response to infection in humans and other animal species have recently been achieved through the use of mouse models of infection. In laboratory mice, years of inbreeding have fixed deleterious alleles, which can be detected as susceptibility phenotypes. Two major breakthroughs advanced this field. One was our ability to manipulate the mouse genome using transgenic and gene knockout technologies. This ‘reverse genetics’ approach allows direct testing of specific genes for their role in host resistance or susceptibility to infection. The other was our ability to clone genes responsible for natural variation in susceptibility to infection among inbred strains of mice, through molecular genetics. This ‘forward genetics’ approach can uncover novel mechanisms of host defense that are crucial to effective and protective responses to infection.
To date more than 30 mouse loci (Table 1) and many fewer human genes (Table 2) have been associated with the outcome of virus infection 1, 2. Each locus controls infection by a single virus family or strain. This is probably related to the vast diversity of virus life cycles and the likelihood that the product of each host resistance gene interacts with unique molecules encoded by individual viruses (Fig. 1).
Table 1.
Mouse loci affecting susceptibility to viral infectionsa
| Virus | Locus | Locus definition | Chromosome locationb | Gene product | Mode of actionc |
|---|---|---|---|---|---|
| Coronaviridae | |||||
| Mouse hepatitis virus-2 | Hv1 | Provides viral entry receptor | |||
| Mouse hepatitis virus-4 | Hv2 | Controls resistance of peritoneal macrophage to infection | 7 (5.5 cM) | CEACAM1 | Provides viral entry receptor |
| Retroviridae | |||||
| Ecotropic murine leukemia virus (MuLV) | Fv1 | Influences susceptibility to FV-induced erythroleukemia | 4 (76.5 cM) | Endogenous gag-related gene | Blocks viral preintegration complex |
| Spleen focus-forming virus (SFFV) | Fv2 | Controls resistance to spleen focus formation | 9 (60 cM) | STK | Truncated STK results in expansion of infected cell in the primary phase of erythroleukemia |
| Ecotropic MuLV | Fv4/Akvr1 | Controls resistance to Friend MuLV | 12 (38 cM) | Ecotropic Env protein | Blocks ecotropic virus receptor |
| Mink cell focus-forming (MCF) virus | Rmcf | Confers resistance to the polytropic MCF virus | 5 (10 cM) | Polytropic Env protein | Blocks polytropic virus receptor |
| Friend virus (FV) | Rfv1 | Influences susceptibility to FV-induced disease | 17 | H2-D | Induces an efficient cytotoxic T lymphocyte (CTL) response |
| Rfv2 | Modulates FV-induced disease | 17 | H2-linked | ||
| Rfv3 | Influences susceptibility to FV-induced disease | 15 | Produces vigorous antivirus antibody response | ||
| Moloney murine leukemia virus (MoMLV) | Rmv1 | Increases IgG-specific anti-MoMLV response | 17 | H2-linked | Controls antibody responses against infection |
| Rmv2 | Increases IgG-specific anti-MoMLV response | 17 | H2-linked | Controls antibody responses against infection | |
| Rmv3 | Increases IgG-specific anti-MoMLV response | 17 | H2-linked | Controls antibody responses against infection | |
| Orthomyxoviridae | |||||
| Influenza virus and Thogoto virus | Mx1 | Rescues lethal infection | 16 (71.2 cM) | GTPase | Blocks primary transcription and facilitates the degradation of the viral ribonucleoprotein complexes |
| Rhabdoviridae | |||||
| Vesicular stomatitis virus | Mx2 | Rescues lethal infection | 16 (71.2 cM) | GTPase | Blocks trafficking of the viral ribonucleoprotein complexes |
| Flaviviridae | |||||
| Flavivirus | Flv | Controls virus replication of flavivirus infection in mouse brain | 5 (67 cM) | OAS1B | Induces RNA degradation by activating RNase L |
| Herpesviridae | |||||
| Murine cytomegalovirus | Cmv1 | Controls viral replication on spleen | 6 (63.29 cM) | Ly49H | Recognizes and kills viral-infected cells |
| H2 | Serves as receptor for mouse cytomegalovirus (MCMV) | 17 | H2 | Provides viral entry receptor | |
| Herpes simplex virus | Rhs1 | Controls early primary infection | 6 | ||
| γ-Herpesvirus | Trbv4c1 | Results in increased Vbeta4+ T cells | 17 (23 cM) | ||
| Trbv4c2 | Results in increased Vbeta4+ T cells | 6 (0.5 cM) | |||
| Picornaviridae | |||||
| Theiler's virus | Tmevd1 | Confers resistance to Theiler's murine encephalomyelitis virus (TMEV)-induced demyelinating disease | 6 (22 cM) | ||
| Tmevd2 | Confers resistance to TMEV-induced demyelinating disease | 3 (43 cM) | |||
| Tmevd3 | Confers resistance to TMEV-induced demyelinating disease | 14 | |||
| Tmevd4 | Confers resistance to TMEV-induced demyelinating disease | 14 | |||
| Tmevd5 | Confers resistance to TMEV-induced demyelinating disease | 11 (52 cM) | |||
| Tmevp1 | Controls susceptibility to persistent infection | 17 | H2-Db | Induces an efficient CTL response | |
| Tmevp2 | Controls susceptibility to persistent infection | 10 (67 cM) | |||
| Tmevp3 | Controls susceptibility to persistent infection | 10 (60 cM) | |||
| Poxviridae | |||||
| Mousepox virus | Rmp1 | Rescues lethal infection | 6 | NK cell mediated | |
| Rmp2 | Rescues lethal infection | 2 (23.5 cM) | |||
| Rmp3 | Rescues lethal infection | 17 (19.5 cM) | H2-Db | ||
| Rmp4 | Controls virus replication in spleen and liver | 1 (79 cM) | |||
| Togaviridae | |||||
| Sindbis virus | Nsv1 | Reduces levels of viral RNA in the brain | 2 |
Based on information available in Mouse Genome Informatics at Jackson Laboratory (http://www.informatics.jax.org).
Chromosome number, with map distance to centromere in centimorgans (cM) in parentheses.
The mechanisms of action of some genes are hypothetical.
Table 2.
Host genes primarily associated with susceptibility to viral infections in humansa
| Virus | Affected gene | OMIMb | Chromosome location | Polymorphisms | Consequences | Refs |
|---|---|---|---|---|---|---|
| Retroviridae | ||||||
| HIV | CCR5 | 601373 | 3p21 | Deletion | Resistance to HIV infection | [10] |
| CCR2 | 601267 | 3p21 | Point mutation | Delayed progression to AIDS | [10] | |
| CXCL12 3′UTR | 600835 | 10q11.1 | Point mutation | Delayed progression to AIDS | [10] | |
| MBL2 | 154545 | 10q11.2–q21 | Point mutation | Increased susceptibility to HIV infection | [70] | |
| IL10 promoter | 124092 | 1q31–q32 | Point mutation | Accelerated progression to AIDS | [71] | |
| Hepadnaviridae | ||||||
| Hepatitis B | TNF-α promoter | 191160 | 6p21.3 | Point mutation | Increased hepatitis B virus (HBV) persistence | [41] |
| MBL2 | 154545 | 10q11.2–q21 | Point mutation | Increased HBV persistence | [72] | |
| VDR | 601769 | 12q12–q14 | Point mutation | Decreased HBV persistence | [73] | |
| Herpesviridae | ||||||
| Epstein–Barr virus (EBV) | SH2D1A | 308240 | Xq25 | Various | Development of X-linked lymphoproliferative disease (XLP) following infection by EBV | 74, 75 |
Mutations in several genes associated with immunodeficiency, such as severe combined immunodeficiency (SCID), also result in increased susceptibility to various pathogens, including viruses, and are not listed here.
Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/Omim).
Fig. 1.
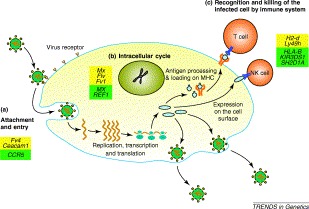
A typical virus life cycle. Despite the diversity of genomic organization and mechanisms of replication found among virus families, most viruses pass through several common stages during the infection process. Host resistance factors that determine the outcome of virus infection exist at each of the following stages: 1, viral attachment and entry into the host cell; 2, virus multiplication inside the host cell; 3, recognition and lysis of virus-infected cells. Mouse genes affecting susceptibility are shown in yellow boxes, human genes are shown in green boxes.
In this review we illustrate the contribution of mouse genetics to our understanding of mechanisms of host resistance to virus infection. These mechanisms might manifest themselves as: (1) barriers to infection at the host cell membrane; (2) intracellular host responses to infection; or (3) recognition or destruction of infected cells. Viruses used as examples are described in Table 3 .
Table 3.
Viruses discussed in this review
| Virus | Official virus species name | Family | Comments |
|---|---|---|---|
| MHV | Mouse hepatitis virus | Coronaviridae | One of the most common causes of epizootics in laboratory mouse colonies |
| Moloney murine leukemia virus (MoMLV) and Friend murine leukemia virus (MuLV) | Gammaretrovirus | Retroviridae | Provides mouse model for a mechanism of oncogenic transformation by RNA tumor virus |
| HIV-1 | Human | Retroviridae | Member of the subfamily Lentivirinae |
| immunodeficiency virus 1 | Associated with the acquired immunodeficiency syndrome (AIDS) | ||
| Influenza virus | Influenza virus | Orthomyxoviridae | Causes an infection of the respiratory tract that affects millions of people every year. There are three types of influenza viruses, A, B and C. Influenza A can infect humans and other animals whereas influenza B and C infect humans |
| MVE virus | Murray Valley encephalitis virus | Flaviviridae | Causes various symptoms from mild to severe with permanent impaired neurological functions, sometimes fatal |
| YF virus | Yellow fever virus | Flaviviridae | Causes yellow fever, a viral hemorrhagic fever. The virus can result in epidemics with mortality rates of up to 60% |
| WN virus | West Nile virus | Flaviviridae | Causes fatal encephalitis and lesions of diffuse inflammation and neuronal degeneration in humans |
| Murine cytomegalovirus (MCMV) | Murid herpesvirus 1 | Herpesviridae | Mouse model of human cytomegalovirus infection and disease |
| HBV | Hepatitis B virus | Hepadnaviridae | Infects human hepatocytes |
| Associated with acute and chronic hepatitis in humans | |||
| Persistent infection is associated with chronic liver disease, which can lead to the development of cirrhosis and hepatocellular carcinoma | |||
| HCV | Hepatitis C virus | Flaviviridae | Infects human hepatocytes |
| Persistence of HCV occurs in ≈80% of infections | |||
| Persistent infection is associated with chronic liver disease, which can lead to the development of cirrhosis and hepatocellular carcinoma | |||
| Theiler's murine encephalomyelitis virus (TMEV) | Theilovirus | Picornaviridae | Causes a chronic persistent demyelinating infection of the white matter in miceProvides a highly relevant mouse model of multiple sclerosis |
1. Barriers to infection at the host cell membrane
The first step in the life cycle of a virus is attachment to a receptor on the cell surface and delivery of the viral genome into the interior. Usually the virus takes advantage of a host molecule with an unrelated function, such as an adhesion molecule or complement regulator, for its own benefit. Receptors are recognized as important determinants of virus host range and tissue tropism; and some host resistance/susceptibility loci encode molecules that are expressed on the cell surface.
For example, SJL mice are 10 000 times more resistant to a lethal dose of mouse hepatitis virus (MHV) – a murine coronavirus – than C57BL/6 or BALB/c mice [3]. Resistance is due to allelic variation in the gene encoding carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) [4]. Susceptible strains carry the Ceacam1a allele, which encodes the principal MHV receptor, CEACAM1a. Resistant strains, such as SJL, are homozygous for the Ceacam1b allele, which encodes a 27-amino-acid substitution in the N-terminal virus-binding domain of CEACAM1a [4]. An immunoreceptor tyrosine-based inhibitory motif (ITIM) (see Glossary) is present in the cytoplasmic domain of CEACAM1, also suggesting a potential immunomodulatory function for this molecule during MHV infection. Although no role for human CEACAM1a has been described during viral infection of humans [5], a possible immunomodulatory function is consistent with the observation of a CEACAM1-mediated suppression of CD4+ T-cell activation during infection by the pathogenic bacteria Neisseria gonorrhoeae, Neisseria meningitidis and Haemophilus influenzae. These human pathogens bind to domain 1 of CEACAM1, very close to the site recognized by MHV [6].
Another example of natural host resistance is the restriction of ecotropic Murine leukemia virus (MuLV) infection by the mouse Fv4 gene. Binding of retroviral Env glycoproteins to cellular receptors triggers fusion of viral and cellular membranes, and allows the virus nucleocapsid to enter the cell. The Fv4 (or Akvr) locus is a defective endogenous ecotropic provirus that encodes an Env protein, which also has a fusion defect 7, 8. It has been proposed that the Env protein encoded by Fv4 blocks MuLV infection by interacting with the cellular receptor and restricting the amount of receptor available for exogenous retrovirus attachment (Box 1 ) [9].
Box 1. Box 1. Resistance to retroviral infection.
An important element of resistance to retroviral infection is provided by endogenous retroviral elements. Some retroviral elements in the mammalian genome are transcriptionally active but carry deletions and point mutations that render them unable to replicate. Some of their gene products, such as the FV4 and FV1 proteins, interfere with infection by their exogenous relatives thus providing a selective advantage to their hosts. For example, Fv4 encodes an Env protein that interferes with ecotropic murine leukemia virus (MuLV) infection (see main text) at the level of viral entry. Examples of this phenomenon can be found in mice, chickens and cats, suggesting that Env-mediated retroviral interference is commonly used for limiting virus spread [76]. Gene therapy based on the principle of receptor interference has demonstrated that introduction of Fv4-Env-transduced bone marrow cells into a thymectomized host confers resistance to Friend leukemia virus-induced leukemogenesis [77]. These findings suggest that a similar approach could be used as a therapeutic strategy to inhibit infections by other retroviruses in vivo, including immunodeficiency virus. For example, transfer of a gene encoding a normally processed but fusion-defective retroviral Env protein into susceptible cells would interfere with viral entry and potentially reduce the infectiousness of viruses emerging from the cell.
The Fv1 locus 78, 79 encodes a retrovirus capsid-like protein [80] that restricts MuLV infection at a post-entry stage, before integration of the provirus [81]. Because all retroviruses replicate in very similar ways, it is conceivable that similar restriction mechanisms could be found in humans. Interestingly, although an Fv1 ortholog was not detected in nonmurine species, a mechanism of preintegration interference, resistance factor 1 (REF1), has been demonstrated in human cell lines [82]. At the time of writing, the REF1 gene has not been cloned, but there is speculation that, like Fv1, REF1 might consist of endogenous retroviral sequences. A recent study suggests the existence of a REF1-like restriction in humans and nonhuman primates that determines lentivirus tropism [83]. Consistent with this proposal, the target for this restriction is within the capsid protein of HIV [83].
An example of an alternative mechanism of resistance to retroviral infection is provided by Fv2, which determines the progression of Friend virus complex-induced erythroleukemia. Fv2 is identical to Ron, which encodes STK, a member of the MET family of receptor tyrosine kinases. Susceptible mice express a positive-acting truncated version of STK lacking most of the extracellular domain, which is associated with proliferation of SFFV-infected erythroblasts [84].
In humans, resistance to human immunodeficiency virus (HIV) is also mediated by a barrier at the cell surface. CD4 is the primary or ‘attachment’ receptor for HIV, but CD4 is necessary but not sufficient for the productive entry of HIV into target cells. The identification of CCR5 as a coreceptor for HIV prompted genetic screening of individuals that escape HIV disease despite high-risk behavior [10]. Individuals carrying a homozygous 32 bp deletion in the coding sequence of CCR5 (CCR5Δ32) are extremely resistant to the ‘R5’ strain of HIV because the deletion results in a frame shift and generates a nonfunctional receptor [10]. The CCR5Δ32 mutation is found predominantly in the Caucasian population and is absent in Africans, American Indians and East Asians [11]. It has been speculated that this distribution is consistent with resistance to an agent that predates HIV and caused enormous mortality. One candidate is Yersinia pestis, the causative agent of bubonic plague, although other pathogens targeting the macrophage/monocyte lineage cannot be excluded [12].
2. Intracellular inhibitors of virus replication
Delivery of a viral genome to the interior of an infected host cell does not guarantee that an infection will be successful, because potent defense mechanisms act at the intracellular level. One of the initial cellular responses to viral infection is the production of type I interferons (IFNs). IFNs, in turn, stimulate the expression of several gene products, including the double-stranded RNA-activated protein kinase (PKR) and RNase L, which leads to the establishment of an antiviral state in cells, characterized by a general inhibition of protein synthesis.
The IFN-induced MX1 protein is one of the best-studied determinants of innate immune responses to viral infection. The inbred mouse strain A2G is resistant to doses of mouse-adapted influenza virus (an orthomyxovirus) that are lethal for other inbred strains [13]. Resistance in these mice is determined by a single dominant locus, Mx1, whose gene product, MX1, rapidly accumulates in the nuclei of cells following influenza virus infection, and blocks virus spread [14]. Susceptible mice produce no functional MX protein due to either a nonsense mutation (CBA/J) or gene deletion (BALB/c) [15]. The Mx1 product belongs to the dynamin superfamily of large GTPases conserved in all vertebrates. In mice there are actually two Mx gene products, MX1 and MX2. MX1 protein blocks transcription of influenza virus, probably via interaction with the PB2 subunit of the influenza virus polymerase [16]. By contrast, the MX2 protein is cytoplasmic and has been shown to inhibit viruses that replicate in the cytoplasm, such as vesicular stomatitis virus (VSV) and members of the family Bunyaviridae [14]. MX proteins inhibit virus replication by interfering with the transport of viral nucleocapsids, and by sorting them to locations where they become unavailable for the generation of new virus particles [14], possibly to promyelocytic leukemia protein nuclear bodies (PML NBs) [17], which are known to be a site of proteasome-mediated degradation.
Humans also possess MX genes, MXA and MXB, but only the MXA gene product has antiviral properties. However, in contrast to its mouse MX1 ortholog, the MXA GTPase accumulates in the cytoplasm of IFN-treated cells, where it inhibits not only the replication of orthomyxoviruses, VSV and bunyaviruses but also the replication of other RNA viruses such as measles virus, coxsackievirus B4 and Semliki Forest virus [14].
A mechanism of resistance to flaviviruses, which also appears to be IFN-mediated, was recently revealed by the cloning of Flv, which confers resistance (Flv r) or susceptibility (Flv s) to flavivirus infection. Virus titers in the brains of resistant mice infected with Murray Valley encephalitis virus, yellow fever virus or West Nile virus are orders of magnitude lower than in susceptible animals. Viral yields in tissue cultured from resistant or susceptible animals are also dramatically different, indicating that the Flv gene product acts intracellularly on flavivirus replication [18]. Genetic mapping localized Flv1 to chromosome 5 [19] and, more recently, it was demonstrated that Flv susceptibility is associated with a nonsense mutation in a member of the 2′,5′-oligoadenylate synthetase (OAS) family, Oas1b 20, 21. OAS proteins are induced by IFNs and play a central role in producing the antiviral state by binding double-stranded RNA and catalyzing the synthesis of 2′,5′-oligoadenylates (2-5A), which, in turn, interact with and activate RNase L to degrade single-stranded viral and cellular RNAs. Whereas Oas1b genes from resistant mice encode full-length proteins, those from susceptible mice encode proteins truncated at the C-terminus that might not form active synthetases. In contrast to the single OAS1 gene found in humans, there are eight closely linked Oas1 genes in the murine genome 20, 22, but only Oas1b is associated with resistance to flaviviruses.
3. Recognition/destruction of infected cells
In addition to resistance imparted by barriers to virus entry or by intracellular antiviral defense mechanisms, cytotoxic cells can control the level of viral replication in an animal (Box 2 ). Host recognition of virus-infected cells is mediated by two players: natural killer (NK) cells and cytotoxic T cells.
Box 2. Box 2. Cell-mediated immunity against viruses.
In vertebrates, host defense against virus infection is mediated by innate and adaptive immunity. Innate immunity constitutes the first line of defense, providing a rapid response through germ-line encoded proteins that pre-exist or are induced within hours of infection. Adaptive immunity is a slower, yet highly specific response mediated by B cells and T cells that confers effective and long-lasting protection against infection, and is characterized by immunological memory. Major histocompatibility complex (MHC) molecules are highly polymorphic glycoproteins encoded by MHC class I and II genes, which are mainly involved in the presentation of peptide antigens to T cells. MHC class I molecules bind peptides derived from proteins synthesized in the cytosol, such as viral proteins, and present them to cytotoxic CD8+ T cells. By contrast, MHC class II molecules are loaded with peptides derived from exogenous antigens engulfed within intracellular vesicles, for presentation to CD4+ T cells. MHC class I molecules also play an important role in modulating the cytotoxic activity of natural killer (NK) cells.
NK cells are a component of the innate system, so named because of their propensity to kill certain neoplastic and virus-infected cells without prior sensitization. The cytotoxic activity of NK cells is regulated by signals elicited by inhibitory receptors containing immunoreceptor tyrosine-based inhibitory motifs (ITIMs), or stimulatory receptors associated with immunoreceptor tyrosine-based activation motifs (ITAM)-bearing adaptor molecules. Inhibitory receptors interact with MHC class I molecules and prevent cell killing of healthy cells by autologous NK cells. In situations where MHC class I is downregulated, such as during tumorigenesis or viral infection, reduced inhibitory signals result in NK-cell-mediated lysis [85]. Activating NK-cell receptors recognize stress-induced or pathogen-encoded MHC class I-like proteins and stimulate killing of cells expressing these molecules [85].
3.1. Role of NK cells in cytomegalovirus infection
Herpesviruses avoid recognition and activation of the adaptive immune system by downregulating major histocompatibility complex (MHC) class I molecules on infected cells. However, class I downregulation renders the infected cell susceptible to recognition by NK cells expressing inhibitory receptors, in accordance with the ‘missing self’ hypothesis of Karre et al. [23]. Viruses such as mouse cytomegalovirus (MCMV) have upped the ante in their battle against the immune system by deploying MHC class I homologs, such as m144, which confer resistance to the innate immune response by inhibiting NK-cell-mediated attack (Box 2) [24]. Strains of the C57BL background carry a dominant resistance allele, Cmv1 r, that restricts viral replication in target organs, whereas other mouse strains carry a recessive susceptibility allele, Cmv1 s, that allows rapid proliferation of the virus [25]. Cmv1 r encodes an activating NK-cell receptor, Ly49H 26, 27, 28, which is absent in susceptible strains. Mouse strains express different repertoires of Ly49 molecules, which are part of a large family of polymorphic receptors expressed on overlapping populations of NK cells. Upon binding of MHC class I ligands, different Ly49 molecules deliver either activating or inhibitory signals that modulate NK-cell function [29]. Recent studies have revealed that Ly49H recognizes MCMV-infected cells via direct interaction with the viral antigen, m157, which shares structural homology with MHC class I molecules and is expressed on the surface of infected cells 30, 31. m157 also binds to an inhibitory receptor, Ly49I, expressed in susceptible strain 129 mice, suggesting that m157 could have evolved as a mechanism to escape NK-cell killing by targeting inhibitory receptors [30]. In resistant mice, Ly49H might have evolved as a countermeasure against virus-encoded MHC class I homologs, providing an overriding activating signal to the NK cell, and promoting the elimination of MCMV-infected cells (Fig. 2a). Considering the prevalence of human cytomegalovirus (HCMV) in the human population, we expect that there are likely to be HCMV-specific activating receptors present on human leukocytes that might function in a manner analogous to Ly49H.
Fig. 2.
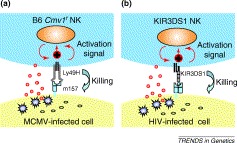
Direct recognition of virus-infected cells by natural killer (NK)-cell receptors. (a) NK-cell recognition and killing of mouse cytomegalovirus (MCMV)-infected cells in mice. The recognition of the m157 protein by the activating Ly49H receptor provides the first example of direct recognition of a virally encoded molecule by an NK-cell receptor. Ly49H associates with the adaptor molecule DAP12 through a charged arginine residue in its transmembrane domain. DAP12 possesses an immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic domain that serves as a docking site for Syk and Zap70 protein tyrosine kinases. Upon Ly49H binding to m157, a cascade of tyrosine phosphorylation is initiated leading to cellular activation and perforin-mediated killing of the virus-infected cell. (b) Proposed NK-cell recognition and killing of HIV-infected cells in humans. Human killer-cell immunoglobulin-type receptor (KIR) is considered to be a functional homolog of mouse Ly49. An epistatic interaction between KIR3DS1 and HLA-B delays progression to AIDS, suggesting that HLA-B behaves as a ligand for KIR3DS1 [32]. The peptide presented on HLA-B that is responsible for this interaction remains to be identified. Because KIR3DS1 receptor is also associated with the adaptor molecule DAP12, the intracellular signaling cascade leading to cellular activation and killing of virus-infected cells seems to be similar to that triggered by mouse Ly49H.
3.2. Role of the MHC in efficient T-cell recognition
The MHC (Box 2) is a set of genes, present in all vertebrates, with immunological and nonimmunological functions. MHC class I genes play a crucial role in combating viral infection in mice (Fig. 3a). Congenic mice with intra-MHC recombinations provide an important tool for dissecting the contribution of MHC class I subregions to virus infection. For example, comparisons of T-cell responses in H2 congenic mice that differ in their recovery from Friend leukemia virus infection were used to localize Friend leukemia virus resistance loci Rfv1 and Rfv2. Rfv1 mapped to the H2D subregion and determined T-cell activation. Rfv2 mapped to the IA subregion and determined unresponsiveness of T cells in a proliferation assay [35]. Resistance to acute lethal infection of MCMV is also controlled by genes linked to H2, with the k haplotype being more resistant than b or d. Resistance was associated with genes of subregions K/IA and D. Interestingly in this model, transfection of macrophages with sequences encoding MHC molecules such as H2-Kd, Dd or Kb renders these cells, which are major reservoirs for MCMV, sensitive to MCMV infection [37], consistent with a role for MHC molecules as MCMV receptors.
Fig. 3.

Roles of the major histocompatibility complex (MHC) in combating virus infections. (a) Mouse susceptibility loci in the MHC are shown. Studies using congenic mice having intra-MHC recombination identified the contribution of specific MHC alleles to virus infection. Mostly, MHC class I genes play a crucial role against virus infection of mice. Only a small number of genes in the MHC and their relative positions on chromosome 17 are shown. (b) Genetic associations of the human MHC with susceptibility to virus infection. Only a small number of genes in the MHC, with their relative positions on chromosome 6, are shown. The genes encoding class I are depicted in red, class II in yellow and class III in green. TNF is the gene encoding tumor necrosis factor. C2 and C4 are complement genes.
In humans, association analysis between MHC and specific viral infections has also suggested a differential role of specific alleles in susceptibility to HIV, hepatitis B virus (HBV) and hepatitis C virus (HCV) (Fig. 3b). For example, HLA-DRB1*1302 is associated with resistance to chronic HBV infection in both African and European populations 42, 43 whereas HLA-B*35-Cw*04 is associated with the rapid progression of AIDS in Caucasian populations [38].
Although polymorphisms in the classical MHC class I and class II genes are known to be related to antigen presentation, it is important to note that there are other genes within the MHC class III region, such as components of the complement cascade (C2, C4), cytokines – tumor necrosis factor (TNF)-α and -β – and proteins involved in antigen processing (Lmp2, Tap1), which also have an important function in immunity [51]. Therefore, disease resistance or susceptibility that maps to the MHC must be interpreted with caution, to differentiate the role of antigen presentation from the other roles of MHC-associated genes.
4. Studies of complex models of susceptibility to viral infection
Although advances in genomic analysis have paralleled the discovery of host susceptibility traits that are determined by single alleles, most differences in host susceptibility to viral infection are the result of interactions among different genes with multiple alleles. The study of infectious disease under complex genetic control in humans is complicated by a variety of factors including genetic heterogeneity, low penetrance and environmental factors. The tools currently available to the mouse geneticist make the identification and isolation of disease susceptibility genes much more attainable. An example is the response to Theiler's murine encephalomyelitis virus (TMEV), a rodent model for multiple sclerosis [52]. In genetically susceptible mice, certain TMEV strains cause persistent infection, inflammation, and a chronic demyelinating disease. One of the loci determining Theiler's murine encephalomyelitis virus persistence (Tmevp1), maps to the MHC region and corresponds to the H2D gene [52]. Mice transgenic for the H2D b allele acquired resistance to persistent virus infection, suggesting that the gene effect was at the level of peptide presentation by H2Db [53]. Using the amount of viral RNA in the spinal cords of persistently infected mice as a phenotype, and a genetic dissection strategy based on genome scans, a second locus, Tmevp2, was localized near the type II interferon (IFN-γ) gene on distal chromosome 10 [54]. Analysis of a panel of congenic mice that inherited different portions of mouse chromosome 10 revealed that Tmevp2 was actually two distinct loci, now termed Tmevp2 and Tmevp3, and excluded the IFN-γ gene from the candidate region [55]. Recently, a strong candidate for Tmevp3, named Tmevpg1, has been identified as a noncoding RNA expressed in immune cells infiltrating the central nervous system of resistant mice, where an inverse relationship with IFN-γ mRNA levels has been observed [56], suggesting a role for this RNA in regulating IFN-γ synthesis. Consistent with this observation, the same reciprocal pattern of RNA expression was observed for TMEVPG1, the human counterpart, and IFN-γ in human NK cells and T cells.
Several complementary avenues promise significant acceleration in the identification of genes that determine resistance or susceptibility in complex models of virus infection. The well-defined sets of recombinant congenic strains (RCS) [57], consomic (chromosome substitution) strains [58] and advanced intercrossed lines [59] that are now available can be used to map a locus of interest and to characterize the specific phenotypic component of disease susceptibility under genetic control. Also, traditional genetic mapping techniques are now complemented by high-throughput methods for studying gene function and regulation. For example, high-density oligonucleotide arrays [60] or cDNA arrays [61] allow for gene expression monitoring on a genome-wide scale and offer an opportunity to establish functional links between genotype and phenotype. A strategy that combines congenic mapping with microarray expression profiling would aid in the identification of novel susceptibility genes and biochemical pathways not previously known to be involved in disease etiology. Furthermore, the availability of the human and mouse genome sequences represents the ultimate breakthrough in quantitative trait loci (QTL) studies by placing the identification of candidate genes within a defined genetic interval only a (computer) mouse click away.
One of the current limitations of using inbred strains of mice to study susceptibility to viral infection is the limited extent of genetic variation, mainly due to a small number of original progenitor strains [62]. Therefore, although these resources are valuable, they do not permit the identification of all possible resistance or susceptibility loci. Wild-derived inbred strains of mice are an important source of additional resistance alleles [63] that is just beginning to be exploited, as demonstrated by the characterization of Flv. Chemical mutagenesis, a method that has been successfully applied to the study of other model organisms, from bacteria to Drosophila, is another promising strategy for creating novel susceptibility alleles in the mouse. The alkylating agent N-ethyl-N-nitrosourea (ENU) introduces point mutations and creates random variation throughout the genome [64]. This is highly advantageous for defining host susceptibility phenotypes because it represents an unbiased method for identifying previously unrecognized physiological pathways because no assumptions are made about the relevant genes. ENU mutagenesis yields models of simple inheritance that are readily accessible to analysis by positional cloning [64].
5. Conclusions and future perspectives
The effort to understand the genetic basis of susceptibility to viral disease is driven by three considerations: (1) the increased public awareness of the toll imposed by viruses on the host; (2) the increase in susceptible human populations because of longer life expectancy, frequently accompanied by chronic illness, and the consequences of advances in medical technology, including immunosuppressive therapies for organ transplantation or treatment of malignancy; and (3) the need to develop new therapies for infections caused by multidrug-resistant microorganisms and by microorganisms for which there is currently no treatment.
Genetic studies of host resistance to viral infection have provided fundamental insights into cell biology, viral pathogenesis, and molecular and cellular immunology. We have provided examples of three levels at which the genetics of the host can determine the outcome of a viral infection. Mouse genetics has shown the importance of allelic variation of virus receptor genes in determining the severity of viral disease. However, in addition to virus–receptor interaction, a complex multicomponent process is required for virus entry into the host cell. Disrupting functional interactions crucial to virus entry via genetic manipulation of virus receptors or associated molecules, or by using inhibitory small molecules, represent promising approaches to antiviral therapy. Therapies based on the principle of receptor interference for viruses other than retroviruses (Box 1) could be explored.
Type I IFN induces an intracellular antiviral response against many different RNA and DNA viruses. Mouse genetics has provided a starting point for dissecting intracellular mechanisms of host defense, and has revealed that several IFN-inducible gene products have inhibitory effects on a much more restricted number of viruses than expected. The precise nature (sequence or structural) of determinants recognized by effector proteins such as MX or OAS1B remain to be characterized. The identification of additional effector molecules and viral targets must continue to be an important area of active research, particularly in view of the existence of orthologous human genes.
Mouse genetics has also demonstrated that recognition and destruction of virus-infected cells by NK cells is mediated by specific interactions between activating NK-cell receptors and viral target molecules. Viruses pose a particular problem for specific recognition because all the components of the virus are synthesized and assembled in the host cell. Important questions to be answered include: (1) Do other activating NK-cell receptors have specialized functions in virus recognition? (2) Is the MHC class I fold a recognition-associated structure? (3) What is the extent of the repertoire of target molecules for NK-cell-activating receptors? In addition, studies of mouse NK-cell receptors, such as the Ly49 family, have provided a conceptual framework for understanding human NK-cell biology. Because no functional human Ly49 counterpart has been identified, prime candidates for the recognition of viral pathogens by human NK cells are members of the killer-cell immunoglobulin-like receptor (KIR) family [65]. Although the ligands of activating KIR receptors remain to be identified, it has been proposed that activating KIR molecules have evolved to recognize infectious agents (Fig. 2b) [32].
It is suspected that several common and/or chronic human diseases under complex genetic control are triggered by a viral infection. This idea is supported by experimental evidence derived from mouse models for initiation and exacerbation of atherosclerosis following MCMV infection [66], for diabetes [67] or dilated cardiomyopathy [68] following coxsackievirus infection, and for multiple sclerosis-like disease following TMEV [33] or MHV [69] infection. Identifying genes that control susceptibility to acute and chronic viral infection in these models is a crucial step in understanding the development of these complex pathologies.
Comparisons between mouse and human mechanisms of host resistance or susceptibility to viral infection have increased our awareness not only of their differences but also, more importantly, of their similarities. In the future, the use of comparative genomic approaches in animal model systems will provide more comprehensive knowledge of the impact of viruses on human health.
Acknowledgements
We are grateful to Christine di Donato for critical reading of the manuscript. Our laboratories are supported by grants from the Canadian Institutes of Health Research (CIHR) and Natural Sciences and Engineering Research Council of Canada (NSERC). Seung-Hwan Lee is supported by a CIHR doctoral scholarship and Silvia M. Vidal is a Junior Scientist of CIHR.
Glossary
Glossary
- Advanced intercross lines
: mouse lines generated by producing an F2 generation between two inbred strains and then intercrossing in each subsequent generation, but avoiding sibling matings. This provides increasing probability of tightly linked genes recombining.
- Congenic strain
: a mouse strain that has been bred to be identical to an inbred strain, except for a selected differential chromosomal segment. Congenic strains are derived by backcrossing to a parental inbred strain for at least ten generations while selecting for heterozygosity at a particular locus.
- Consomic strain
: a variation on a congenic strain in which recombinants between two inbred strains are backcrossed to produce a strain that carries a single chromosome from one strain on the genetic background of the other.
- Epistasis
: the interaction between alleles at different loci that allows an allele at one locus to alter the effects of alleles at a different locus.
- Immunoreceptor tyrosine-based activation motif (ITAM)
: a cytoplasmic tyrosine-containing motif that is the site of tyrosine phosphorylation. These motifs are associated with tyrosine kinases and other phosphotyrosine-binding proteins involved in cellular activation.
- Immunoreceptor tyrosine-based inhibitory motif (ITIM)
: a cytoplasmic tyrosine-containing motif. In contrast to ITAMs, phosphorylation of the tyrosine residues within ITIMs recruits the Src-homology-2-domain-containing tyrosine phosphatase SHP-1 and/or SHP-2, transducing a negative signal that inhibits cellular activation.
- Interferons (IFNs)
: a group of immunoregulatory proteins that are produced by cells infected with a virus and have the ability to inhibit viral growth. IFNs are classified as type I (IFN-α/β), which have antiviral properties, and as type II (IFN-γ), which is known as immune interferon. Despite the use of different receptors, certain IFN-α/β and IFN-γ functions are shared because the signal transduction pathways activated through these receptors partly overlap.
- Murine leukemia virus (MuLV)
: there are four subgroups of naturally occurring MuLVs based on receptor usage on mouse cells: ecotropic MuLVs, which are able to infect only mouse cells utilizing the cationic amino acid transporter as a receptor; amphotropic MuLVs, which are able to infect mouse cells as well as those of other species (including human) by binding to an inorganic phosphate transporter protein; polytropic MuLVs, which can infect cells from mouse as well as nonmurine species using yet another cellular receptor protein, thought to be a G-protein-coupled receptor; and xenotropic MuLVs, which use receptors from cells of most species except mice.
- Quantitative trait loci (QTLs)
: the locations of genes that affect a trait that is measured on a quantitative (linear) scale, such as body weight or blood pressure in animals, as identified by statistical analysis. These traits are typically affected by more than one gene, and also by the environment.
- Recombinant congenic strain
: a variation on recombinant inbred strains in which the initial outcross is followed by several generations of backcrossing before inbreeding.
- Retroviral interference
: the phenomenon by which prior infection of cells with a retrovirus confers strong resistance to infection of the same cells by a retrovirus that utilizes the same receptor; however, cells remain susceptible to viruses that use a different receptor. This process results from masking or downregulation of the receptor due to interaction with the glycoprotein of the established virus.
References
- 1.Casanova J.L. Forward genetics of infectious diseases: immunological impact. Trends Immunol. 2002;23:469–472. doi: 10.1016/s1471-4906(02)02289-5. [DOI] [PubMed] [Google Scholar]
- 2.Cooke G.S., Hill A.V. Genetics of susceptibility to human infectious disease. Nat. Rev. Genet. 2001;2:967–977. doi: 10.1038/35103577. [DOI] [PubMed] [Google Scholar]
- 3.Stohlman S.A., Frelinger J.A. Resistance to fatal central nervous system disease by mouse hepatitis virus strain JHM. I. Genetic analysis. Immunogenetics. 1978;6:277–281. [Google Scholar]
- 4.Holmes K.V., Dveksler G.S. Specificity of coronavirus/receptor interactions. In: Wimmer E., editor. Cellular Receptors for Animal Virues. Cold Spring Harbor Laboratory Press; 1994. pp. 403–443. [Google Scholar]
- 5.Yeager C.L. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature. 1992;357:420–422. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan K. Crystal structure of murine sCEACAM1a[1,4]: a coronavirus receptor in the CEA family. EMBO J. 2002;21:2076–2086. doi: 10.1093/emboj/21.9.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dandekar S. Molecular characterization of the Akvr-1 restriction gene: a defective endogenous retrovirus-borne gene identical to Fv-4r. J. Virol. 1987;61:308–314. doi: 10.1128/jvi.61.2.308-314.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeda H., Sugimura H. Fv-4 resistance gene: a truncated endogenous murine leukemia virus with ecotropic interference properties. J. Virol. 1989;63:5405–5412. doi: 10.1128/jvi.63.12.5405-5412.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor G.M. Fv-4: identification of the defect in Env and the mechanism of resistance to ecotropic murine leukemia virus. J. Virol. 2001;75:11244–11248. doi: 10.1128/JVI.75.22.11244-11248.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berger E.A. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- 11.Martinson J.J. Global distribution of the CCR5 gene 32-basepair deletion. Nat. Genet. 1997;16:100–103. doi: 10.1038/ng0597-100. [DOI] [PubMed] [Google Scholar]
- 12.Stephens J.C. Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes. Am. J. Hum. Genet. 1998;62:1507–1515. doi: 10.1086/301867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindenmann J. Resistance of mice to mouse adapted influenza A virus. Virology. 1962;16:203–204. doi: 10.1016/0042-6822(62)90297-0. [DOI] [PubMed] [Google Scholar]
- 14.Haller O., Kochs G. Interferon-induced mx proteins: dynamin-like GTPases with antiviral activity. Traffic. 2002;3:710–717. doi: 10.1034/j.1600-0854.2002.31003.x. [DOI] [PubMed] [Google Scholar]
- 15.Staeheli P. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol. Cell. Biol. 1988;8:4518–4523. doi: 10.1128/mcb.8.10.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stranden A.M. Function of the mouse Mx1 protein is inhibited by overexpression of the PB2 protein of influenza virus. Virology. 1993;197:642–651. doi: 10.1006/viro.1993.1639. [DOI] [PubMed] [Google Scholar]
- 17.Engelhardt O.G. Interferon-induced antiviral Mx1 GTPase is associated with components of the SUMO-1 system and promyelocytic leukemia protein nuclear bodies. Exp. Cell Res. 2001;271:286–295. doi: 10.1006/excr.2001.5380. [DOI] [PubMed] [Google Scholar]
- 18.Darnell M.B., Koprowski H. Genetically determined resistance to infection with group B arboviruses. II. Increased production of interfering particles in cell cultures from resistant mice. J. Infect. Dis. 1974;129:248–256. doi: 10.1093/infdis/129.3.248. [DOI] [PubMed] [Google Scholar]
- 19.Urosevic N. Development and characterization of new flavivirus-resistant mouse strains bearing Flv(r)-like and Flv(mr) alleles from wild or wild-derived mice. J. Gen. Virol. 1999;80:897–906. doi: 10.1099/0022-1317-80-4-897. [DOI] [PubMed] [Google Scholar]
- 20.Perelygin A.A. Positional cloning of the murine flavivirus resistance gene. Proc. Natl. Acad. Sci. U. S. A. 2002;99:9322–9327. doi: 10.1073/pnas.142287799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mashimo T. A nonsense mutation in the gene encoding 2′-5′-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11311–11316. doi: 10.1073/pnas.172195399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eskildsen S. Gene structure of the murine 2′-5′-oligoadenylate synthetase family. Cell. Mol. Life Sci. 2002;59:1212–1222. doi: 10.1007/s00018-002-8499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karre K. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 24.Farrell H.E. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature. 1997;386:510–514. doi: 10.1038/386510a0. [DOI] [PubMed] [Google Scholar]
- 25.Scalzo A.A. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J. Exp. Med. 1990;171:1469–1483. doi: 10.1084/jem.171.5.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S.H. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat. Genet. 2001;28:42–45. doi: 10.1038/ng0501-42. [DOI] [PubMed] [Google Scholar]
- 27.Brown M.G. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- 28.Daniels K.A. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J. Exp. Med. 2001;194:29–44. doi: 10.1084/jem.194.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson S.K. The ever-expanding Ly49 gene family: repertoire and signaling. Immunol. Rev. 2001;181:79–89. doi: 10.1034/j.1600-065x.2001.1810106.x. [DOI] [PubMed] [Google Scholar]
- 30.Arase H. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 31.Smith H.R. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. U. S. A. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin M.P. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat. Genet. 2002;31:429–434. doi: 10.1038/ng934. [DOI] [PubMed] [Google Scholar]
- 33.Brahic M., Bureau J.F. Genetics of susceptibility to Theiler's virus infection. Bioessays. 1998;20:627–633. doi: 10.1002/(SICI)1521-1878(199808)20:8<627::AID-BIES5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 34.Brownstein D.G. Serial backcross analysis of genetic resistance to mousepox, using marker loci for Rmp-2 and Rmp-3. J. Virol. 1992;66:7073–7079. doi: 10.1128/jvi.66.12.7073-7079.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Britt W.J., Chesebro B. H-2D control of recovery from Friend virus leukemia: H-2D region influences the kinetics of the T lymphocyte response to Friend virus. J. Exp. Med. 1983;157:1736–1745. doi: 10.1084/jem.157.6.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Debre P. Genetic control of sensitivity to Moloney leukemia virus in mice. III. The three H-2 linked Rmv genes are immune response genes controlling the antiviral antibody response. Eur. J. Immunol. 1980;10:914–918. doi: 10.1002/eji.1830101205. [DOI] [PubMed] [Google Scholar]
- 37.Price P. Are MHC proteins cellular receptors for CMV? Immunol. Today. 1994;15:295–296. doi: 10.1016/0167-5699(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 38.Carrington M. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748–1752. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- 39.Kaslow R.A. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med. 1996;2:405–411. doi: 10.1038/nm0496-405. [DOI] [PubMed] [Google Scholar]
- 40.Jeffery K.J. HLA alleles determine human T-lymphotropic virus-I (HTLV-I) proviral load and the risk of HTLV-I-associated myelopathy. Proc. Natl. Acad. Sci. U. S. A. 1999;96:3848–3853. doi: 10.1073/pnas.96.7.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hohler T. A tumor necrosis factor-alpha (TNF-alpha) promoter polymorphism is associated with chronic hepatitis B infection. Clin. Exp. Immunol. 1998;111:579–582. doi: 10.1046/j.1365-2249.1998.00534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hohler T. HLA-DRB1*1301 and *1302 protect against chronic hepatitis B. J. Hepatol. 1997;26:503–507. doi: 10.1016/s0168-8278(97)80414-x. [DOI] [PubMed] [Google Scholar]
- 43.Thursz M.R. Association between an MHC class II allele and clearance of hepatitis B virus in the Gambia. N. Engl. J. Med. 1995;332:1065–1069. doi: 10.1056/NEJM199504203321604. [DOI] [PubMed] [Google Scholar]
- 44.Thio C.L. Class II HLA alleles and hepatitis B virus persistence in African Americans. J. Infect. Dis. 1999;179:1004–1006. doi: 10.1086/314684. [DOI] [PubMed] [Google Scholar]
- 45.Thursz M. Influence of MHC class II genotype on outcome of infection with hepatitis C virus. The HENCORE group. Hepatitis C European Network for Cooperative Research. Lancet. 1999;354:2119–2124. doi: 10.1016/s0140-6736(99)91443-5. [DOI] [PubMed] [Google Scholar]
- 46.Minton E.J. Association between MHC class II alleles and clearance of circulating hepatitis C virus. Members of the Trent Hepatitis C Virus Study Group. J. Infect. Dis. 1998;178:39–44. doi: 10.1086/515599. [DOI] [PubMed] [Google Scholar]
- 47.Alric L. Genes of the major histocompatibility complex class II influence the outcome of hepatitis C virus infection. Gastroenterology. 1997;113:1675–1681. doi: 10.1053/gast.1997.v113.pm9352872. [DOI] [PubMed] [Google Scholar]
- 48.Cramp M.E. Association between HLA class II genotype and spontaneous clearance of hepatitis C viraemia. J. Hepatol. 1998;29:207–213. doi: 10.1016/s0168-8278(98)80005-6. [DOI] [PubMed] [Google Scholar]
- 49.Kuzushita N. Influence of HLA haplotypes on the clinical courses of individuals infected with hepatitis C virus. Hepatology. 1998;27:240–244. doi: 10.1002/hep.510270136. [DOI] [PubMed] [Google Scholar]
- 50.Aikawa T. HLA DRB1 and DQB1 alleles and haplotypes influencing the progression of hepatitis C. J. Med. Virol. 1996;49:274–278. doi: 10.1002/(SICI)1096-9071(199608)49:4<274::AID-JMV3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 51.Sequencing Consortium, M.H.C. Complete sequencing and gene map of a human major histocompatibility complex (MHC) Nature. 1999;401:921–923. doi: 10.1038/44853. [DOI] [PubMed] [Google Scholar]
- 52.Monteyne P. The infection of mouse by Theiler's virus: from genetics to immunology. Immunol. Rev. 1997;159:163–176. doi: 10.1111/j.1600-065x.1997.tb01014.x. [DOI] [PubMed] [Google Scholar]
- 53.Azoulay A. FVB mice transgenic for the H-2Db gene become resistant to persistent infection by Theiler's virus. J. Virol. 1994;68:4049–4052. doi: 10.1128/jvi.68.6.4049-4052.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bureau J.F. Mapping loci influencing the persistence of Theiler's virus in the murine central nervous system. Nat. Genet. 1993;5:87–91. doi: 10.1038/ng0993-87. [DOI] [PubMed] [Google Scholar]
- 55.Bihl F. Two loci, Tmevp2 and Tmevp3, located on the telomeric region of chromosome 10, control the persistence of Theiler's virus in the central nervous system of mice. Genetics. 1999;152:385–392. doi: 10.1093/genetics/152.1.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vigneau S. Tmevpg1, a candidate gene for the control of Theiler's virus persistence, could be implicated in the regulation of gamma interferon. J. Virol. 2003;77:5632–5638. doi: 10.1128/JVI.77.10.5632-5638.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fortin A. Recombinant congenic strains derived from A/J and C57BL/6J: a tool for genetic dissection of complex traits. Genomics. 2001;74:21–35. doi: 10.1006/geno.2001.6528. [DOI] [PubMed] [Google Scholar]
- 58.Nadeau J.H. Analysing complex genetic traits with chromosome substitution strains. Nat. Genet. 2000;24:221–225. doi: 10.1038/73427. [DOI] [PubMed] [Google Scholar]
- 59.Mott R., Flint J. Simultaneous detection and fine mapping of quantitative trait loci in mice using heterogeneous stocks. Genetics. 2002;160:1609–1618. doi: 10.1093/genetics/160.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lipshutz R.J. High density synthetic oligonucleotide arrays. Nat. Genet. 1999;21:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- 61.Schena M. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 62.Wade C.M. The mosaic structure of variation in the laboratory mouse genome. Nature. 2002;420:574–578. doi: 10.1038/nature01252. [DOI] [PubMed] [Google Scholar]
- 63.Guenet J.L., Bonhomme F. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet. 2003;19:24–31. doi: 10.1016/s0168-9525(02)00007-0. [DOI] [PubMed] [Google Scholar]
- 64.Balling R. ENU mutagenesis: analyzing gene function in mice. Annu. Rev. Genomics Hum. Genet. 2001;2:463–492. doi: 10.1146/annurev.genom.2.1.463. [DOI] [PubMed] [Google Scholar]
- 65.Barten R. Divergent and convergent evolution of NK-cell receptors. Trends Immunol. 2001;22:52–57. doi: 10.1016/s1471-4906(00)01802-0. [DOI] [PubMed] [Google Scholar]
- 66.Bruggeman C.A. Does cytomegalovirus play a role in atherosclerosis? Herpes. 2000;7:51–54. [PubMed] [Google Scholar]
- 67.Ramsingh A.I. Coxsackieviruses and diabetes. Bioessays. 1997;19:793–800. doi: 10.1002/bies.950190909. [DOI] [PubMed] [Google Scholar]
- 68.Hill S.L., Rose N.R. The transition from viral to autoimmune myocarditis. Autoimmunity. 2001;34:169–176. doi: 10.3109/08916930109007381. [DOI] [PubMed] [Google Scholar]
- 69.Glass W.G. Mouse hepatitis virus infection of the central nervous system: chemokine-mediated regulation of host defense and disease. Viral Immunol. 2002;15:261–272. doi: 10.1089/08828240260066215. [DOI] [PubMed] [Google Scholar]
- 70.Garred P. Susceptibility to HIV infection and progression of AIDS in relation to variant alleles of mannose-binding lectin. Lancet. 1997;349:236–240. doi: 10.1016/S0140-6736(96)08440-1. [DOI] [PubMed] [Google Scholar]
- 71.Shin H.D. Genetic restriction of HIV-1 pathogenesis to AIDS by promoter alleles of IL10. Proc. Natl. Acad. Sci. U. S. A. 2000;97:14467–14472. doi: 10.1073/pnas.97.26.14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thomas H.C. Mutation of gene of mannose-binding protein associated with chronic hepatitis B viral infection. Lancet. 1996;348:1417–1419. doi: 10.1016/s0140-6736(96)05409-8. [DOI] [PubMed] [Google Scholar]
- 73.Bellamy R. Tuberculosis and chronic hepatitis B virus infection in Africans and variation in the vitamin D receptor gene. J. Infect. Dis. 1999;179:721–724. doi: 10.1086/314614. [DOI] [PubMed] [Google Scholar]
- 74.Coffey A.J. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat. Genet. 1998;20:129–135. doi: 10.1038/2424. [DOI] [PubMed] [Google Scholar]
- 75.Sayos J. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395:462–469. doi: 10.1038/26683. [DOI] [PubMed] [Google Scholar]
- 76.Best S. Endogenous retroviruses and the evolution of resistance to retroviral infection. Trends Microbiol. 1997;5:313–318. doi: 10.1016/S0966-842X(97)01086-X. [DOI] [PubMed] [Google Scholar]
- 77.Kitagawa M. A gene therapy model for retrovirus-induced disease with a viral env gene: expression-dependent resistance in immunosuppressed hosts. Leukemia. 2001;15:1779–1784. doi: 10.1038/sj.leu.2402279. [DOI] [PubMed] [Google Scholar]
- 78.Lilly F. Susceptibility to two strains of Friend leukemia virus in mice. Science. 1967;155:461–462. doi: 10.1126/science.155.3761.461. [DOI] [PubMed] [Google Scholar]
- 79.Odaka T., Yamamoto T. Inheritance of susceptibility to Friend mouse leukemia virus. 11. Spleen foci method applied to test the susceptibility of crossbred progeny between a sensitive and a resistant strain. Jpn. J. Exp. Med. 1965;35:311–314. [PubMed] [Google Scholar]
- 80.Best S. Positional cloning of the mouse retrovirus restriction gene Fv1. Nature. 1996;382:826–829. doi: 10.1038/382826a0. [DOI] [PubMed] [Google Scholar]
- 81.DesGroseillers L., Jolicoeur P. Physical mapping of the Fv-1 tropism host range determinant of BALB/c murine leukemia viruses. J. Virol. 1983;48:685–696. doi: 10.1128/jvi.48.3.685-696.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Towers G. A conserved mechanism of retrovirus restriction in mammals. Proc. Natl. Acad. Sci. U. S. A. 2000;97:12295–12299. doi: 10.1073/pnas.200286297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cowan S. Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11914–11919. doi: 10.1073/pnas.162299499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Persons D.A. Fv2 encodes a truncated form of the Stk receptor tyrosine kinase. Nat. Genet. 1999;23:159–165. doi: 10.1038/13787. [DOI] [PubMed] [Google Scholar]
- 85.Cerwenka A., Lanier L.L. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 2001;1:41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]