

Abstract
Breda virus (BRV), a member of the genus torovirus, is an established etiological agent of diarrhea of cattle, which is found as two separate serotypes, BRV-1 and BRV-2. In this study, a 7.5 kb fragment of the BRV-1 genome that bracketed the genes for the structural proteins of BRV was amplified by long RT-PCR and the amplicon purified and sequenced directly. Sequence analysis revealed the presence of four open reading frames (ORF) corresponding to the peplomer (S), envelope (M), and nucleocapsid (N) genes, and an ORF for a novel 1.2 kb gene located between the M and N genes. This new gene was identical in nucleotide sequence to the hemagglutinin-esterase (HE) gene of BRV-2. With the exception of this new ORF, BRV-1 manifests 80% nucleotide sequence identity with the torovirus prototype, Berne virus (BEV) in the 7.5 kb region from the 3′ end of the genome that contains the genes for the structural proteins. A 504 base segment containing the ORF for the BRV-1 N gene was amplified by RT-PCR, and cloned into an Escherichia coli expression system. The resulting protein was purified by SDS-PAGE and used to immunize guinea pigs. Hyperimmune serum was reactive with bovine torovirus (BTV) and human torovirus (HTV) antigens. By immunoelectron microscopy, it was shown to aggregate broken but not intact torovirus particles from BTV-positive fecal specimens. By immunoblot, the hyperimmune serum reacted specifically with the 20 kD N proteins of both BTV and HTV, as well as with the expressed N protein. BRV-1 and BRV-2 immune sera from gnotobiotic calves, but not human convalescent sera from HTV-infected patients, reacted with the expressed N protein by immunoblot. These findings were applied to the design of a dot blot assay that could specifically detect BTV and HTV from fecal specimens.
Keywords: Torovirus, Breda virus, Nucleocapsid gene, Long PCR
1. Introduction
Breda virus belonging to the genus torovirus of the family Coronaviridae, order Nidovirales, was first isolated in 1982 from the stools of neonatal calves with diarrhea in Breda, IA (Woode et al., 1982). This virus was found to be morphologically similar and antigenically related to the torovirus prototype, Berne virus, which is a recognized infectious agent of horses (Weiss et al., 1983). Breda viruses are relatively pleomorphic particles that can be difficult to recognize by electron microscopy, and cannot as yet be propagated in cell culture. Hence, serological methods have generally been used for the routine identification of this agent (Brown et al., 1987, Koopmans et al., 1989). On the basis of results from hemagglutination inhibition (HI) tests, enzyme-linked immunosorbant assays (ELISA), and immunoelectron microscopy (IEM), two serotypes of BRV have been recognized. Breda virus serotype 1 (BRV-1) represents the original isolate from Iowa, and serotype 2 (BRV-2) includes a second isolate from a colostrum-deprived calf in Iowa, and an isolate from a diarrheic calf in Ohio (Woode et al., 1985).
Although the biochemical and serological properties of BRV have been well described, little has been elucidated about the molecular characteristics of this virus. To date, the 3′ non-coding region of the BRV-1 genome has been partially sequenced (Koopmans et al., 1991), and more recently, an additional 3 kb upstream of the poly (A) tail of the BRV-2 genome has been sequenced. The latter findings revealed the presence of a novel 1.2 kb hemagglutinin-esterase gene located between the M and N genes that is not present in BEV, although the nucleotide sequence at the 3′ end of this gene was shown to be highly similar to the X pseudogene of BEV (Cornelissen et al., 1997). The above findings provided a basis for the recent study by Duckmanton et al. (1998)who used RT-PCR to show that bovine torovirus is a major etiological agent of diarrhea in calves.
In the present study, an amplicon encompassing the entire 3′ end of the BRV-1 genome which encodes the viral structural proteins was obtained by long RT-PCR and sequenced. From this data, the BRV-1 N gene which codes for the nucleocapsid protein was cloned and expressed in Escherichia coli. Anti-sera to this recombinant protein were shown to be reactive with bovine and human toroviruses.
2. Materials and methods
2.1. Specimens and sera
The stool specimen from a gnotobiotic calf infected with a purified preparation of the BRV (code GC-32) and antisera to BRV-1 and BRV-2 from experimentally infected gnotobiotic calves were obtained from Dr G. Woode, Texas A&M and Dr M. Hardy, Montana State University. Bovine torovirus-positive fecal specimens from diarrheic calves, control specimens from asymptomatic calves, and rotavirus-positive specimens from diarrheic calves were obtained from the Animal Health Laboratory in Guelph, Ontario as described previously (Duckmanton et al., 1998). Patient specimens containing human torovirus and acute/convalescent paired sera from patients whose stools were diagnosed positive for HTV by electron microscopy (EM) were obtained from the Virology Laboratory at the Hospital for Sick Children, Toronto, Ontario as previously described (Duckmanton et al., 1997).
2.2. Primers
Oligonucleotide primers (synthesized by ACGT Corp., Toronto, Canada) for amplification of the 7.5 kb fragment of the BRV-1 genome encompassing the genes for the viral structural proteins were designed based on the known sequence of the BEV genome. The sense primer (5′ CTCTGGATTAATTCAGGAGGTGCCGTTGTTGTGTC 3′) was deduced from the 5′ end of the peplomer gene (EMBL accession number X52506), and the antisense primer (5′ TCTAGATGGTTACACACAGTGGAGCCAGAGGCAAG 3′) was deduced from the 3′ non-coding region upstream of the poly (A) tail (DDBJ accession number D00563). To complete the sequence of the 5′ end of the S gene, a primer derived from the 3′ end of the POL1b gene, 5′CAGCTGGTGTGAATACCTCCTCTTCGGAGGT3′ was used as the sense primer.
Oligonucleotide primers (synthesized by ACGT Corp., Toronto, Canada) for amplification of the 504 base fragment containing the ORF of the BRV-1 N gene were designed based on the sequence of the BRV-1 N gene obtained in this study. The sense primer
(5′ TCGCGGATCCATGAATTCTATGCTTAAATCCAAATG 3′), designed in the 5′ end of the N gene ORF, contained a BamHI restriction site upstream of the start codon for cloning purposes. The antisense primer (5′ CCGTGCGAGCTCGAGTATTCATTACCACCT 3′), designed in the 3′ end of the N gene ORF, contained an XhoI restriction site in place of the stop codon.
2.3. RNA extraction
As described previously (Duckmanton et al., 1998) the BRV-1 positive fecal specimen was diluted in an equal volume of phosphate-buffered saline (w/v) and clarified by repeat centrifugation at 9000 and 12 000×g for 15 min at 4°. Viral RNA was extracted from the partially purified supernatant using TRIzol Reagent (Gibco BRL, Gaithersburg, MD) according to the manufacturer’s protocol and the RNA pellets were resuspended in 10 μl of DNase free, RNase free double distilled water (5 prime 3 prime Inc., Boulder, CO) containing 10% 100 mM dithiothreitol, and 5% (v/v) 20–40 U/μl RNasin (Promega, Madison, WI). Aliquots of RNA were stored at −80°.
2.4. Long RT-PCR
The RT and PCR reactions were performed using the approach of Tellier et al., 1996a, Tellier et al., 1996b. The reaction mixtures were set up in yet another isolated room, using dedicated micropipetters and aerosol-resistant tips. Reactions were then performed in a third room designated for PCR amplification. For the RT reaction, an RNA aliquot was thawed on ice, incubated for 2 min at 65°, and chilled on ice. The RNA was added to 10 μl of the RT mixture containing 4 μl of 5× 1st Strand Synthesis Buffer (Gibco BRL), 0.5 μl RNasin (20–40 U/μl) (Promega), 1 μl of 100 mM dithiothreitol (Promega), 1 μl of a 10 mM stock solution of deoxynucleotide triphophates (dNTPs) (Pharmacia, Piscataway, NJ), 2.5 μl of a 10 μM primer stock solution, and 1 μl (200U) of Superscript II reverse transcriptase (Gibco BRL). The reaction mixture was incubated at 42° for 1 h, 1 μl of RNase H (1–4 U/μl) (Gibco BRL) and 1 μl of RNase T1 (900–3000 U/μl) (Gibco BRL) were then added, and the reaction mixture was further incubated at 37° for 20 min.
For the PCR reaction, 2 μl of the RT reaction was added to the PCR mixture containing 40 mM Tricine–KOH (pH 9.2 at 25°), 15 mM KOAc, 3.5 mM Mg(OAc)2, 75 μg/ml bovine serum albumin (10× KlenTaq PCR reaction buffer; Clontech, Palo Alto, CA), 250 μM each dNTP, 10 pmol of each primer, and 1 μl of 50× Advantage KlenTaq polymerase mix (Clontech). The total volume of the PCR reaction in thin walled PCR tubes (Stratagene, La Jolla, CA) was 50 μl. The reaction mixture was overlaid with 40 μl of mineral oil and amplified in a Robocycler thermal cycler (Stratagene, La Jolla, CA). To amplify the 7.5 kb BRV-1 genome fragment, the reaction underwent denaturation at 99° for 35 s, annealing at 67° for 30 s, and elongation steps of 68° for 8 min for the first 25 cycles, and 68° for 12 min for the last ten cycles. The same RT-PCR protocol was used to amplify the BRV-1 N gene fragment, but the cycling program was modified as follows. Denaturation at 99° for 35 s, annealing at 67° for 30 s, and elongation at 68° for 4 min for 35 cycles. Reactions were analyzed by electrophoresis on a 0.7% agarose gel, subsequently stained with ethidium bromide, and viewed under an UV transilluminator (Shielding from UV was provided by illuminating the gel tray through two Plexiglas trays to minimize photo nicking).
2.5. DNA sequencing
As described previously (Duckmanton et al., 1997, Duckmanton et al., 1998), the PCR products were excised from agarose gel, and purified using the Jetsorb system (Genomed, Frederick, MD). Purified amplicons were then sequenced directly using the protocol for direct label incorporation in the fmol DNA sequencing system (Promega) according to the manufacturer’s recommendations. Sequence data were analyzed using the computer programs GCG version 8 (Genetics Computer Group Inc., Madison, WI), and Gene Runner version 3.04 (Hastings on the Hudson, NY).
2.6. Cloning and expression
The PCR product containing the BRV N gene was purified using the Wizard PCR Preps DNA Purification System (Promega). Amplicons were then ligated to a pET28a(+) cloning vector (Novagen, Madison, WI), and transformed into INVαF′ One Shot cells (Invitrogen, Carlsbad, CA) as per the manufacturer’s recommendations. Plasmids containing the PCR insert were purified from LB broth cultures using the QIAGEN Plasmid Mini Kit (QIAGEN, Chatsworth, CA), and sequenced using the fmol DNA sequencing system (Promega) as described above. Purified clones whose inserts were intact and in frame were transformed into BL21(DE3) expression host cells (Novagen) as per the manufacturer’s recommendations. The transformed cells were grown in broth cultures to which IPTG was added to induce expression of the N protein. Time course samples containing total cell protein were removed from induced cultures at 2 and 3 h post-induction. Crude soluble and insoluble cell fractions were prepared from total cell protein cultures by the addition of 100 μg/ml of lysozyme (Pharmacia) and 1/10 volume of 1% Triton X-100 (Sigma Chemicals, St. Louis, MO) to the samples. Each sample was sonicated with a microtip to shear the DNA, and centrifuged at 12 000×g for 15 min at 4°.
The soluble and insoluble cell fractions were subjected to SDS-PAGE on a 12% resolving and 4% stacking gel. Proteins present in the SDS-PAGE gel were transferred electrophoretically to a polyvinylidene fluoride (PVDF) nylon membrane (Millipore, Bedford, MA) for 90 min at 100 V. The membranes were washed and blocked overnight in a solution of 5% skim milk in Tris-buffered saline containing 0.5% Tween-20 (TBST). The membranes were then incubated for 1 h at room temperature in a 1:3000 dilution of T7*Tag Antibody-Alkaline Phosphatase Conjugate (Novagen) in TBST. The membranes were washed and developed using one caplet of 5-bromo-4-chloro-3-indolyl phosphate-nitro blue tetrazolium (BCIP-NBT; SigmaFAST, Sigma Chemicals) dissolved in 10 ml of water. Color developed at room temperature within 5–10 min.
2.7. Preparation of guinea pig antisera to BRV-1
The purified insoluble cell fraction was subjected to preparative SDS-PAGE on a 12% resolving gel and a 4% stacking gel. The gel was stained with Coomassie brilliant blue R250. The band corresponding to the 20 kD N protein was excised and soaked in distilled water for 2 h to remove the residual destaining solution. The band was cut into small pieces, resuspended in an equal amount of distilled water (w/v), mixed 1:1 with Freund’s incomplete adjuvant and injected subcutaneously into two young adult male guinea pigs once a week for three successive weeks. The guinea pigs were then injected once with a 1:1 suspension of the purified insoluble cell fraction pellet containing the N protein with Freund’s incomplete adjuvant. Pre- and post-immunization sera, designated gpPIαN and gpHIαN respectively, were collected. The sera were heat inactivated at 56° for 30 min, aliquoted, and stored at −20°.
In an effort to test the specificity of the guinea pig sera, bovine and human stool specimens, purified by centrifugation through ammonium acetate cushions (Duckmanton et al., 1997) were subjected to SDS-PAGE. These included three BTV-positive and two BTV-negative control fecal specimens, four HTV-positive fecal specimens, and the insoluble fraction of the E. coli expressing the N protein. Proteins were transferred electrophoretically to a PVDF membrane, and blocked as described above. Membranes were incubated for 3 h at room temperature in a 1:2000 dilution of either gpPIαN or gpHIαN sera in 1% skim milk in TBST. The membranes were washed and incubated for 2 h at room temperature in a 1:3000 dilution of alkaline phosphatase conjugated-rabbit anti-guinea pig IgG (RαGP; Sigma Chemicals) in 1% skim milk in TBST. Purified N protein control samples were also tested for reactivity with four human acute/convalescent paired sera from patients whose stools had been diagnosed positive for HTV by EM as described previously (Duckmanton et al., 1997), and bovine anti-BRV-1 (bαBRV-1), or bovine anti-BRV-2 (bαBRV-2) pre- and post-immune sera from BRV infected gnotobiotic calves. Sera were used at a dilution of 1:2000 followed by 1:3000 dilution of alkaline phosphatase conjugated murine anti-human IgG or alkaline phosphatase conjugated goat anti-bovine IgG (Sigma Chemicals). Following further washing, the membranes were developed using BCIP-NBT as described above.
2.8. Immunoelectron microscopy
Immunoelectron microscopy was performed as described previously (Duckmanton et al., 1997) using the gpPIαN and gpHIαN sera diluted at 1:50 with 1% ammonium acetate. The mixtures were centrifuged at 9000×g for 15 min at room temperature and the resuspended pellets stained with 2% phosphotungstic acid, and examined by negative contrast electron microscopy (EM) on a Philips EM 300 at a magnification of 50 000×. Control specimens included a purified bovine rotavirus-positive sample, and two purified samples from calves with diarrhea in which no viruses were detected by EM and which were also negative for bovine torovirus by RT-PCR.
2.9. Dot blot
Stool specimens, characterized for their virus content were examined for immunoreactivity with the gpPIαN and gpHIαN sera by dot blot analysis. The specimens were diluted in an equal volume of phosphate-buffered saline (w/v) and clarified by centrifugation at 9000×g and 12 000×g for 15 min at 4°. A 15 μl volume of each supernatant was vacuum spotted onto a PVDF membrane and allowed to dry for 2–5 min. The membranes were blocked for 30 min in a solution of 5% skim milk in TBST. The membranes were then incubated for 1 h at room temperature in a 1:1000 dilution of either gpPIαN or gpHIαN sera in 1% skim milk in TBST. The membranes were washed with TBST and incubated for 45 min at room temperature in a 1:3000 dilution of RαGP (Sigma Chemicals) in 1% skim milk in TBST. Following further washing, the membranes were developed using BCIP-NBT (Sigma Chemicals) as described above.
3. Results
3.1. Long RT-PCR and sequencing of BRV-1 structural genes
Using primers designed from the genome sequence of BEV, an amplicon of 7564 bases was amplified from BRV-1 RNA (Fig. 1 ). The amplicon was excised, purified and used directly for DNA sequencing. To complete the sequence of the 5′ end of the S gene, a small amount of a 7.7 kb amplicon was obtained using the sense primer from the 3′ end of the POL 1b region and the antisense primer described previously. Sequence analysis revealed the presence of four ORFs (Fig. 2 ). The first ORF, 4752 nucleotides in length (nts 2685–7436 upstream from the polyA), codes for a polypeptide of 1584 amino acids which contains domains typical of type I membrane glycoproteins: a 26 residue C-terminal transmembrane anchor, a 19 residue N-terminal signal sequence, and 21 potential N-glycosylation sites. A cleavage site for a ‘trypsin-like’ protease (arginine residues 1003–1007), and two heptad repeat domains (residues 1160–1231 and 1474–1519) were also identified. The nucleotide sequence of the ORF has 76% identity with the peplomer gene of BEV.
Fig. 1.
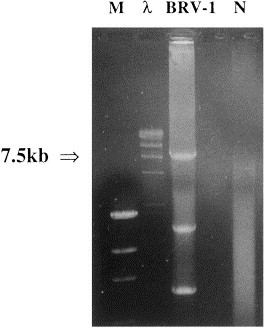
Agarose/ethidium bromide gel electrophoresis of products amplified by long RT-PCR using RNA extracted from a bovine fecal specimen positive for Breda virus-1 (BRV-1) and primers flanking the torovirus nucleocapsid and peplomer genes. Lane marked N represents a negative control (ddH2O). DNA mass ladder (M) and lambda HindIII (λ) were used as molecular size markers.
Fig. 2.
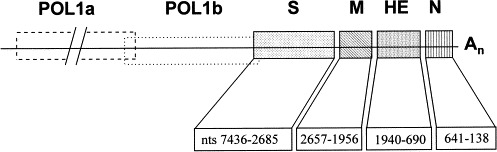
Schematic representation of the BRV-1 genome showing the open-reading frames for the four viral structural genes, the peplomer (S), envelope (M), hemagglutinin-esterase (HE), and nucleocapsid (N). Numbers indicate the genome positions of the first and last nucleotides of each ORF sequenced starting from the 3′ end of the genome. The two ORFs of the polymerase gene (POL1a and POL1b) have not yet been sequenced.
The next downstream ORF, 702 nucleotides in length (nts 1956–2657), codes for a polypeptide of 233 amino acids. The nucleotide sequence of this ORF is 85% identical to the BEV envelope gene which codes for the M protein (den Boon et al., 1991). Analysis of the amino acid sequence showed that the encoded protein contains the characteristics of a class III membrane protein with three possible successive transmembrane α-helices in the N-terminus.
Downstream from the BRV-1 M gene, a third ORF of 1251 nucleotides in length (nts 690–1940) was identified. Nucleotide sequence analysis revealed that the 457 bases at the 3′ end of this ORF exhibit 77% identity with the X pseudogene of BEV. However, there remain 794 bases upstream of this section that are not present in the BEV genome. Together, these two sections make up an ORF whose sequence has 100% identity with the hemagglutinin-esterase gene of BRV-2 (Cornelissen et al., 1997). The BRV-1 ORF codes for a protein of 417 amino acids with the characteristics of a type I membrane glycoprotein: a 14 residue N-terminal signal sequence, a 24 residue C-terminal transmembrane anchor, and seven potential N-glycosylation sites. The putative catalytic site of influenza C virus (ICV) and coronavirus acetylesterases, the F-G-D-S motif, is therefore also conserved in the BRV-1 HE homolog.
The 3′-most ORF is 504 nucleotides in length (nts 138–641), and nucleotide sequence analysis revealed that it manifests 83% sequence identity to the BEV N gene (Fig. 3 ). This ORF codes for a 167 residue polypeptide with a predicted molecular weight (M r) of approximately 19.2 kD, which is consistent with the M r of the BEV nucleocapsid protein. In addition, two clusters of basic amino acid residues are present in the N protein.
Fig. 3.

Nucleotide sequence of the BRV-1 nucleocapsid gene and the 3′ non-coding region of the BRV-1 genome aligned with that of BEV. Identical nucleotides are shown as dots. The predicted amino acid sequence of the BRV-1 N protein is also shown, and initiation (>>>) and termination (<<<) codons are underlined.
Each of the four ORFs is preceded by a short conserved nucleotide sequence (U G/C UUUAG U/A) which may likely represent a transcription initiation signal on the RNA that directs the synthesis of subgenomic mRNAs. With the exception of the 1.2 kb that make up the HE gene, the ORFs of the BRV-1 structural proteins manifested 80% nucleotide sequence identity to the BEV genome. The Genbamk accesion number for this sequence is AF076621.
3.2. Expression of the N gene
The 504 nucleotide N gene ORF, cloned in frame downstream of a T7 promoter in the pET28a (+) cloning vector, was expressed in BL21(DE3) cells following induction with IPTG. SDS-PAGE and immunoblot analysis with the T7*Tag conjugate identified the expressed N protein specifically in the insoluble cell fraction. The expressed N protein in the insoluble pellet was purified by SDS-PAGE and used to immunize young male guinea pigs. Pre- and post-immunization sera were collected and tested for N protein specific reactivity.
3.3. SDS-PAGE and immunoblotting
When partially purified BTV and HTV preparations were analyzed by SDS-PAGE, bands with M r of 9, 20, 32, 35, 42, 66, and 75 kDa were detected for BTV as shown in Fig. 4 a, and bands with M r of 7, 18, 20, 32, 36, 66, 75 kDa were detected for HTV (Fig. 4b). The 20kD bands correspond to the N protein equivalent of BEV (Snijder et al., 1989). The guinea pig sera were tested for specific reactivity with these protein bands by immunoblot using three BTV-positive stool samples, two BTV-negative stool samples, four HTV-positive stool specimens, and a purified BRV-1 N protein positive control. The gpHIαN serum, but not the gpPIαN serum, was found to react specifically with the BTV-positive specimens (Fig. 4a), and to cross react with the HTV-positive specimens (Fig. 4b). Neither of the sera reacted with the BTV-negative specimens. The expressed N protein reacted with the gpHIαN serum, the bαBRV-1 post-immune serum, and the bαBRV-2 post-immune serum, but not with the gpPIαN, the bαBRV-1 pre-immune serum or the bαBRV-2 pre-immune serum. The expressed 20 kDa N protein did not react with any of the human acute/convalescent paired sera (Fig. 4c).
Fig. 4.
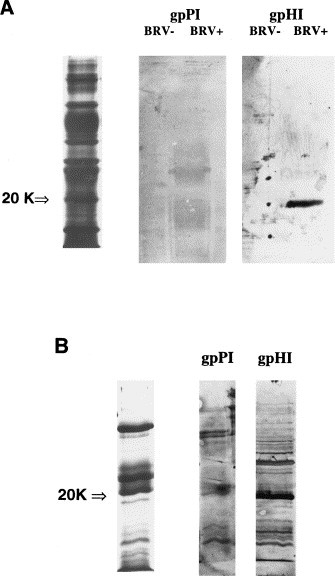
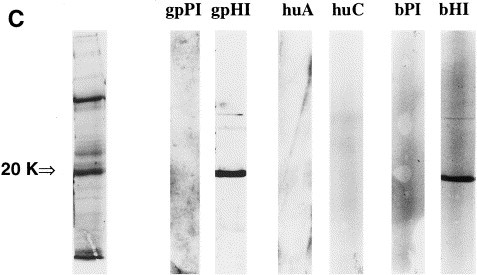
Representative SDS-PAGE gels (left lane) and corresponding immunoblots of guinea pig anti-BRV-1 N protein preimmune (gpPI) and hyperimmune (gpHI) sera with (A) purified bovine torovirus-negative (BTV−) and positive (BTV+) fecal specimens, and (B) a purified human torovirus-positive fecal specimen. (C) SDS-PAGE gel of the expressed BRV-1 N protein from the insoluble E. coli fraction (left) and corresponding immunoblots of gpPI and gpHI sera, human acute (huA) and convalescent (huC) paired sera, and bovine anti-BRV-1 preimmune (bPI) and hyperimmune (bHI) sera.
3.4. Immunoelectron microscopy
The guinea pig antiserum to the BRV-1 N protein was tested by IEM for reactivity with five purified BTV-positive preparations, two BTV-negative preparations, and a preparation containing bovine rotavirus. In all cases it was found that the mixing of gpHIαN serum with the BTV positive specimens resulted in the formation of aggregates of toroviruses shown in Fig. 5 B which consisted of what appeared as partially disrupted toroviruses. Conversely, mixing of gpPIαN serum with BTV positive specimens resulted only in single unaggregated torovirus virus particles (Fig. 5A). No toroviruses were seen in any preparations containing BTV negative specimens.
Fig. 5.
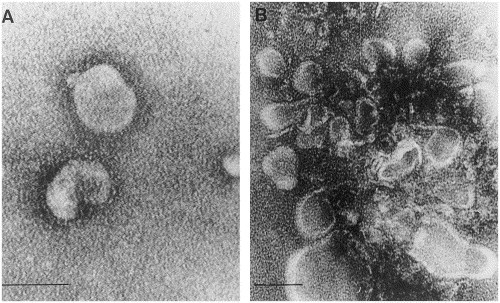
Immunoelectron microscopy of a purified bovine torovirus-positive preparation with (a) guinea pig anti-BRV-1 N protein preimmune serum showing intact unaggregated toroviruses and (b) guinea pig anti-BRV-1 N protein hyperimmune serum showing aggregated partly disrupted particles. Bars=100 nm.
3.5. Dot blot
The above gpHIαN and gpPIαN sera were used to design a dot blot system to detect torovirus in stool specimens. A panel of stool specimens previously characterized as positive or negative for torovirus by electron microscopy and reverse transcription-PCR was used in this process. Of 20 specimens that were previously shown to be positive for BTV, 19 were reactive by dot blot with the gpHIαN serum and not with the gpPIαN serum. None of the BTV negative or bovine rotavirus positive specimens were reactive. However, five specimens positive for HTV were reactive with the gpHIαN serum and not with the gpPIαN serum. Fig. 6 illustrates representative dot blots of BTV positive, HTV positive, bovine rotavirus positive and BTV negative stool specimens with the gpHIαN and gpPIαN sera.
Fig. 6.
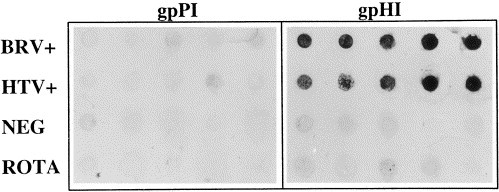
Representative dot blot of five bovine torovirus-positive (BTV+), five human torovirus-positive (HTV+), five bovine virus-negative (NEG), and five bovine rotavirus-positive (ROTA) fecal specimens using guinea pig anti-BRV-1 N protein preimmune (gpPI) and hyperimmune (gpHI) sera and rabbit anti-guinea pig antiserum.
4. Discussion
Berne virus and Breda virus have been well characterized at the biochemical and serological levels, and as such have been shown to be morphologically similar and antigenically related (Weiss et al., 1983). However, our current knowledge of the molecular aspects of toroviruses is based on BEV since it is the only torovirus that has successfully been grown in cell culture. The organization of the BEV genome and its replication strategy has been described by Snijder et al. (1990a)who showed that the BEV genome is a single-stranded positive sense RNA of approximately 26–28 kb in length. The organization of this genome is similar to that of avian infectious bronchitis virus, mouse hepatitis virus and equine arterivirus with which there is evidence of common ancestry (Horzinek et al., 1987, Snijder and Horzinek, 1993, Snijder et al., 1994). Based on the genome organization and replication strategies of these viruses, the coronaviruses, toroviruses, and arteriviruses have recently been assigned to a new order, the Nidovirales (Cavanagh, 1997, DeVries et al., 1997). Sequencing of the 15 kb from the 3′ end of the BEV genome has revealed the presence of five ORFs that encode the genes for the polymerase 1a (POL1a), the polymerase 1b (POL1b), the peplomer, the envelope, and the nucleocapsid protein (Snijder et al., 1990a). One putative nonfunctional ORF (X) located between the M and N genes has also been identified (Snijder et al., 1990a, Snijder et al., 1991).
By comparison, the BRV genome has been less extensively characterized. The sequence of 269 nts upstream of the poly (A) tail of the BRV-1 genome has been determined and shown to be 93% identical to that of BEV (Koopmans et al., 1991). Furthermore, the discovery of a novel 1.2 kb gene located between the genes for the nucleocapsid and membrane proteins of BRV-2 has shed new light on the organization of the BRV genome (Cornelissen et al., 1997). However, there remained a need to further explore the molecular aspects of this virus both to allow for the better understanding of the relatedness of these agents and to develop torovirus-specific detection assays such as RT-PCR, or to express viral proteins for use in serological assays.
In this study, long RT-PCR has been used successfully to amplify 7.5 kb at the 3′ end of the BRV-1 genome. This method has been used previously to amplify the full-length or nearly full-length genomes of hepatitis A virus, and hepatitis C virus (Tellier et al., 1996a, Tellier et al., 1996b). Sequence analysis of the BRV-1 amplicon revealed the presence of four ORFs that bear strong sequence similarities to corresponding ORFs of BEV, and have the capacity to code for all of the viral structural proteins. Sequencing the amplicon directly is advantageous since it inherently provides the consensus sequence, and RNA viruses are typically quasi-species (Holland et al., 1992). In addition, cloning of long viral sequences may lead to the selection of defective clones if the viral sequence is toxic to E. coli (Forns et al., 1997).
The torovirus peplomer protein is considered to be involved in viral infectivity (Snijder et al., 1990b), and has been shown to be recognized by neutralizing antibodies (Horzinek et al., 1986). The molecular properties of the BRV-1 S gene resemble those of the peplomer gene of BEV and coronaviruses (Snijder et al., 1990b, Snijder et al., 1994). That is, the nucleotide sequence of the S gene and its corresponding amino acid sequence are highly conserved, especially in the C-terminal half of the protein. The BRV-1 peplomer protein displays the characteristics of a type I membrane glycoprotein, and as such is potentially variably N-glycosylated.
The presence of a predicted cleavage site for a trypsin-like protease is consistent with the S protein being subject to post-translational cleavage during viral replication. As has been predicted for BEV (Snijder et al., 1990b), cleavage between amino acids 1002 and 1003 deduced from the 3′ end of the BRV-1 genome most likely generates the precursor to the mature S protein. Host-dependent cleavage by a trypsin-like protease has been known to occur during the maturation of membrane proteins in a number of virus families (Cavanagh et al., 1986). In fact, a similar post-translational cleavage event occurs during the replication of BEV (Snijder et al., 1990b).
The presence of two heptad repeats in the amino acid sequence of the S protein is indicative of a coiled-coil protein structure. In this conformation, α-helical domains are stabilized by the interaction of regularly spaced hydrophobic residues that form the interface between two α-helices (Cohen and Parry, 1986). As for the S proteins of BEV and coronaviruses, the two heptad repeats of BRV-1 are unequal in length. It has been proposed that the major heptad repeat may be involved in the generation of the intra-chain coiled-coil secondary structure of the S protein, as well as inter-chain interactions involved in S protein oligomerization (Cavanagh, 1983, De Groot et al., 1987, Snijder et al., 1990b). Such interactions may stabilize the elongated BRV-1 S protein in its spike configuration.
BEV and coronaviruses are intracellularly budding RNA viruses, whose maturation is thought to be governed by specific properties of their envelope proteins (Den Boon et al., 1991). The BRV-1 M protein shows numerous similarities to the envelope proteins of BEV and coronaviruses (Armstrong et al., 1984, Den Boon et al., 1991). It is a class III membrane protein with three membrane-spanning domains in its N-terminus. Thus, the envelope protein of BRV-1, like that of BEV, is very likely to be essential for viral assembly.
It has been previously suggested that the amino acid sequence of the BEV X pseudogene shows a striking similarity to the 3′ end of the ORFs for coronavirus HE protein and the HE-1 subunit of IVC (Snijder et al., 1991). The ORF for the BRV-2 HE gene, was found to manifest 30% predicted amino acid sequence identity with the HE proteins of both coronavirus and IVC (Cornelissen et al., 1997). The nucleotide and amino acid sequences of the BRV-1 HE gene are identical to those of the BRV-2 HE gene. Hence, the BRV-1 HE displays the properties of an N-glycosylated protein, and may also possess acetylesterase activity since it contains the putative F-G-D-S acetylesterase catalytic site that is conserved among the HE proteins of coronaviruses, ICV, and BRV-2. This HE gene homolog has also been found to be present in the HTV genome (Duckmanton et al., unpublished data). Thus, to date, BEV is the only torovirus that was not found to contain the HE gene and it has been suggested that BEV lost the 5′-most portion of its HE gene during tissue culture adaptation (Snijder et al., 1991) and the HE gene may be present in the natural BEV virion.
The nucleotide sequence and predicted amino acid sequence of the BRV-1 N protein also closely resemble those of BEV. The amino acid sequence contains two clusters of basic amino acid residues, which are also present in BEV. These may play a role in the binding of nucleic acid during assembly of the torovirion (Snijder et al., 1989). In addition, the 3′ non-coding region of the BRV genome was found to be conserved among the toroviruses (Koopmans et al., 1991, Duckmanton et al., 1997). It has been proposed that part of this non-coding sequence, excluding the poly (A) tail, may likely play a role in the initiation of negative-strand RNA synthesis during viral transcription. Analysis of this region of the torovirus genome revealed the presence of a large stem-loop secondary structure that may serve as one of the recognition signals involved in negative-strand RNA synthesis (Snijder et al., 1989). The N protein gene of a porcine torovirus has recently been reported. The porcine N gene is smaller, having 489 bp and coding for a 18.7 kDa protein whose amino acid sequence has 66.9% identity with BEV (Kroneman et al., 1998).
Using the expressed N protein to develop antisera in guinea pigs allowed us to examine the degree of cross-reactivity between toroviruses at the N protein level. By immunoblot, the gpHIαN serum reacted specifically with a 20 kDa protein in BTV-positive fecal specimens as well as HTV-positive specimens, demonstrating antigenic cross-reactivity among these toroviruses at the N gene level. This suggests a high degree of sequence homology between the HTV and BTV N genes, which is consistent with our previous observation that the nucleotide sequence in the 3′ end of the HTV genome encompassing a portion of the N gene has been shown to be 92% identical to BRV-1 in this area (Koopmans et al., 1991, Duckmanton et al., 1997). Cross-reactivity between toroviruses has previously been demonstrated with BEV preparations found to react with bovine convalescent sera by IEM (Beards et al., 1986), and BRV antigens and BEV antibodies found to be reactive by immunofluorescence microscopy assays (IFA), ELISA, and IEM (Beards et al., 1986, Weiss and Horzinek, 1987). In addition, HTV particles were found to be coated and aggregated after addition of calf sera containing antibodies to BRV (Beards et al., 1986). Lastly, human stool specimens documented to contain HTV particles by electron microscopy were also shown to be reactive in a torovirus-specific ELISA based on BRV antiserum (Koopmans et al., 1993).
It was therefore surprising to note that no such cross reactivity could be demonstrated between the expressed BRV N protein and human convalescent sera from torovirus pateints, even though these sera were previously shown to react with torovirus-positive human fecal specimens by HI and IEM (Duckmanton et al., 1997). However, this result is consistent with immunoblot analyses done previously using these human acute/convalescent paired sera, whereby the convalescent serum was only found to react specifically with a 75–90 kD band representing the peplomer protein. This suggests that, following a torovirus infection, the major immune response in humans is to the torovirus peplomer protein and not to other structural proteins such as the nucleocapsid. This type of response has also been demonstrated in the coronaviruses (Schmidt and Kenny, 1981, Battaglia et al., 1987). Conversely, the convalescent serum from gnotobiotic calves infected with BRV was reactive with the expressed BRV N protein. This could reflect a stronger response to the N protein by the calf immune system particularly since it is to the homologous virus.
By IEM, the gpHIαN serum was shown to specifically aggregate broken BRV-1 particles but not intact particles under conditions where the control pre-immune serum from the same guinea pig did not produce any aggregates. This is consistent with the fact that the nucleocapsid protein is internal to the virion and only broken particles whose nucleocapsid protein had been exposed could be reactive in this assay. These observations support previously reported concepts of virus structure and the location of the N protein (Weiss et al., 1983, Snijder et al., 1989).
Lastly, the dot blot assays demonstrated that the gpHIαN serum reacted specifically with BTV-positive and HTV-positive fecal specimens, but not with virus-negative controls or rotavirus-positive specimens. As shown by EM and IEM, many BRV-1 particles in stool samples are partly disrupted and their nucleocapsids exposed. Thus, once these agents are bound to a nylon membrane, it is possible to specifically detect them using designated antisera to the N protein. The speed and facility of the dot blot give it good potential as a, practical immunospecific method for the diagnosis of bovine torovirus in veterinary practice. The strong cross-reactivity of the serum with HTV-positive specimens demonstrated by immunoblot and by this assay shows that the dot blot may also be useful for the diagnosis of torovirus in the human population.
Acknowledgements
The authors would like to thank Dr G.N. Woode at Texas A&M and Dr\ M. Hardy of Montana State University for providing the BRV-1-infected bovine fecal specimen, and the bαBRV-1 and bαBRV-2 antisera used for immunoblot analysis. We would also like to thank Drs Éva Nagy and Susy Carman from the Animal Health Laboratory, Guelph, Ontario for providing the bovine torovirus-positive fecal specimens from diarrheic calves, control specimens from asymptomatic calves, and rotavirus-positive specimens from diarrheic calves. This research was supported by a grant from the Medical Research Council of Canada to M.P. and a grant from the Research Institute, Hospital for Sick Children, to R.T..
References
- Armstrong J, Niemann H, Smeekens S, Rottier P, Warren G. Sequence and topology of a model intracellular membrane protein, E1 glycoprotein, from a coronavirus. Nature. 1984;308:751–752. doi: 10.1038/308751a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia M, Passarani N, Di Matteo A, Gerna G. Human enteric coronaviruses: further characterization and immunoblotting of viral proteins. J. Inf. Diseases. 1987;155:140–143. doi: 10.1093/infdis/155.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beards G.M, Brown D.W, Green J, Flewett T.H. Preliminary characterization of torovirus-like particles of humans: comparison with the Berne virus of horses and Breda virus of calves. J. Med. Virol. 1986;20:67–78. doi: 10.1002/jmv.1890200109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D.W, Beards G.M, Flewett T.H. Detection of Breda antigen and antibody in humans and animals by enzyme-immunoassay. J. Clin. Microbiol. 1987;25:637–640. doi: 10.1128/jcm.25.4.637-640.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Coronavirus IBV: structural characterization of the spike protein. J. Gen. Virol. 1983;64:2577–2583. doi: 10.1099/0022-1317-64-12-2577. [DOI] [PubMed] [Google Scholar]
- Cavanagh D, Davis P.J, Pappin D.J.C, Binns M.M, Boursnell M.E.G, Brown T.D.K. Coronavirus IBV: partial amino terminal sequencing of spike polypeptide S2 identifies the sequence Arg-Arg-Phe-Arg-Arg at the cleavage site of the spike precursor polypeptide of IBV strains Beaudette and M41. Virus Res. 1986;4:133–143. doi: 10.1016/0168-1702(86)90037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch.Virol. 1997;142(63):29–633. [PubMed] [Google Scholar]
- Cohen C, Parry D.A.D. Helical coiled coils—a widespread motif in proteins. Trends Biochem. Sci. 1986;11:245–248. [Google Scholar]
- Cornelissen L.A.H.M, Wierda C.M.H, van der Meer F.J, Herrewegh A.A.P.M, Horzinek M.C, Egberink H.F, de Groot R.J. Hemagglutinin-esterase, a novel structural protein of torovirus. J. Virol. 1997;71:5277–5286. doi: 10.1128/jvi.71.7.5277-5286.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot R.J, Luytjes W, Horzinek M.C, van der Zeijst B.A.M, Spaan W.J.M, Lenstra J.A. Evidence for a coiled-coil structure in the spike proteins of coronaviruses. J. Mol. Biol. 1987;196:963–966. doi: 10.1016/0022-2836(87)90422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Boon J.A, Snijder E.J, Locker J.K, Horzinek M.C, Rottier P.J.M. Another triple-spanning envelope protein among intracellularly budding RNA viruses: the torovirus E protein. Virology. 1991;182:655–663. doi: 10.1016/0042-6822(91)90606-C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries A.A.F, Horzinek M.C, Rottier P.J.M, De Groot R.J. The genome organization of the Nidovirales: similarities and differences between arteri-, toro-, and coronaviruses. Semin. Virol. 1997;8:33–47. doi: 10.1006/smvy.1997.0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckmanton L, Luan B, Devenish J, Tellier R, Petric M. Characterization of torovirus from human fecal specimens. Virology. 1997;239:158–168. doi: 10.1006/viro.1997.8879. [DOI] [PubMed] [Google Scholar]
- Duckmanton L, Carman S, Nagy E, Petric M. Detection of bovine torovirus in fecal specimens of calves with diarrhea from Ontario farms. J. Clin. Microbiol. 1998;36:1266–1270. doi: 10.1128/jcm.36.5.1266-1270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forns X, Bukh J, Purcell R.H, Emerson S.U. How Escherichia coli can bias the results of molecular cloning: preferential selection of defective genomes of hepatitis C virus during the cloning procedure. Proc. Natl. Acad. Sci. USA. 1997;94:13909–13914. doi: 10.1073/pnas.94.25.13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland J.J, De La Torre J.C, Steinhauer D.A. RNA virus populations as quasispecies. Curr. Topics Microbiol. Immunol. 1992;176:1–20. doi: 10.1007/978-3-642-77011-1_1. [DOI] [PubMed] [Google Scholar]
- Horzinek M.C, Ederveen J, Kaeffer B, De Boer D, Weiss M. The peplomers of Berne virus. J. Gen. Virol. 1986;67:2475–2483. doi: 10.1099/0022-1317-67-11-2475. [DOI] [PubMed] [Google Scholar]
- Horzinek M.C, Flewett T.H, Saif L.F, Spaan W.J, Weiss M, Woode G.N. A new family of vertebrate viruses: Toroviridae. Intervirology. 1987;27:17–24. doi: 10.1159/000149710. [DOI] [PubMed] [Google Scholar]
- Koopmans M, van den Boom U, Woode G.N, Horzinek M.C. Seroepidemiology of Breda virus in cattle using ELISA. Vet. Res. 1989;19:233–243. doi: 10.1016/0378-1135(89)90069-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans M, Snijder E, Horzinek M.C. cDNA probes for the diagnosis of bovine torovirus (Breda virus) infection. J. Clin. Microbiol. 1991;29:493–497. doi: 10.1128/jcm.29.3.493-497.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans M, Petric M, Glass R, Monroe S.S. Enzyme-linked immunosorbent assay reactivity of torovirus-like particles in fecal specimens form humans with diarrhea. J. Clin. Microbiol. 1993;31:2738–2744. doi: 10.1128/jcm.31.10.2738-2744.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroneman A, Cornelissen L.A.H.M, Horzinek M.C, De Groot R.J, Egberink H.F. Identification and characterization of a porcine torovirus. J. Virol. 1998;72:3507–3511. doi: 10.1128/jvi.72.5.3507-3511.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt O.W, Kenny G.E. Immunogenicity and antigenicity of human coronaviruses 229E and OC43. Infect. Immun. 1981;32:1000–1006. doi: 10.1128/iai.32.3.1000-1006.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J, den Boon J.A, Spaan W.J.M, Verjans G.M.G.M, Horzinek M.C. Identification and primary structure of the gene encoding the Berne virus nucleocapsid protein. J. Gen. Virol. 1989;70:3363–3370. doi: 10.1099/0022-1317-70-12-3363. [DOI] [PubMed] [Google Scholar]
- Snijder E.J, Horzinek M.C, Spaan W.J. A 3′ co-terminal nested set of independently transcribed mRNAs is generated during Berne virus replication. J. Virol. 1990;64:331–338. doi: 10.1128/jvi.64.1.331-338.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J, den Boon J, Spaan W.J, Weiss M, Horzinek M.C. Primary structure and post-translational processing of the Berne virus peplomer protein. Virology. 1990;178:355–363. doi: 10.1016/0042-6822(90)90332-L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J, den Boon J.A, Horzinek M.C, Spaan W.J.M. Comparison of the genome organization of toro- and coronaviruses: evidence for two non-homologous RNA recombination events during Berne virus evolution. Virology. 1991;180:448–452. doi: 10.1016/0042-6822(91)90056-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J, Horzinek M.C. Toroviruses: replication, evolution and comparison with other members of the coronavirus-like superfamily. J. Gen. Virol. 1993;74:2305–2316. doi: 10.1099/0022-1317-74-11-2305. [DOI] [PubMed] [Google Scholar]
- Snijder E.J, Horzinek M.C, Spaan W.J. The coronavirus-like superfamily. In: Laude H, Vautherot J.F, editors. Coronaviruses: Molecular Biology and Pathogenesis. Plenum; New York: 1994. [Google Scholar]
- Tellier R, Bukh J, Emerson S.U, Purcell R.H. Amplification of the full-length hepatitis A virus genome by long reverse transcription-PCR and transcription of infectious RNA directly from the amplicon. Proc. Natl. Acad. Sci. USA. 1996;93:4370–4373. doi: 10.1073/pnas.93.9.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier R, Bukh J, Emerson S.U, Miller R.H, Purcell R.H. Long PCR and its application to hepatitis viruses: amplification of hepatitis A, hepatitis B, and hepatitis C virus genomes. J. Clin. Microbiol. 1996;34:3085–3091. doi: 10.1128/jcm.34.12.3085-3091.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M, Steck F, Horzinek M.C. Purification and partial characterization of a new enveloped RNA virus (Berne virus) J. Gen. Virol. 1983;64:1849–1858. doi: 10.1099/0022-1317-64-9-1849. [DOI] [PubMed] [Google Scholar]
- Weiss M, Horzinek M.C. The proposed family toroviridae: agents of enteric infections. Arch. Virol. 1987;92:1–15. doi: 10.1007/BF01310058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woode G.N, Reed D.E, Runnels P.L, Herrig M.A, Hill T.H. Studies with an unclassified virus isolated from diarrheic calves. Vet. Microbiol. 1982;7:221–240. doi: 10.1016/0378-1135(82)90036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woode G.N, Saif M, Quesada M, Winnand N.J, Pohlenz J.F, Kelso Gourley N. Comparative studies on three isolates of Breda virus of calves. Am. J. Vet. Res. 1985;46:1003–1010. [PubMed] [Google Scholar]