

Abstract
Peptide microarrays bear the potential to discover molecular recognition events on protein level, particularly in the field of molecular immunology, in a manner and with an efficiency comparable to the performance of DNA microarrays. We developed a novel peptide microarray platform for the detection of antibodies in liquid samples. The system comprises site-specific solution phase coupling of biotinylated peptides to NeutrAvidin, localized microdispensing of peptide–NeutrAvidin conjugates onto activated glass slides and a fluorescence immuno sandwich assay format for antibody capture and detection. Our work includes synthetic peptides deduced from amino acid sequences of immunodominant linear epitopes, such as the T7 phage capsid protein, Herpes simplex virus glycoprotein D, c-myc protein and three domains of the Human coronavirus 229E polymerase polyprotein. We demonstrate that our method produces peptide arrays with excellent spot morphology which are capable of specific and sensitive detection of monoclonal antibodies from fluid samples.
Keywords: Antibody diagnostics, Fluorescence immuno assay, Linear epitope, Peptide microarray, Site-specific immobilization
1. Introduction
Miniaturization of standard analytical techniques is a prominent task of biomedical research and development. Special attention is given to microarray technology. Although efficiency and performance of DNA microarray technology have been developed to industrial standards, comparable analytic tools on protein are not available to date. Synthetic low-molecular weight peptides have outstanding properties for use as versatile probes in chip-based analysis. They can be assembled in defined orientation and high density and fully automated synthesis makes them an economically attractive alternative to recombinant proteins.
Peptide microarrays have the potent capacity to provide proteome specific information in a fast and efficient manner compared to traditional assays, e.g. microtiter plate formats. Although promising, this technology has to date been implemented into few studies of both basic and applied research [1], [2], [3]. For one reason, peptide probes are essentially restricted to mimicking linear polypeptide structures. The utility of peptide chips is limited when complex three-dimensional conformations of the parental protein are crucial for a given biological function [4]. Also, a large part of proteome information can be indirectly accessed on the well-established DNA or RNA microarray level. Hence, the peptide chip application, i.e. content, is decisive. More practically, the complexity of peptide sequences, their heterogeneity in chemical and physical properties and the diversity of mechanisms mediating their biological function has restricted the breakthrough of peptide microarray technology, yet.
It is particularly felt that new effective control measures and good diagnostic tools are required for the rapid and reliable detection of viral infections and the monitoring of a patient's immune status. As a result of permanent antigen contact, the human immune system comprises billions of B-lymphocytes. The enormous spectrum of antigen specificities is determined by the amino acid composition and the structure of each antigen receptor, i.e. antibody. Encoded by less than 200 gene segments, the diversity of the immunoglobulin repertoire is the result of a complex genetic mechanism, somatic recombination, leading to random combination of immunoglobulin gene segments and the expression of highly variable proteins. Almost any substance can give rise to an immune reaction with secretion of many different antibodies, each of which with a unique specificity and affinity [5]. Clearly, screening the human antibody repertoire for specific molecules is practically impossible with microarray methods working on DNA or mRNA level.
The most commonly used method is based on the detection of antigen specific antibodies from serum samples by conventional ELISA assays. This format offers sensitivity, specificity and automation, and progress was made by replacing structural and non-structural viral proteins with shorter recombinant immunodominant domains [6]. A drawback of the ELISA is the lack of parallelity, i.e., sample multiplexing, which renders the method relatively expensive. Transfer of these immunological assays from microtiter plates into microarray formats increases the frequencies of analytical processes and provides increased reliability of results. The miniaturization is also reflected in material savings and cost reduction.
Currently, peptide microarrays have come into focus for discovery oriented basic biological science as well as new diagnostic tools for clinical applications. Until now, the implementation of peptide microarrays has been reported for the quantitative evaluation of protein kinase activity [7], [8], [9] for the simultaneous detection of pathogen infections [10], [11], epitope screening experiments [12] and for protease specificity determination [13]. A couple of other studies took advantage of the well-established SPOT technique for in situ synthesis of peptide libraries on cellulose membranes. Among other applications, these arrays were used for the identification of antibody epitopes and mimotopes [14] and identification of peptides and amino acids from the antibody paratope that contribute to antigen binding [15].
The purpose of this work is to describe the development of a peptide microarray platform for specific and sensitive screening of antibodies from fluid samples. We present a novel and particularly advantageous method for printing of functional peptide arrays using site-specific pre-coupling of biotinylated synthetic peptides to NeutrAvidin (NA) and direct spotting of peptide–NA conjugates (PNAC) onto activated glass surfaces. The introduced peptide chip format is a fluorescence linked immuno sandwich assay (FLISA) and comprises six different monoclonal anti-virus and anti-phage antibodies (mAb) and their cognate peptide antigens. A schematic outline of the assay is shown in Fig. 1 . The work includes accurate optimization of the technique and we demonstrate the efficiency of the peptide microarray for antibody detection from buffered solutions and human sera.
Fig. 1.
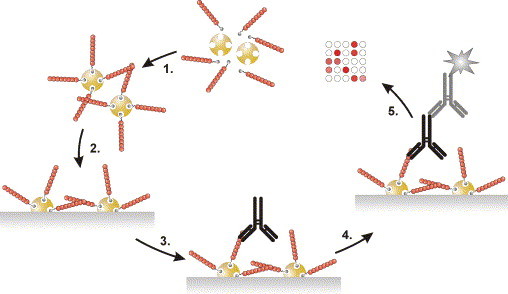
Schematic outline of the peptide microarray immuno assay. (1) Solution phase pre-coupling of biotinylated peptides to NeutrAvidin. (2) Localized microdispensing of PNACs onto activated glass surfaces. (3) Incubation with primary antibodies (i.e. sample). (4) Incubation with fluorescently labeled secondary antibody. (5) Fluorescence imaging and data analysis. Molecules are drawn out of scale.
2. Experimental Section
2.1. Buffers
Hundred and fifty millimolars of phosphate buffered saline (PBS): 137 mM sodium chloride, 2.7 mM potassium chloride, 2 mM potassium di-hydrogen phosphate, 10 mM di-sodium hydrogen phosphate (AppliChem, Darmstadt, Germany, www.applichem.de), pH 7.4. Ten millimolars of PBS: 13.7 mM sodium chloride, 0.27 mM potassium chloride, 0.2 mM potassium di-hydrogen phosphate, 1 mM di-sodium hydrogen phosphate (AppliChem), pH 7.6. PBS-T: 150 mM PBS pH 7.4 + 0.05% (v/v) Tween 20 (Sigma-Aldrich Chemie GmbH, Steinheim, Germany, www.sigmaaldrich.com). Fifty millimolars of HEPES: 50 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid), 50 mM potassium chloride (AppliChem), pH 7.5.
2.2. Antibodies
Monoclonal mouse anti-T7-Tag antibody (T7-mAb) monoclonal mouse anti-HSV-Tag antibody (HSV-mAb) and biotinylated anti-T7-Tag antibody (B-T7-mAb) was purchased from Novagen Inc. (Madison, WI, USA, www.novagen.com). Monoclonal mouse anti-Myc antibody (Myc-mAb) was a product of Oncogene Science (Cambridge, MA, USA, www.apoptosis.com). Monoclonal anti-Pol, anti-Hel and anti-Con antibodies (Pol-mAb, Hel-mAb, Con-mAb) were produced as described by Grötzinger et al. [16]. Cy5-conjugated polyclonal goat anti-mouse antibody (Cy5-GAM) was from Jackson ImmunoResearch (Cambridgeshire, UK, www.jacksonimmuno.com).
2.3. Peptides and synthesis
Amino acid building blocks (O-pentafluorophenyl esters) were products of peptides&elephants GmbH (Potsdam-Nuthetal, Germany). Rink amide AM resin, N-biotinyl-N′-Fmoc-ethylenediamine-MPB AM resin, benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), 1-hydroxybenzotriazole (HOBt), Fmoc-aminohexanoic acid (AHX) and d-biotin-N-succinimidyl ester (Biotin-oSU) came from Merck Biosciences AG (Darmstadt, Germany, www.emdbiosciences.com). Acetonitrile, acetic acid, diethyl ether, dimethyl formamide (DMF), dithiothreitol (DTT), 4-methyl morpholin (NMM), N-methyl pyrrolidone (NMP), piperidine, trifluoroacetic acid (TFA), triisopropyl silane (TIPS) and HPLC-water were bought from Carl Roth GmbH (Karlsruhe, Germany, www.carl-roth.de). 5(6)-Carboxy-tetramethylrhodamin-N-succinimidyl ester (TAMRA) came from Fluka GmbH (Seelze, Germany, www.sigmaaldrich.com).
Peptides were synthesized on a LIPS® 96 peptide synthesizer (peptides&elephants) as previously described [17]. Briefly, peptides were synthesized in resin pre-loaded MultiPep 96® microtiter plates (peptides&elephants) in 2 μmol scale using standard Fmoc strategy. The steps for each amino acid coupling were as follows: removal of temporary Fmoc protection groups by three cycles of incubation for 5 min with 20% piperidine in DMF (v/v) followed by five cycles of washing with DMF, double coupling of 8 μmol activated amino acid solution (0.2 M in NMP) for 45 min and removal of excess amino acids by five cycles of washing with DMF. For fluorescence labeling of peptide 7, 5(6)-carboxy-tetramethylrhodamin-N-succinimidyl ester (TAMRA) was coupled to the ɛ-amino group of the terminal lysine after selective removal of the Mtt-protection group by five repeats of washing in 100 μl 2% TIPS, 2% TFA (v/v) in DCM. A solution of TAMRA/PyBOP/NMM (molar ratios 1/1/2) in DMF was added to the resin with two-fold molar excess of TAMRA to the peptide and incubated for 12 h at room temperature. The coupling was done in duplicate. The resin was washed in DMF till colorlessness of the solvent, five times in DCM and dried. Essentially the same protocol was used for coupling of the aminohexane spacer (AHX). For N-terminal modification of peptides with biotin-OSu, 8 μmol of the respective reagent (0.2 M in NMP) was mixed with 16 μmol HOBt (0.4 M in NMP). The coupling was done in two extended cycles with 12 h each. Products were washed five times with DMF and additional seven times with DCM. Permanent protection groups were removed and peptides were released from the resin by five cycles of incubation with 150 μl 91% TFA, 4% TIPS, 3% DTT and 2% H2O (v/v) for 2.5 h. Peptides were lyophilized, re-solved in 100 μl TFA and precipitated by addition of 1 ml ice-cold hexane–diethylether solution (50/50). After centrifugation, the supernatants were removed and the peptides were dried at room temperature. Finally, peptides were dissolved in a mixture of 50% H2O, 25% acetonitrile and 25% acetic acid, analyzed by mass spectroscopy (Finnigan Surveyor MSQ Plus, Thermo Finnigan, Bremen, Germany, www.thermofinnigan.com) and lyophilized. The peptides used in this work are listed in Table 1 and Fig. 2 . Peptide stock solutions were prepared in 95% H2O, 4.9% acetonitrile and 0.1% acetic acid.
Table 1.
Peptides used in this work
| No. | Name | Origin | Sequence (N → C)a | pI/GRAVYb |
|---|---|---|---|---|
| 1 | T7-Tag | T7 phage minor capsid protein | MASMTGGQQMGTN | 5.28/−0.492 |
| 2 | HSV-Tag | Herpes simplex virus glycoprotein D | TQPELAPEDPEDS | 3.39/−1.669 |
| 3 | Myc | c-myc protein | EEQKLISEEDLLR | 4.25/−1.100 |
| 4 | Pol | DKDDAFYIVKRCI | 6.03/−0.292 | |
| 5 | Hel | Human coronavirus polymerase polyprotein | IVFTDDKLSNMRI | 5.96/+0.100 |
| 6 | Con | NKTSLPTNIAFEL | 6.00/−0.115 | |
| 7 | TAMRA | Random sequence | K*ELPDPQAEDEPS | n.d. |
Fig. 2.
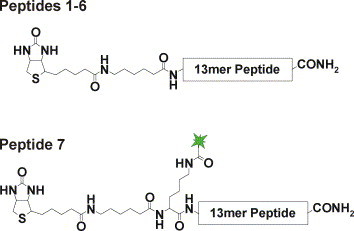
Chemical structure of synthetic peptide probes used in this work.
2.4. Optimization of solution phase coupling of biotinylated peptides to NeutrAvidin
NeutrAvidin (Perbio Science Deutschland GmbH, Bonn, Germany, www.perbio.com) was labeled with Dy-633 NHS-ester (Dyomics GmbH, Jena, Germany, www.dyomics.com) according to the manufacturer's instructions. Aliquots of labeled NeutrAvidin in HPLC-grade H2O (2 mg ml−1 final concentration) were incubated with 2–100-fold molar excess of TAMRA-labeled and biotinylated peptide (No. 7) at +4 °C over night. Peptide–NA conjugates were separated from excess peptide by extensive washing on Ultrafree-0.5 centrifugal filter devices with 30 kDa NMWL (Millipore GmbH, Schwalbach, Germany). Molar peptide/NA ratios (PNR) conjugates in the retentate were determined at 555 and 637 nm wavelength in a Specord 200 photometer (Analytik Jena AG, Jena, Germany, www.analytik-jena.de) using the following equation:
with a correction factor CF of 0.22 for the absorption of Dy-633 NA at 555 nm and a degree of labeling (DOL) of 0.57.
2.5. Evaluation of protein concentration, chip substrate, spotting buffer and surface coupling kinetics
Dy-633 labeled NA was diluted to 0.1–1 mg ml−1 in 10 mM PBS pH 7.6 [20] or 50 mM HEPES pH 7.5 with supplementations of 0.5–1% trehalose or 5–10% glycerol (AppliChem). Solutions were spotted with the sciFLEX Arrayer piezoelectric dispenser (Scienion AG, Berlin, Germany, www.scienion.de) at dew point temperature onto amine and aldehyde coated glass slides (Genetix GmbH, München, Deutschland, www.genetix.com) and epoxy slides (Eppendorf GmbH, Hamburg, Deutschland, www.eppendorf.com). Printed slides were incubated in moist chambers for 0–96 h at room temperature. The surface coupling of NA was stopped by drying the slides for 10 min at 30 °C and subsequent blocking with 10% (w/v) skim milk (AppliChem) in 150 mM PBS-T pH 7.4 for 1 h at room temperature with slow rotation. Slides were rinsed with deionized water and dried in nitrogen. Surface immobilized NA was measured with the 428 ArrayScanner (Affymetrix, Santa Clara, CA, USA, www.affymetrix.com) and quantified using the ImaGene V5.5 software (Biodiscovery, El Segundo, CA, USA, www.biodiscovery.com). The fluorescence intensity of the spots was related to the density of the NA-layer on the chip surface using a calibration curve prepared with defined concentrations of the same Dy-633 NA conjugate.
2.6. Long-term stability and functionality of NeutrAvidin immobilizates
Dy-633 labeled NA was printed in 0.4 mg ml−1 concentration (10 mM PBS pH 7.6) with the sciFLEX Arrayer at dew point temperature onto amine and aldehyde coated glass slides (Genetix GmbH). Slides were incubated in moist chambers for 24 h at room temperature. Excess NA was removed by washing for 1 h in PBS-T + 10% skim milk. Next, the slides were immersed in buffered saline pH 7.4 and incubated at 37 °C. After periods of 0–144 h, two slides of each surface type were withdrawn and probed with biotinylated and Dy-547 labeled anti-T7-mAb (2 μg ml−1 in PBS-T, 2 h, room temperature). The slides were washed three times in PBS-T, rinsed in deionized water and dried in nitrogen. Surface bound NA and biotin-mAb was measured in the Cy5 and Cy3 channel of the Affymetrix 428 ArrayScanner and quantified using the ImaGene V5.5 software. The amount of immobilized NA and mAb at the point of time t i is expressed as a percentage of initial relative fluorescence (RFU0) to the time t 0 on the respective surface:
2.7. Antibody detection assays
Peptide–NA conjugates of peptides 1–6 were made by incubation of 2 mg ml−1 NA with five-fold molar excess of peptide for 12 h at +4 °C. Without further purification, PNACs were spotted with the sciFLEX Arrayer in 0.4 mg ml−1 concentration in 10 mM PBS onto amine coated glass slides at dew point temperature. Printed slides were incubated in moist chambers for 24 h at room temperature, dried and blocked by 1 h washing in PBS-T + 10% skim milk with slow rotation. Dilutions of the six monoclonal antibodies (1 μg ml−1 final concentration each) were spiked into 150 mM PBS-T pH 7.4 + 3% skim milk or into human serum (Sigma-Aldrich Chemie GmbH), 1:50 diluted in PBS-T. Chip incubation was 3 h for PBS samples and over night at +4 °C for serum samples. After washing, incubation with 10 μg ml−1 Cy5-GAM in PBS-T + 3% skim milk for 2 h at room temperature was maintained. A second cycle of stringent washing, rinsing in deionized water and drying in nitrogen followed. The slides were scanned and analyzed. Fluorescence signals were evaluated by determination of the ratio between PNAC spots and NA reference spots (signal/background ratio, SBR):
The limit of detection (LOD) for each type of PNAC was determined using slides incubated only with PBS or diluted serum and Cy5-GAM:
3. Results and discussion
Aim of our work is the development of peptide microarrays for the specific and sensitive detection of antibodies from fluid samples, i.e. human serum. Our approach is based on synthetic peptide probes with biotin linkers for site-specific immobilization via biotin–avidin affinity on activated glass substrates. When full protein sequences are reduced to short peptides, e.g. an immunodominant linear epitope, a high percentage of the remaining functionalities can contribute to the peptide–protein interaction and, consequently, modification of these functions reduces the native affinity. Immobilization strategies are required which react orthogonal to multivalent amino acid residues. Site-specific coupling of peptides to solid supports was reported using covalent methods [8], [9], [10], [11], [21] and taking advantage of the biotin–avidin interaction [21]. To our experience, however, the biotin–avidin affinity binding, although well characterized and frequently used, is unexpectedly challenging on the chip surface with nanoliter volumes of peptide solutions. In particular, the spotting buffer has to be optimized for individual physico-chemical properties of peptides and steric interference on the surface can drastically reduce the coupling efficiency. To address this problem, we established a novel strategy using solution phase pre-coupling of biotinylated peptides to NeutrAvidin and direct spotting of conjugates onto activated surfaces. This method is particularly advantageous, since the peptides contribute to less than 10% of the mass of conjugates and therefore, the physico-chemical properties of the PNACs are almost exclusively determined by the NeutrAvidin, uniforming the handling of entire peptide libraries. To optimize the peptide/protein ratio for the pre-coupling, we tested the NA saturation using different molar excesses of fluorescence labeled biotinylated peptide with constant protein concentration of 2 mg ml−1 and over night incubation. The results are presented in Fig. 3 . The diagram shows the inset molar peptide excess in relation to the resulting NA saturation. A five-fold molar peptide excess almost leads to saturation of the NA binding capacity of four biotin molecules. Higher excesses further drive the coupling to saturation, but reduce the efficiency of the reaction. The results underline the reliability of solution phase coupling of biotinylated molecules to avidin. Using the presented reaction conditions, a molar peptide/protein ratio of 5:1 is suggested for efficient pre-coupling of biotinylated peptides to NA. The small amount of free peptide in solution after coupling eliminates the need for separation of unbound molecules prior to spotting. In addition, higher proportions of peptide solvent can drastically reduce the binding of NA to the chip surface (data not shown).
Fig. 3.
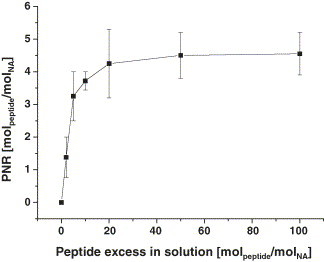
Optimization of the solution phase coupling of biotinylated peptides to NeutrAvidin. TAMRA labeled peptide and Dy-633 labeled NA were incubated in the specified molar ratios for 12 h at +4 °C. The PNR was photometrically determined after removal of excess peptide in solution. Mean values of duplicate determination.
In principle, the immobilization of PNACs on glass surfaces can occur covalently or by simple adsorption. Various types of surface chemistries can be considered, with aldehyde and epoxy surfaces for covalent and amino surfaces for adsorptive coupling being the most popular, since pre-fabricated slides are readily available. We tested the specified chip substrates in terms of suitability for immobilization of PNACs. Simultaneously, the optimum protein concentration for the microdispensing and a suitable spotting buffer was to be determined. The results of these experiments are depicted in Fig. 4 . Fig. 4a shows images of the surfaces after printing of different PNAC concentrations and removal of unbound molecules, the diagram in Fig. 4b displays the relative fluorescence intensity of the spots. Fig. 4c shows the reaction course of adsorptive NA immobilization onto amino silane for a selection of different buffer compositions. Fluorescence intensities are related to the density of NA molecules per square millimeter. The physisorption of NA on amino silane surfaces leads to the highest density of the protein layer with simultaneously good spot morphology and low spot-to-spot and chip-to-chip deviations. The aldehyde and epoxy surfaces exhibit lower binding capacities and result in unfavorable spot morphology, however, surface tailored spotting buffers may improve this effect. Increasing the spotted protein concentration from 0.1 to 0.4 mg ml−1 results in a strong gain in surface density while this effect is significantly lower with further increase from 0.4 to 0.6 mg ml−1. 0.8 and 1.0 mg ml−1 protein concentration lead to spotting artefacts, e.g. comets and smearings, and are not considered for data evaluation. For the adsorption of NA (0.4 mg ml−1) to amino silane, highest protein densities of approximately 1010 molecules/mm2 are achieved using pure 10 mM PBS buffer for spotting. The reaction reaches saturation state after 18–24 h. All buffer supplements lead to a slower reaction rate and 20–50% reduced protein binding. Our findings are consistent with previous reports that adsorptive binding of proteins leads to the potentially highest density of surface layers, but this reaction can be relatively slow due to the rate of protein arrangement and partial unfolding on the surface [22]. The current protein density using 0.4 mg ml−1 NA corresponds to approximately 30% of the theoretical monolayer, assuming an idealized globular NA molecule with a footprintsize of 20 nm2 [23]. With an average of 1.5 solution exposed binding sites per NA molecule, the surface contains at least 30 fmol peptide probes per square millimeter readily available for antibody capture.
Fig. 4.
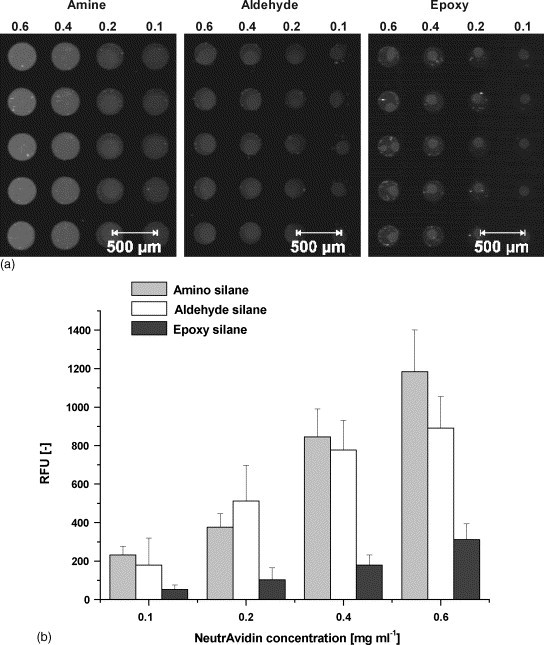
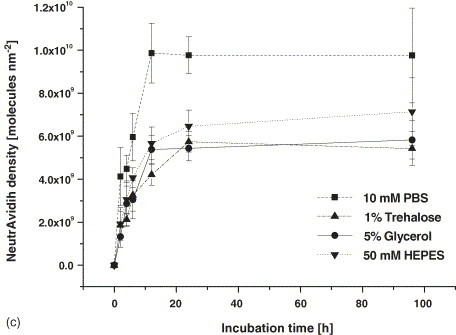
Evaluation of chip surface, spotting buffer and coupling kinetic. (a) Fluorescence images of Dy-633 NA immobilized on amine, aldehyde and epoxy coated glass slides in grey scale. NA was spotted in the specified concentrations (mg ml−1) in 10 mM PBS pH 7.6 and incubated for 24 h. Unbound NA was removed by washing for 1 h in PBS-T + 10% skim milk. Fluorescence images were taken with the Affymetrix 428 ArrayScanner at 5 dB voltage gain. (b) Quantitative spot analysis. Relative fluorescence units (RFU) represent the mean of 120 Spots in 12 arrays per slide type and NA concentration. (c) Time course of adsorptive surface coupling of NA on amine coated glass slides. NA was spotted in 0.4 mg ml−1 concentration in the specified buffers. Fluorescence intensities of spots (mean of 80 spots in 8 arrays per buffer and time) were related to protein density via a calibration curve.
Protein adsorbates, however, are less stable than covalently linked immobilizates. Continued desorption of PNACs in the course of microarray processing, e.g. incubation and washing, can cause significant decrease of the entire assay sensitivity as a result of reduced binding capacity in the spots and simultaneous competition of antibodies with free peptide ligands in solution. To address this issue, we determined the rate of desorption of NA immobilizates from amine coated glass slides during incubation in physiological medium and compared the results with the desorption from aldehyde coated slides. Fluorescently labeled NA was spotted in 0.4 mg ml−1 onto both amine and aldehyde coated glass slides and incubated in buffered solution at 37 °C for 0–144 h. At specified points of time, the remaining NA density in the spots as well as the activity of the adsorbates, i.e. the biotin binding capacity, was determined. Fig. 5 shows the outcome of the investigation. The diagram displays the percentage of surface detected NA amount and activity in relation to the period of incubation. On either surface, a considerable desorption of NA is observed during the first 24 h of incubation, associated with a comparable loss of biotin binding capacity. Further incubation leads to marginal desorption. The protein density adjusts at about 15–20% of the initial layer. Hence, protein adsorbates on amino silane are sufficiently stable for microarray applications which normally involve physiological conditions and short terms of incubation and washing. Interestingly, immobilizates on aldehyde surfaces that have the capacity for covalent binding desorb in a highly similar way to the adsorbates. Most likely, reversion of initially formed Schiff's base intermediates between primary amines of NA and aldehyde groups on the surface results in release of the protein. Mild reduction of the Schiff's Base, however, yields a stable covalent bond and may eliminate the observed effect.
Fig. 5.
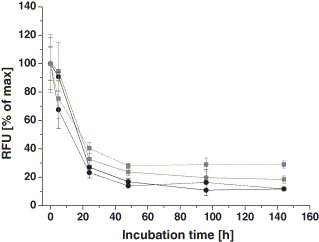
Long-term stability and activity of NA immobilizates. Dy-633 labeled NA was spotted in 0.4 mg ml−1 concentration onto amine (solid lines) and aldehyde (dotted lines) coated glass slides. Printed slides were incubated in buffered saline pH 7.4 for the specified times at 37 °C. The biotin binding capacity was afterwards tested by incubation of slides with 2 μg ml−1 Dy-547 labeled and biotinylated anti-T7 mAb for 2 h at room temperature. Black lines and circles represent the relative amount of surface immobilized NA, grey lines and squares the amount of captured biotinylated mAb. Data are mean values of 50 spots in 2 arrays.
To prove the suitability of our peptide microarray concept for antibody detection, we spiked the six model monoclonal antibodies into PBS buffer and diluted human serum with 1 μg ml−1 final concentration of each mAb. The solutions were analyzed on peptide microarrays using 3 h incubation at room temperature for the PBS sample and over night incubation at +4 °C for diluted serum. The results of these experiments are displayed in Fig. 6 . Fig. 6a shows an exemplary fluorescence image of the peptide microarray after antibody incubation in grey scale, Fig. 6b the quantitative evaluation of signal/background ratios. All antibodies are successfully detected well above the detection limit. The peptide microarray exhibits clearly defined and very homogeneous spots. Signal-to-background ratios of the antibody detection range from 36 (T7-mAb) to 125 (Hel-mAb). Importantly, no unspecific signals are observed when the arrays are selectively incubated with only one type of mAb (data not shown). Our results confirm the benefits of utilizing conjugates of peptide probes and a carrier protein for the chip production. The peptide specific physico-chemical characteristics, e.g. pI and hydrophobicity, are leveled by those of NeutrAvidin, leading to unified properties of the spotted molecules and clearly improved spot morphology, thus paving the way for standardized protocols. The differences in quantitative detection are presumably the outcome of unequal antibody affinities to the cognate peptide antigens. In addition, the native mAb affinity can be considerably altered by the surface presentation of the peptide antigen, e.g. the accessibility of the binding site and the immobilization direction. This is an important issue of our current research and optimization efforts.
Fig. 6.
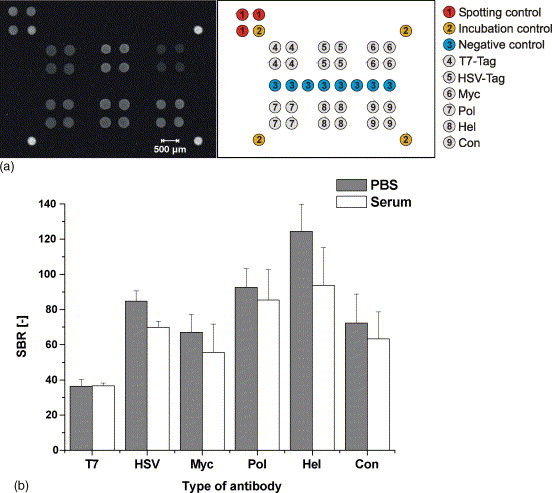
Exemplary fluorescence image and data interpretation of antibody detection with the peptide microarray. (a) PNACs of the six peptide probes, spotting control (Dy-633 labeled NA) and NA reference spots were spotted in 0.4 mg ml−1 concentration, the incubation control (mouse IgG) in 0.2 mg ml−1 concentration in 10 mM PBS pH 7.6 onto amine coated glass slides. The slides were incubated 3 h at room temperature (PBS) or over night at +4 °C (1:50 diluted human serum) with a mixture of mAbs (1 μg ml−1 each) and 2 h with Cy5-GAM (10 μg ml−1) in PBS-T at room temperature. Fluorescence images were taken with the Affymetrix 428 ArrayScanner at 25 dB PMT adjustment. The scheme in the right panel displays the chip layout. (b) Quantitative fluorescence data interpretation. Signal/background ratios represent mean values of 32 spots in 8 arrays. The limit of detection was 2.7 for Myc-mAb, 2.5 for Pol-mAb and below 1.5 for other mAbs.
4. Conclusions
Peptide microarrays provide an attractive approach for highly parallel antibody profiling of serum samples. In this work, we have described a novel method for site-specific immobilization of peptide probes on activated glass slides taking advantage of the fast and efficient solution phase pre-coupling of biotinylated peptides to NeutrAvidin followed by localized microdispensing of peptide–NeutrAvidin conjugates. Our method produces microarrays with excellent spot morphology and eliminates the need for laborious, expensive and often technical complex slide surface preparation. The technique of site-specific pre-coupling of peptides and NeutrAvidin is particularly advantageous to unify the physico-chemical properties of heterogeneous peptide libraries for standardized spotting and chip production methods. We conclude that our combination of fully automated peptide library synthesis and the utilization of peptides as variable probes in microarray applications shows good promise for implementation in clinical immune diagnostics.
Acknowledgement
This work was supported by the German Federal Ministry of Education and Research (BMBF) Grant No. 03|1313A and B within the framework of the InnoRegio BioHyTec Berlin-Brandenburg initiative.
Biographies
Heiko Andresen was born in 1975 in Hamburg, Germany. He studied biotechnology at the Technical University Berlin, Germany and completed parts of his studies at the Royal Institute of Technology, Stockholm, Sweden. The subject of his diploma thesis was the development of an electrochemical DNA biosensor for the detection of bacterial pathogens. After his graduation as civil engineer in 2003, he began his PhD thesis at the Fraunhofer Institute of Biomedical Engineering in Potsdam, Germany.
Carsten Grötzinger was born in 1964 in Berlin, Germany. He studied biochemistry and molecular biology at the Humboldt University Berlin, Germany. The subject of his diploma work at the Institute of Virology of the Charité Medical School was the characterization of HIV antibody epitopes. After research for his doctoral thesis at the Institute of Virology and Immune Biology of the University of Würzburg, Germany on molecular biology of human coronaviruses he returned to Berlin. His work as laboratory head at the Charité Biomedical Research Center is currently focussing on the development of fluorescent peptide conjugates for diagnosis and therapy.
References
- 1.Reimer U., Reineke U., Schneider-Mergener J. Peptide arrays: from macro to micro. Curr. Opin. Biotechnol. 2002;13:315–320. doi: 10.1016/s0958-1669(02)00339-7. [DOI] [PubMed] [Google Scholar]
- 2.Min D.H., Mrksich M. Peptide arrays: towards routine implementation. Curr. Opin. Chem. Biol. 2004;8:554–558. doi: 10.1016/j.cbpa.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Panicker R.C., Huang X., Yao S.Q. Recent advances in peptide-based microarray technologies. Comb. Chem. High Throughput Screen. 2004;7:547–556. doi: 10.2174/1386207043328517. [DOI] [PubMed] [Google Scholar]
- 4.Frank R. High-density synthetic peptide microarrays: emerging tools for functional genomics and proteomics. Comb. Chem. High Throughput Screen. 2002;5:429–440. doi: 10.2174/1386207023330165. [DOI] [PubMed] [Google Scholar]
- 5.Janeway C.A., Travers P., Walport M., Shlomchik M. 5th ed. Garland Publishing; New York: 2001. Immunobiology. [Google Scholar]
- 6.Nakagiri I., Ichihara K. ELISA for anti-HCV antibody employing a shorter synthetic core region peptide. J. Virol. Meth. 1995;52:195–207. doi: 10.1016/0166-0934(94)00164-c. [DOI] [PubMed] [Google Scholar]
- 7.MacBeath G., Schreiber S.L. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 8.Houseman B.T., Huh J.H., Kron S.J., Mrksich M. Peptide chips for the quantitative evaluation of protein kinase activity. Nat. Biotechnol. 2002;20:270–274. doi: 10.1038/nbt0302-270. [DOI] [PubMed] [Google Scholar]
- 9.Lesaicherre M.L., Uttamchandani M., Chen G.Y., Yao S.Q. Antibody-based fluorescence detection of kinase activity on a peptide array. Bioorg. Med. Chem. Lett. 2002;12:2085–2088. doi: 10.1016/s0960-894x(02)00378-5. [DOI] [PubMed] [Google Scholar]
- 10.Melnyk O., Duburcq X., Olivier C., Urbes F., Auriault C., Gras-Masse H. Peptide arrays for highly sensitive and specific antibody-binding fluorescence assays. Bioconjug. Chem. 2002;13:713–720. doi: 10.1021/bc015584o. [DOI] [PubMed] [Google Scholar]
- 11.Duburcq X., Olivier C., Malingue F., Desmet R., Bouzidi A., Zhou F., Auriault C., Gras-Masse H., Melnyk O. Peptide–protein microarrays for the simultaneous detection of pathogen infections. Bioconjug. Chem. 2004;15:307–316. doi: 10.1021/bc034226d. [DOI] [PubMed] [Google Scholar]
- 12.Pellois J.P., Zhou X., Srivannavit O., Zhou T., Gulari E., Gao X. Individually addressable parallel peptide synthesis on microchips. Nat. Biotechnol. 2002;20:922–926. doi: 10.1038/nbt723. [DOI] [PubMed] [Google Scholar]
- 13.Salisbury C.M., Maly D.J., Ellman J.A. Peptide microarrays for the determination of protease substrate specificity. J. Am. Chem. Soc. 2002;124:14868–14870. doi: 10.1021/ja027477q. [DOI] [PubMed] [Google Scholar]
- 14.Reineke U., Ivascu C., Schlief M., Landgraf C., Gericke S., Zahn G., Herzel H., Volkmer-Engert R., Schneider-Mergener J. Identification of distinct antibody epitopes and mimotopes from a peptide array of 5520 randomly generated sequences. J. Immunol. Meth. 2002;267:37–51. doi: 10.1016/s0022-1759(02)00139-4. [DOI] [PubMed] [Google Scholar]
- 15.Laune D., Molina F., Ferrieres G., Villard S., Bes C., Rieunier F., Chardes T., Granier C. Application of the Spot method to the identification of peptides and amino acids from the antibody paratope that contribute to antigen binding. J. Immunol. Meth. 2002;267:53–70. doi: 10.1016/s0022-1759(02)00140-0. [DOI] [PubMed] [Google Scholar]
- 16.Grötzinger C., Heusipp G., Ziebuhr J., Harms U., Suss J., Siddell S.G. Characterization of a 105-kDa polypeptide encoded in gene 1 of the human coronavirus HCV 229E. Virology. 1996;222:227–235. doi: 10.1006/viro.1996.0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kreuzer O.J., Birringer M., Zarse K., Henkel J., Bier F.F., Thürk M., Zinsser W. In: Novel Tools and Applications in Parallel Peptide Synthesis. Peptide Revolution: Genomics, Proteomics & Therapeutics. Chorev M., Sawyer T.K., editors. American Peptide Society; 2003. [Google Scholar]
- 18.Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A. In: Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook. Walker John M., editor. Humana Press; 2005. [Google Scholar]
- 19.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 20.Delehanty J.B., Ligler F.S. Method for printing functional protein microarrays. Biotechniques. 2003;34:380–385. doi: 10.2144/03342mt02. [DOI] [PubMed] [Google Scholar]
- 21.Lesaicherre M.L., Uttamchandani M., Chen G.Y., Yao S.Q. Developing site-specific immobilization strategies of peptides in a microarray. Bioorg. Med. Chem. Lett. 2002;12:2079–2083. doi: 10.1016/s0960-894x(02)00379-7. [DOI] [PubMed] [Google Scholar]
- 22.Welle A., Grunze M., Tur D. Plasma protein adsorption and platelet adhesion on poly[bis(trifluoroethoxy)phosphazene] and reference material surfaces. J. Colloid Interf. Sci. 1998;197:263–274. doi: 10.1006/jcis.1997.5238. [DOI] [PubMed] [Google Scholar]
- 23.Cheek B.J., Steel A.B., Torres M.P., Yu Y.Y., Yang H. Chemiluminescence detection for hybridization assays on the flow-thru chip, a three-dimensional microchannel biochip. Anal. Chem. 2001;73:5777–5783. doi: 10.1021/ac0108616. [DOI] [PubMed] [Google Scholar]