

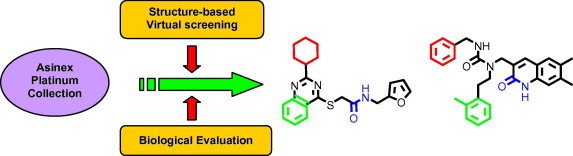
Graphical abstract
Novel inhibitors of the SARS-3CLpro were identified using a combination of structure-based virtual screening and biological evaluation.
Keywords: Structure-based virtual screening, Gold, Asinex, SARS-3CLpro inhibitor
Abstract
Severe acute respiratory syndrome is a highly infectious upper respiratory tract disease caused by SARS-CoV, a previously unidentified human coronavirus. SARS-3CLpro is a viral cysteine protease critical to the pathogen’s life cycle and hence a therapeutic target of importance. The recently elucidated crystal structures of this enzyme provide an opportunity for the discovery of inhibitors through rational drug design. In the current study, Gold docking program was utilized to conduct extensive docking studies against the target crystal structure to develop a robust and predictive docking protocol. The validated docking protocol was used to conduct a structure-based virtual screening of the Asinex Platinum collection. Biological evaluation of a screened selection of compounds was carried out to identify novel inhibitors of the viral protease.
1. Introduction
Severe acute respiratory syndrome (SARS) is a highly infectious upper respiratory tract disease which reached epidemic status in 2003.1, 2, 3, 4, 5 The first reported outbreak of the disease occurred in 2002 in the Guangdong province of China. Within the span of a year the disease had spread over to 32 countries in Asia, North America, and Europe. The disease infected nearly 8000 people worldwide with an average mortality rate of around 10%. The etiological agent of the disease was identified as a previously unknown human coronavirus christened as the SARS coronavirus (SARS-CoV). It is an enveloped positive sense RNA virus from the coronaviridae family containing a single chain RNA genome of ∼29,700 nucleotides, the largest viral RNA genome reported to date.6, 7 Although there have been no reported occurrences of SARS infections since 2004, the disease should still be treated as a high health risk because of its high virulence and contagious nature. Historical evidence on several viral diseases, such as influenza, suggests that recurrences of epidemic outbreaks, caused by the wild-type or mutated variants of the virus, are common. These recurrences are usually spread over a long period of time and may occur in regions which are geographically distant to each other. Therefore, there is an urgent need for the understanding of the etiology, pathology, and possible therapeutic targets against this virus. Knowledge gained from these studies can not only be used in the design of therapies against the current form of the virus but may be carried over in the future to other mutated variants or other pathogenic viruses from the same family.
The SARS-CoV genome contains two open reading frames connected by a ribosomal frame shift which encode for two large replicase polyproteins pp1a (∼450 kDa) and pp1ab (∼750 kDa) which function in the viral replication and transcription processes. The polyproteins are processed by viral proteases8, 9 to generate the functional components of a multiprotein complex known as viral replicase–transcriptase.10, 11 While most coronaviruses utilize three proteases for proteolytic processing the SARS-CoV is known to encode only two proteases for this purpose. These two proteases include a papain-like cysteine protease (PLP2pro)12 and a Chymotrypsin-like cysteine protease known as 3C-like protease (3CLpro). The 3CLpro enzyme13, 14, 15, 16, 17, 18 is also called as the main protease (Mpro) since it plays a pivotal role in the processing of the viral polyproteins and controlling of the replicase complex activity. The enzyme is indispensable to the viral replication and infection processes, thereby making it an ideal target for the design of antiviral therapy. The availability of multiple crystal structures19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 of the enzyme with co-crystallized ligands makes the target amenable to structure-based drug design.
Protein crystallography as well as biomolecular NMR has played a major role in the drug discovery efforts against this enzyme. The first crystal structure of the SARS-3CLpro dimer19 with a peptidic CMK inhibitor was elucidated in 2003 and since then over twenty crystal structures of the enzyme have been elucidated. These crystal structures include those of the enzymes apo19, 20, 21, 28 form as well as those with peptidic19, 20, 23, 26, 27, 29 and small molecule inhibitors24 bound to the enzyme’s active site. Crystal structures have also helped in elucidating the pH dependent conformational changes in the active site of the enzyme,28 induced-fit effects27 during ligand binding as well as the mechanism of irreversible inactivation26 by specific classes of peptidic inhibitors.
Current drug design efforts30, 31 against this enzyme can be broadly classified into two categories: peptidic and small molecule based inhibitors. Figure 1 shows some of the latest peptidic29, 32, 33, 34 and small molecule24, 35, 36 based inhibitors devised using the structural information and the substrate specificity profile of the SARS-3CLpro enzyme. The peptidic inhibitors were designed by attaching a reactive ‘warhead’ type agent to a peptide mimicking the natural substrate. These warhead groups include Michael acceptors,23, 29, 34 aldehydes,29, 37 epoxy ketones,32 halo methyl ketones,33 and a few others.38 These inhibitors act through a two-step procedure wherein they first bind and form a non-covalent complex with the enzyme such that the warhead group is located in close vicinity of the catalytic residue. This is followed by a nucleophilic attack by the catalytic cysteine and covalent adduct formation. The other category includes the non-peptidic inhibitors containing small molecule scaffolds. These inhibitors have been discovered using various techniques such as structure based virtual screening,24, 36, 39, 35 pharmacophore based screening40 as well as high through screening41 methodologies.
Figure 1.
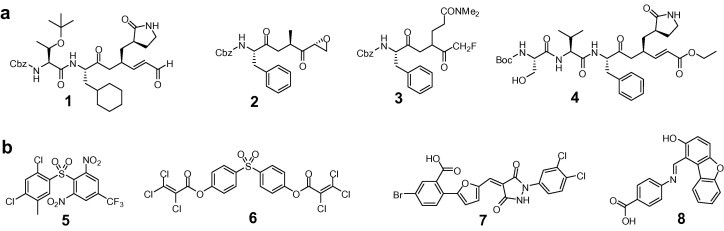
Structures of some of the recently reported (a) peptidic and (b) non-peptidic small molecule based inhibitors identified using the structural and substrate specificity information of SARS-3CLpro.
As part of an effort to discover small molecule based inhibitors of SARS-3CLpro, we conducted a structure-based virtual screening42, 43 against the SARS-3CLpro enzyme. Structure based virtual screening involves the in silico evaluation of a database of molecules against the experimentally determined structure or a comparative model of the protein target using a docking program. Evaluation of the generated docking poses is carried out using a metric such as a scoring function to identify putative binders. Compared to the high throughput screening of an entire compound library, only a small set of molecules selected on the basis of the screening strategy is evaluated in a biological assay to identify inhibitors of the target. In the present study, the Asinex Platinum collection was screened against the SARS-3CLpro enzyme crystal structure using the Gold docking program. Biological evaluation of a selected set of compounds in a SARS-3CLpro inhibition assay led to the identification of novel hits with activity in the low micromolar range which could be further optimized to develop potent antiviral therapies against SARS-CoV infections.
2. Results and discussion
2.1. Analysis of SARS-3CLpro binding site requirements
The 33.8 kDa SARS-3CLpro enzyme is a functional homodimer and shows three structural domains with an antiparallel β-barrel-shaped structure akin to the serine protease Chymotrypsin. The active site is located in a groove between the domains I and II and is a fairly solvent exposed and shallow cavity. The enzyme is an atypical cysteine protease, containing a catalytic diad (His41-Cys145) instead of a triad. Multiple crystal structures of SARS-3CLpro with co-crystallized inhibitors (1UK4,19 2AMD,20 PDB code) have been elucidated. The co-crystallized peptidic Michael-acceptor based inhibitor 9IN, from the SARS-3CLpro structure (2AMD, PDB code), is structurally analogous to the Cbz protected hexapeptide-CMK (chloro methyl ketone) inhibitor (1UK4, PDB code) and undergoes similar binding site interactions. Therefore, the co-crystallized pose of 9IN (Fig. 2 a and b) is used here to describe the relevant binding site interactions and the enzyme’s subsite specificity. Mutation studies44 have been carried out on the substrates of related coronavirus proteases to understand the specificity at each of the binding subsites and the effect of these mutations on the substrate binding and catalytic efficiency of the enzyme. It was found that the mutations at the P1 position produced the most significant loss in enzyme activity and the corresponding S1 sub site is very specific to a Glutamine residue. The other subsites of interest were S1′, S2, and S4 and the preferred substrate amino acid side chains for these sites were S1′ (alanine/serine), S2 (leucine), and S4 (valine/serine). In case of the 9IN co-crystallized pose, the β-carbon of the Michael acceptor forms a covalent bond with the sulfur of the catalytic cysteine (Cys145). The cyclized ketoglutamine group occupies the S1 subsite and acts as an entropically favored surrogate for the glutamine side chain of the native substrate. The carbonyl oxygen of the ketoglutamine group undergoes a hydrogen bonding interaction with His163 while the ring amide NH forms a hydrogen bond donor interaction with the backbone carbonyl oxygen of Phe140. The side chain of Glu166 is also in close vicinity to form a hydrogen bond interaction with the ring amide NH group after minor rotameric adjustment. An interaction of this nature between the nitrogen of the P1-glutamine side chain or its surrogate and the side chain carboxylate oxygen of Glu166 is seen in the crystal structure of SARS-3CLpro inhibited by an aza peptide epoxide22 (2A5I, PDB code) as well as a peptidic aldehyde29 (2GX4, PDB code) based inhibitor. The S2 (His41, Met49, Tyr54) and S4 (Met 130, Pro163, Gln192) subsites are primarily hydrophobic in nature and are occupied by a leucine and valine residue, respectively. The terminal ethyl group of the ligand occupies the S1′ site and undergoes hydrophobic interactions with Thr25 and Leu27. Additional hydrogen bonds are noticed between the peptidic backbone of the ligand and the residues Glu166, Gln189, and Thr190. Overall, the presence of hydrophobic groups at the S1′, S2, and S4 sites and a hydrogen bond acceptor at the S1′ site can be considered as critical pharmacophoric requirements (Fig. 2c) for the binding of putative inhibitors.
Figure 2.
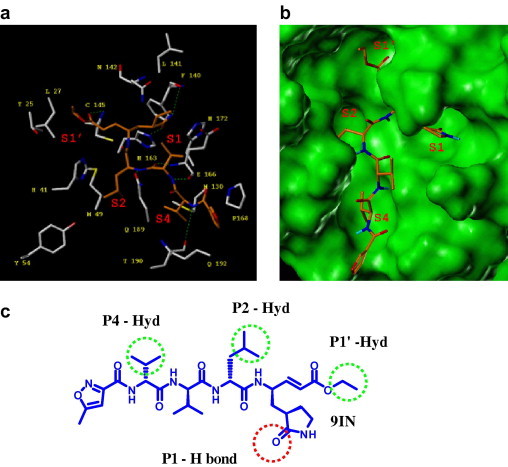
(a) Stick representation of the binding site residues (white) of SARS-3CLpro (2AMD, PDB code) with the co-crystallized peptidic ligand 9IN (orange). Hydrogen bonding interactions are shown in green dotted lines. (b) Surface representation (green) of the binding site of the SARS-3CLpro (2AMD, PDB code) structure along with the co-crystallized ligand 9IN (orange stick representation) showing the enzyme specificity sites. (c) Chemical structure of the co-crystallized ligand 9IN showing the P1′, P2, and P4 hydrophobic groups in green circles and the P1 hydrogen bonding interaction (ketoglutamine amide carbonyl) in red circle.
2.2. First phase screening
When the study was initiated only one crystal structure (1UK4, PDB code) of the SARS-3CLpro with a covalently bound Cbz protected hexapeptide-CMK inhibitor was available. The ligand has a large number of rotatable bonds and improperly resolved coordinates for the terminal portion of the ligand where the catalytic cysteine sulfur forms a covalent adduct with the ligand. Therefore, it was not feasible to conduct a pose validation study using this ligand structure. A series of phthalhydrazide based peptidic analogues45 (Supplementary information, Fig. S1) with inhibitory activity against the SARS-3CLpro had been reported and was utilized in a validation study using Gold 2.2 to select the binding site definition and scoring function utilized for binding pose calculation. Amongst the thirteen ketoglutamine based analogues four had IC50 values of 10 μM or less (pIC50 ⩾ 5, IC50 ⩽ 10 μM), four had IC50 values ranging from 10 to 100 μM while five had activities greater than 100 μM (pIC50 ⩽ 4, IC50 ⩾ 100 μM). Typically, a molecule with a higher binding affinity would have a better interaction and geometrical profile as compared to that of molecule with a weaker binding affinity. A scoring function should be able to evaluate the quality of the binding poses and separate the poses of the actives (IC50 ⩽ 10 μM) and the inactives (IC50 ⩾ 100 μM) based on their docking scores. The docking calculations were conducted using the Gold standard mode speed up which allows for 100,000 genetic operations per docking pose. The Goldscore function was evaluated for its predictive ability and was found to provide a score based separation of the actives and the inactives (Supplementary information, Table S1). The separation becomes more pronounced if one outlier (molecule 4), which was awarded a lower docking score. was discounted from the comparison. This validated protocol was utilized in the actual screening.
Docking of large virtual screening databases is a computationally expensive exercise and the level of calculations utilized for the docking process is a critical factor governing the computational time required for the screening process. One approach we have successfully used previously46, 47 toward minimizing the overall down time without compromising the quality is to carry out a multi-stage cascade docking involving a less intensive and faster docking protocol at the early stages of the screening and a gradual increment in the computational complexity as we proceed toward the final stages. Typically, at the early stages of screening, the aim is to look for shape complementarity of the docked molecules against the binding site and elimination of molecules with low probability of attaining a proper binding pose within the active site. On the contrary, the final stage of the screening process involves the selection of deserving hits based on their complementarity with the binding site interactions and the geometrical qualities of the binding pose itself. While the elimination stage can be handled by a less intensive docking calculation, a more rigorous protocol incorporating exhaustive conformational sampling is a critical requirement for the final stages of the screening. In this case, a three stage docking protocol of increasing computational complexity (Fig. 3 ) was implemented using the Gold 2.2 docking program.
Figure 3.
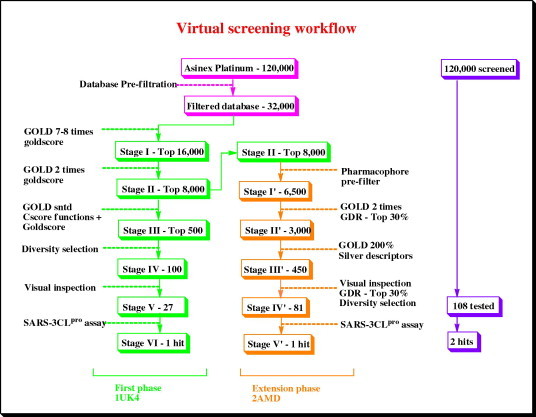
A flowchart depicting the cascading virtual screening approach utilized in the study.
The choice of the virtual screening database plays a major role in terms of the area of synthetic chemical space covered during the search, novelty of the scaffolds identified through the screening, and feasibility for the synthetic modifications of the identified hits. The Asinex Platinum collection used for this study is a ‘drug-like’ collection of more than 100,000 compounds containing more than 500 scaffolds which are unique to this database. Furthermore, around 87% of the compounds in this database have 3–4 point diversity which provides an enhanced opportunity for synthetically tractable modifications to explore structure–activity relationships. Additionally, most of the compounds in this database are synthesized inhouse, making them resuppliable at short notice and allows for better quality control of the supplied product.
In the pre-filtration stage of the virtual screening, the initial database comprising of ∼120,000 molecules was passed through various drug-like and ADME filters (see methods section) to generate a curtailed database of ∼32,000 molecules. This pre-filtered database was submitted to the first stage of the cascade docking at the Gold 7–8 times speed up mode. In this setting, the number of genetic operations performed for the generation of a single docking pose is reduced by a factor of 7/8 times compared to the Gold standard mode, leading to a shorter calculation time. Ten docking poses were generated for every ligand and the top ranking pose based on Goldscore was selected for comparison across multiple ligands. The ligands were ranked based on Goldscore and the top 16,000 were selected for the second stage of the cascade docking using the Gold 2 times speed up mode. The top 8000 molecules (based on Goldscore) from this stage were selected in a similar fashion and submitted for the final stage of the cascade docking run using the Gold standard mode (highest computational complexity) settings. The top ranked poses (based on Goldscore) were rescored using the Cscore module of Sybyl 6.9 which allows evaluation using five different scoring functions Fscore, Gscore, Pmfscore, Dscore, and Chemscore. The poses were reranked by a cumulative score obtained by summing the six different scores including Goldscore and the top 500 molecules were selected for the next stage. A clustering analysis of the top 500 molecules was conducted to identify the major chemical classes present and ∼100 molecules comprising the top ranked molecule from every cluster were selected for visual inspection.
The group of top-ranking structurally diverse molecules obtained through the clustering analysis were visually inspected based on their (1) ability to occupy the key substrate specificity sites S1′, S1, S2, and S4, (2) geometric quality of the ligand binding pose, (3) hydrophilic/lipophilic mismatches, and (4) complementarity of the key interacting features. Finally, 27 molecules were selected on the basis of visual inspection and purchased for biological evaluation. The identity and purity (>90% purity) of the compounds were checked using HPLC and 1H NMR data provided by the vendor.
Conducting biological dose–response studies for a large selection of compounds is a labor intensive process and involves significant use of valuable resources. It would be judicial if these efforts are limited to the evaluation of candidate compounds which have a higher probability of being active in the low micromolar range and are more suitable for future SAR development. The biological evaluation experiments in the current study were therefore devised as a two-step procedure and involved an initial pre-filtration step. In this step, a preliminary screening of the test compounds was conducted at a single concentration of 10 μM. The compounds exhibiting significant percentage inhibition of enzyme activity at this concentration were then subjected to a detailed dose–response study to determine their IC50. Amongst the compounds evaluated PJ07 (Table 1 ) showed around 30% inhibition in this preliminary screening stage. The compound was subjected to a detailed dose–response study and was found to have an IC50 of 18.2 μM against SARS-3CLpro.
Table 1.
Chemical structure, physiochemical properties, and SARS-3CLpro inhibitory activity of the identified hits
| Compound ID | Chemical structure | MW | C log P | SARS-3CLpro activity |
|
|---|---|---|---|---|---|
| % Inh. at 10 μM | IC50 (μM) | ||||
| PJ07 | 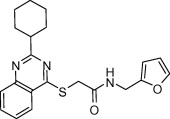 |
381.49 | 4.50 | 30 | 18.2 |
| PJ169 | 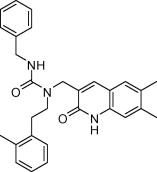 |
453.58 | 4.78 | 40 | 17.2 |
While the biological evaluation of the selected compounds was being carried out, newer crystal structures of the SARS-3CLpro (2AMD, PDB code) with co-crystallized peptidic irreversible inhibitors were published. Unlike the co-crystallized ligand from the previous structure (1UK4, PDB code) where the coordinates for a portion of the inhibitor were inaccurate, the newer co-crystallized ligand structures were fully resolved and involved fewer rotatable bonds making them suitable for ligand pose validation study. Some reports on the discovery of SARS-3CLpro inhibitors had been published and provided a sizeable dataset of actives, useful for conducting a docking enrichment study (Supplementary information, Fig. S2).
2.3. Co-crystallized ligand pose validation study
Pose validation studies were carried out in Gold 3.0.1, using the co-crystallized peptidic ligand (9IN) bound SARS-3CLpro crystal structure (2AMD, PDB code). Variations in the definition of the binding site, spherical radius used for cavity detection, scoring functions utilized for pose selection as well as docking constraints were also evaluated. The docking was carried out at the Gold 200% accuracy level using automatic GA settings. At this accuracy level the docking program performs twice the number of genetic operations compared to the standard mode settings. The automatic GA settings allow the program to vary the total number of GA operations performed, based on the complexity of the ligand being docked. The final docking parameters (binding site definition, scoring function for pose generation) selected through this study were identical to those used in the first phase of screening except for the addition of a constraint set. The constraints (Fig. 4 d) were incorporated into the scoring function in the form of an extra scoring term. Poses with none or some of the constraints satisfied were considered but received lower constraint scores compared to those satisfying all the constraints. The constraint set, which provided the best results, included three hydrophobic constraints corresponding to the hydrophobic groups of 9IN occupying the P1′, P2, and P4 specificity sites and a protein hydrogen bond donor interaction with the N(ε)–H of His163. The top ranked solution, obtained for the ligand using Goldscore function, showed a heavy atom RMSD of 1.59 Ǻ (Fig. 4a) with the actual co-crystallized pose which should be considered good for a ligand of this size and flexibility.
Figure 4.
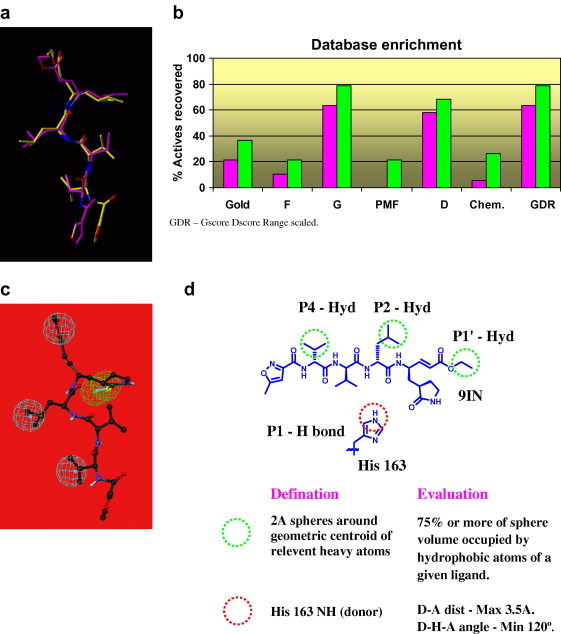
(a) Pose validation study: superimposition of the co-crystallized pose (yellow) of 9IN (2AMD, PDB code) and the top ranked pose (magenta) obtained through docking. (b) Enrichment study: a bar graph showing the percentage of actives recovered at 10% (magenta) and 30% (green) of the ranked database. (c) A receptor based pharmacophore pre-filter generated in Catalyst using the co-crystallized pose of the peptidic ligand 9IN in the SARS-3CLpro crystal structure (2AMD, PDB code). The pharmacophore consists of three hydrophobic features (cyan) mapped onto the P1′, P2, and P4 groups and a hydrogen bond acceptor feature (green) mapped onto the oxygen atom of the P1 site side chain. (d) Definition for the docking constraints and the docking pose based descriptors (Silver) used in the extension phase of screening. Locations of the three hydrophobic constraints/occupancy descriptors are shown in green dotted circles, while the protein hydrogen bond donor constraint/ descriptor is shown in a red dotted circle. The evaluation segments in the schematic refer to the criterions used in Silver to score the docking pose based descriptors.
2.4. Scoring function enrichment study
The final docking protocol from the pose validation study was selected for the enrichment study. Five scoring functions from Cscore as well as Goldscore were evaluated for their ability to enrich the true actives. The enrichment graph (Fig. 4b) shows that amongst the scoring functions evaluated, Gscore followed by Dscore appear to perform better than the others in the 10% and 30% database screened mark. At the 10% database screened mark, the percentage of actives recovered was 64% and 58% for Gscore and Dscore while at the 30% database screened mark, the percentage of actives recovered was 78% and 68%, respectively. Simultaneous high ranking of actives by different scoring functions enhances the confidence level of the docking protocol. Additionally, it may supplement the shortcomings of individual scoring functions in evaluating the binding poses of molecules belonging to a specific structural class. In this case, the two better performing functions, Gscore and Dscore, were combined in the range scaled format to generate a composite GDR score. This composite score showed a behavior similar to Gscore and Dscore with a recovery of nearly 64% and 78% at the 10% and 30% of database screened mark. This docking and scoring protocol was then utilized in the extension phase of the screening study.
2.5. Extension phase screening
The extension phase of screening (Fig. 3) was conducted using the top 8000 molecules from the Gold 2 times speed up stage of the first phase of screening. To curtail the number of molecules to be docked, a pharmacophore pre-filter (Fig. 4c) generated in Catalyst using the co-crystallized pose of 9IN was implemented. It incorporated three hydrophobic features based on the P1′, P2, and P4 hydrophobic groups and one hydrogen bond donor feature based on the carbonyl oxygen of the P1 ketoglutamine side chain. The 6500 molecules passing the pharmacophore prefilter were subjected to docking using the Gold 7-8 times speed up settings. The top 3000 molecules (top ∼30%) ranked on the basis of GDR score were selected for docking using Gold 200%-automatic GA settings.
Visual inspection of the docked poses is by far the most labor intensive and critical stage of a screening study. To enhance the percentage of the docked poses which could be visually inspected an additional filtration a criterion was introduced using the docking pose based descriptors (Fig. 4d). This additional filter was used to conduct a ‘virtual’ visual inspection of the top 3000 poses based on their ability to engage the four pharmacophoric sites critical for binding to the SARS-3CLpro binding pocket. These included three hydrophobic occupancy descriptors for S1′, S2, and S4 sites and a hydrogen bond donor descriptor for the S1 site His163 interaction. Binding poses satisfying three or more of these descriptors were considered to have passed the filtration criteria. About 450 molecules emerging from this filtration were ranked based on the GDR score and visual inspection of the top 150 (top 30% based on GDR score) was conducted using the criterion described previously. Thus, instead of looking at the top 30% (∼1000 poses) of the ranked database which is a physically impossible task we could judiciously reduce it to 150 relevant ones as per our knowledge of the binding site requirements. Finally, a chemically diverse set of 81 compounds was selected for biological evaluation. The compounds were tested at a single concentration of 10 μM and one of the compounds PJ169 (Table 1) showed a SARS-3CLpro inhibition of 40% which was considered favorable for carrying out a dose–response study. The compound was found to have an IC50 of 17.2 μM in inhibiting the SARS-3CLpro enzyme.
Looking back at the screening strategy, it is interesting to note that the hit PJ169 was ranked lower than the top 500 (Stage III) in the first phase of the virtual screening and yet was ranked high enough in the second phase of the screening for visual inspection and selection for biological evaluation. In the second phase, the molecule ranked within the top 30% (GDR score) of the 3000 molecules (Stage II′) but was still far down to be selected for visual inspection. The combination of the score based ranking as well as the docking pose based filters helped us in digging deeper into the deck to identify this novel hit. The knowledge based filter generated using the docking pose based descriptors helped us in curtailing the top 3000 poses to 450 molecules (Stage III′). In essence, the molecule of our interest still had the same score but a large number of poses ranked higher than this one but not fulfilling the descriptor criteria were removed by the filter, thereby boosting PJ169’s position and bringing into a selected group which would be put through actual visual inspection. The first hit PJ07 had been seeded into the molecules being used for virtual screening in the extension phase to add another validation check point for the screening protocol. The molecule was retrieved into the top30% (GDR score) of the 450 molecules in the Stage III′ of the extension phase. This provided further evidence that the identification of hits depended primarily on the filtering criteria used in the screening process and to a lesser extent on the protein structure utilized in the screening process. Additionally, it would also be interesting to know whether the inhibitors identified in this virtual screening show any propensity for covalent adduct formation with the enzyme.
2.6. Analysis of predicted binding poses
Structural analyses of the predicted binding poses (Fig. 4a–d) of these two hits reveal important information about the binding site requirements of the enzyme. In case of PJ07, the pyrimidine ring of the quinazoline moiety is located over the catalytic Cys145 while the phenyl ring occupies the S1′ site, undergoing hydrophobic interactions with Thr25 and Leu27. The cyclohexyl ring extends into the S2 pocket and surrogates for the hydrophobic interactions exhibited by the Leucine side chain of the peptide substrate. The thioacetamide linker occupies the critical S1 pocket wherein the carbonyl oxygen of the amide group forms a critical hydrogen bonding interaction with His163. The terminal furan ring forms a hydrophobic interaction with Leu141 while the aromatic oxygen appears to act as a hydrogen bond acceptor for the backbone NH of Asn142. It is also in the vicinity of the amide side chain of Asn142 and could form an alternative hydrogen bond after torsional adjustments of the linker. The other active molecule, PJ169, also appears to occupy the S1′, S1, and S2 sites. The bicyclic ring system occupies the S1 site wherein the oxygen of the amide carbonyl from the first ring forms a hydrogen bond with His163. The phenyl ring of the bicyclic system forms a hydrophobic interaction with Leu141. The two phenyl containing side chains occupy the S2 and S1′ sites which require hydrophobic interactions. The carbonyl oxygen of the amide linker is in close vicinity to form a hydrogen bond with the backbone NH of Glu166. In both the poses, the catalytic cysteine is blocked from the solvent and is not free for substrate processing.
3. Conclusion
A successful structure-based virtual screening was carried out to identify two novel non-peptidic inhibitors of the SARS-3CLpro enzyme with activity in the low micromolar range. Several procedural modifications incorporated into the computational as well as the biological evaluation stages of the screening led to the formulation of an accurate, time efficient, and economical screening methodology. The cascading docking approach utilized in the screening allowed us to attain a reasonable balance of docking accuracy and computation time. The use of knowledge based filters such as the receptor based pharmacophore pre-filter and docking pose based descriptors in the selection process allowed us to incorporate better control into the screening methodology. The docking pose based descriptors also enhanced the realm for the selection of docked poses based on visual inspection and supplemented the role of the scoring functions in the selection process. The biological evaluation of the compounds was carried out in a two-step process which allowed us to identify inhibitors in the low micromolar activity range using a time-efficient and economic experimental setup. Additionally, the pose validation and enrichment studies conducted on the crystal structure of the target enzyme helped in establishing a rigorous and predictive docking protocol which could be utilized in future structure-based drug design efforts.
The putative binding poses (Fig. 5 ) of the inhibitors, PJ07 and PJ169, identified through the screening appeared to mimic the interactions of the peptidic substrate with the active site of SARS-3CLpro. Their respective binding poses suggest that both the ligands form critical hydrogen bond and hydrophobic interactions with the binding site residues. While the inhibitors show occupancy of the S1′, S1, and S2 pockets the S4 pocket is not occupied by these molecules. Occupancy of the S4 pocket using a hydrophobic group and utilization of some of the additional hydrogen bonding sites such as the backbone of Glu166 and Gln192 side chain provide scope for structure-based modification strategies to enhance the binding affinity. The hits obtained through this screening effort would be utilized to conduct substructure/similarity based search as well as synthetic modifications to identify structurally analogous molecules which could provide enhanced binding site occupancy and improved interaction profiles leading to enhanced activity against the enzyme.
Figure 5.
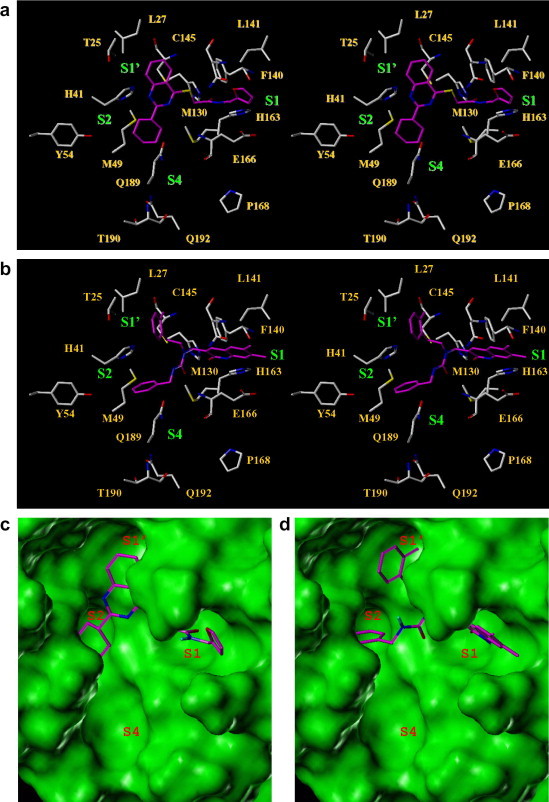
Stereo plots showing the predicted binding poses of (a) PJ07 and (b) PJ169 in the active site of SARS-3CLpro. The carbon atoms of the protein residues are colored in white while those of the ligands are colored in magenta. The corresponding surface views of binding poses of (c) PJ07 and (d) PJ169 showing the shape complementarity of the ligands.
4. Methods
4.1. Screening database and protein preparation
Database pre-filtration of the Asinex Platinum Collection September 2004 (Asinex Ltd Moscow, Russia) was carried out by utilizing background utilities of Sybyl 6.9 (Tripos Inc., St. Louis, MO) incorporated in a c-shell script. The raw database comprising around 120,000 molecules was cleared of salts and mixtures using the dbstripsalt utility. The database was further filtered using the dbslnfilter utility on the basis of drug-like parameters: 200 ⩽ mol wt ⩽ 500, 1 ⩽ hydrogen bond acceptor ⩽ 10, 1 ⩽ hydrogen bond donor ⩽ 5, C log p ⩽ 5, 3 ⩽ Rotatable bonds ⩽ 10, and Aromatic rings ⩽4. Molecules with problematic groups such as metals, N-oxides, aldehydes, chloramines, nitrogen/sulfur mustards, and isocyanides were also removed. The curtailed database was further filtered using the ADME_absorbtion and ADME_solubility models of Cerius2 (Accelrys Inc., San Diego, CA). Molecules with a predicted absorbtion level of 0 (good absorbtion) and predicted solubility level of 2–4 (low-optimal) were selected for the next stage. The filtered database comprised of ∼32,000 molecules which was submitted for 3D-coordinates generation in Concord (Tripos Inc., St. Louis, MO). The reported active molecules (including the ketoglutamine series) used in the docking validation studies were sketched in Sybyl 6.9. Protonation states, bond types, and atom types for the molecules were assigned manually. The structures were refined using 2000 steps of conjugate gradient to a RMS convergence of 0.01 kcal/mol Å using the Tripos force field and Gasteiger–Huckel partial charge method.
The dimeric structures of the SARS-3CLpro were utilized for all virtual screening runs. Both SARS-3CLpro structures used in the study, 1UK4 and 2AMD (PDB code), contain co-crystallized ligands covalently bound to the catalytic cysteine residue. To prepare the protein for docking, the covalent bond between the ligand and the protein residue was deleted. Protonation state, tautomeric state, and hydrogen addition for the protein were carried out using the PPREP (Schrodinger, LLC, Portland, OR) utility while the co-crystallized ligands were handled manually. Manual protonation and tautomeric state modifications of certain key binding site residues were carried out. The Glu166 and His172 residue participates in a salt bridge interaction and was therefore modeled in the charged state. His163 forms a critical hydrogen bond donor interaction with the Glutamine (P1 residue) side chain oxygen of the natural substrate and was modeled in the His-ε protonation state. The catalytic diad Cys145-His41 was modeled in the neutral state. The all hydrogen protein–ligand complex was then submitted to restrained molecular mechanics refinement using the OPLS2001 force field incorporated in the IMPREF (Schrodinger, LLC, Portland, OR) protein structure refinement utility. The final refined structure was used for the docking calculations.
4.2. Docking validation and structure-based virtual screening
Docking studies for the first phase were carried out using Gold 2.2 (CCDC, Cambridge, UK). The binding site was defined using a cavity detection algorithm by searching 10 Ǻ around the sulfur atom of Cys145. The first phase validation study was carried out using the Gold standard mode and the Goldscore function for pose selection. Scoring of the ligand binding poses using multiple scoring functions was conducted using the Cscore module of Sybyl 6.9. Pose validation and enrichment studies in the extension phase of the screening were conducted in Gold 3.0.1 using automatic GA settings-200% accuracy level. The final docking protocol for the extension phase was identical to the first phase in terms of binding site definition and scoring function for pose selection. Hydrophobic constraints (Fig. 4d) were defined as spheres of 2 Ǻ radius defined around the centroid of the heavy atoms of the hydrophobic groups while a protein hydrogen bond donor constraint (Fig. 4d) was defined using the N(ε)–H of His163. The enrichment study was conducted using a dataset comprising of 19 actives (Supplementary information, Fig. S2) and 897 dummy (inactive) molecules taken from the ZINC database (http://zinc.docking.org/). The range scaled scores were calculated using expression 1 implemented in a python script.
| (1) |
where S n(Range scaled) is the range scaled score, S n is the score obtained for a molecule within the docked set, S max is the maximum score obtained for a molecule within the docked set, S min is the minimum score obtained for a molecule within the docked set.
4.3. Pharmacophore pre-filter and docking pose based descriptors
A pharmacophore pre-filter (Fig. 4c) was generated in Catalyst 4.9 (Accelrys Inc., San Diego, CA) using the 2AMD (PDB code) co-crystallized ligand pose. Hydrophobic features were defined as spheres with a tolerance value of 1.5 Ǻ and were located on the geometric centroid of the heavy atoms of the relevant ligand side chains. The hydrogen bond acceptor feature was defined using the side chain carbonyl oxygen of the P1 ketoglutamine group. Each molecule was enumerated by 100 conformers generated in the ‘fast’ mode. Fitting of the conformers to the pharmacophore was handled using the ‘fast fit’ method. Docking pose based descriptors (Fig. 4d) for ‘virtual’ visual inspection were generated using Silver 1.1 (CCDC, Cambridge, UK) using the 2AMD (PDB code) co-crystallized ligand protein complex. Four descriptors (three hydrophobic, one hydrogen bond) were defined and any given pose could attain a score ranging from 0 to 4. The hydrophobic descriptor definition was similar to that of the hydrophobic feature of the pharmacophore pre-filter but consisted of spheres of 2 Å radius. Occupancy of ⩾75% of the sphere volume by the ligand hydrophobic atoms was considered as positive occupancy. The hydrogen bond donor descriptor was defined using the N(ε)–H of His163 and had the geometric criteria of D–H–A angle ⩾120° and D–A distance ⩽3.5 Å. Molecular diversity was calculated using the dissimilarity selection module of Sybyl 6.9.
4.4. Enzyme preparation and bioassay
The methods used for preparation and assay of SARS-3CLpro were those described previously by Bacha et al.48 Briefly, the plasmid-encoded SARS-3CLpro with a polyhistidine tag was expressed in BL21 Star DE3 Escherichia coli competent cells. Four 1-l cultures were grown and induced. Four pellets of approximately 3 g each were harvested and stored at −80 °C. For the purification process the pellets were resuspended in lysis buffer (50 mM potassium chloride (pH 7.8), 400 mM sodium chloride, 100 mM potassium chloride, 10% glycerol, 0.5% Triton X-100, and 10 mM imidazole) and broken by French press. The lysate was centrifuged and the supernatant was collected and loaded on a nickel affinity column (Pharmacia) that had been pre-equilibrated with binding buffer (50 mM sodium phosphate (pH 7.8), 300 mM sodium chloride, and 10 mM imidazole). The column was washed with binding buffer and the protease was eluted with binding buffer plus 300 mM imidazole. The histidine tag was removed by thrombin cleavage. The enzyme showed >95% purity as assessed by SDS–PAGE. The purified protease was concentrated and stored in storage buffer (10 mM sodium phosphate (pH 7.4), 10 mM NaCl, 1 mM TCEP, 0.5 mM EDTA, and 10% glycerol) at −80 °C. The purified enzyme was assayed using a fluorogenic peptide (Dabcyl-TSAVLQSGFR-Edans) and found to be active. The conditions for the assay were 10 mM sodium phosphate (pH 7.4), 10 mM NaCl, 1 mM TCEP, 0.5 mM EDTA, 50 μM fluorogenic peptide, and 5 μM SARS-3CLpro. The K m of the fluorogenic peptide was determined by combining various concentrations of the fluorogenic peptide (0–100 μM) with 1 μM of SARS-3CLpro and was determined to be 10.3 ± 1.9 μM. Compounds were screened for inhibitory activity at 10 μM under the following conditions: 10 mM sodium phosphate (pH 7.4), 10 mM NaCl, 1 mM TCEP, 0.5 mM EDTA, 50 μM fluorogenic peptide, and 5 μM SARS-3CLpro. Increase in fluorescence was measured for 10 min at 25 °C. Compounds showing significant inhibition were utilized in conducting a dose–response study.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2008.01.011.
Supplementary data
References and notes
- 1.Lee N., Hui D., Wu A., Chan P., Cameron P., Joynt G.M., Ahuja A., Yung M.Y., Leung C.B., To K.F., Lui S.F., Szeto C.C., Chung S., Sung J.J. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 2.Poutanen S.M., Low D.E., Henry B., Finkelstein S., Rose D., Green K., Tellier R., Draker R., Adachi D., Ayers M., Chan A.K., Skowronski D.M., Salit I., Simor A.E., Slutsky A.S., Doyle P.W., Krajden M., Petric M., Brunham R.C., McGeer A.J. N. Engl. J. Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- 3.Fouchier R.A., Kuiken T., Schutten M., van Amerongen G., van Doornum G.J., van den Hoogen B.G., Peiris M., Lim W., Stohr K., Osterhaus A.D. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsang K.W., Ho P.L., Ooi G.C., Yee W.K., Wang T., Chan-Yeung M., Lam W.K., Seto W.H., Yam L.Y., Cheung T.M., Wong P.C., Lam B., Ip M.S., Chan J., Yuen K.Y., Lai K.N. N. Engl. J. Med. 2003;348:1977–1985. doi: 10.1056/NEJMoa030666. [DOI] [PubMed] [Google Scholar]
- 5.Drosten C., Preiser W., Gunther S., Schmitz H., Doerr H.W. Trends Mol. Med. 2003;9:325–327. doi: 10.1016/S1471-4914(03)00133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 7.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 8.Ziebuhr J., Heusipp G., Siddell S.G. J. Virol. 1997;71:3992–3997. doi: 10.1128/jvi.71.5.3992-3997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dougherty W.G., Semler B.L. Microbiol. Rev. 1993;57:781–822. doi: 10.1128/mr.57.4.781-822.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 11.Ziebuhr J., Snijder E.J., Gorbalenya A.E. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 12.Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Proc. Natl. Acad. Sci. USA. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen S., Chen L., Tan J., Chen J., Du L., Sun T., Shen J., Chen K., Jiang H., Shen X. J. Biol. Chem. 2005;280:164–173. doi: 10.1074/jbc.M408211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang C., Wei P., Fan K., Liu Y., Lai L. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- 16.Shi J., Wei Z., Song J. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu M.F., Kuo C.J., Chang K.T., Chang H.C., Chou C.C., Ko T.P., Shr H.L., Chang G.G., Wang A.H., Liang P.H. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. PLoS Biol. 2005;3:1742–1752. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu T., Ooi A., Lee H.C., Wilmouth R., Liu D.X., Lescar J. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 2005;61:964–966. doi: 10.1107/S1744309105033257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee T.W., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N. J. Mol. Biol. 2005;353:1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh A.K., Xi K., Ratia K., Santarsiero B.D., Fu W., Harcourt B.H., Rota P.A., Baker S.C., Johnson M.E., Mesecar A.D. J. Med. Chem. 2005;48:6767–6771. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 24.Lu I.L., Mahindroo N., Liang P.H., Peng Y.H., Kuo C.J., Tsai K.C., Hsieh H.P., Chao Y.S., Wu S.Y. J. Med. Chem. 2006;49:5154–5161. doi: 10.1021/jm060207o. [DOI] [PubMed] [Google Scholar]
- 25.Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; Zhang, X.C.; Liao, M.; Bartlam, M.; Rao, Z. J. Virol.2007, Published online ahead of print December 19 2007.
- 26.Yin J., Niu C., Cherney M.M., Zhang J., Huitema C., Eltis L.D., Vederas J.C., James M.N. J. Mol. Biol. 2007;371:1060–1074. doi: 10.1016/j.jmb.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee T.W., Cherney M.M., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N. J. Mol. Biol. 2007;366:916–932. doi: 10.1016/j.jmb.2006.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan J., Verschueren K.H., Anand K., Shen J., Yang M., Xu Y., Rao Z., Bigalke J., Heisen B., Mesters J.R., Chen K., Shen X., Jiang H., Hilgenfeld R. J. Mol. Biol. 2005;354:25–40. doi: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang S., Chen S.J., Hsu M.F., Wu J.D., Tseng C.T., Liu Y.F., Chen H.C., Kuo C.W., Wu C.S., Chang L.W., Chen W.C., Liao S.Y., Chang T.Y., Hung H.H., Shr H.L., Liu C.Y., Huang Y.A., Chang L.Y., Hsu J.C., Peters C.J., Wang A.H., Hsu M.C. J. Med. Chem. 2006;49:4971–4980. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- 30.Yang H., Bartlam M., Rao Z. Curr. Pharm. Des. 2006;12:4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 31.Lai L., Han X., Chen H., Wei P., Huang C., Liu S., Fan K., Zhou L., Liu Z., Pei J., Liu Y. Curr. Pharm. Des. 2006;12:4555–4564. doi: 10.2174/138161206779010396. [DOI] [PubMed] [Google Scholar]
- 32.Goetz D.H., Choe Y., Hansell E., Chen Y.T., McDowell M., Jonsson C.B., Roush W.R., McKerrow J., Craik C.S. Biochemistry. 2007;46:8744–8752. doi: 10.1021/bi0621415. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H.Z., Zhang H., Kemnitzer W., Tseng B., Cinatl J., Jr., Michaelis M., Doerr H.W., Cai S.X. J. Med. Chem. 2006;49:1198–1201. doi: 10.1021/jm0507678. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh A.K., Xi K., Grum-Tokars V., Xu X., Ratia K., Fu W., Houser K.V., Baker S.C., Johnson M.E., Mesecar A.D. Bioorg. Med. Chem. Lett. 2007;17:5876–5880. doi: 10.1016/j.bmcl.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L., Chen S., Gui C., Shen J., Shen X., Jiang H. J. Biomol. Screen. 2006;11:915–921. doi: 10.1177/1087057106293295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dooley A.J., Shindo N., Taggart B., Park J.G., Pang Y.P. Bioorg. Med. Chem. Lett. 2006;16:830–833. doi: 10.1016/j.bmcl.2005.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Al-Gharabli S.I., Shah S.T., Weik S., Schmidt M.F., Mesters J.R., Kuhn D., Klebe G., Hilgenfeld R., Rademann J. Chembiochem. 2006;7:1048–1055. doi: 10.1002/cbic.200500533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J., Pettersson H.I., Huitema C., Niu C., Yin J., James M.N., Eltis L.D., Vederas J.C. J. Med. Chem. 2007;50:1850–1864. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- 39.Chen L., Gui C., Luo X., Yang Q., Gunther S., Scandella E., Drosten C., Bai D., He X., Ludewig B., Chen J., Luo H., Yang Y., Yang Y., Zou J., Thiel V., Chen K., Shen J., Shen X., Jiang H. J. Virol. 2005;79:7095–7103. doi: 10.1128/JVI.79.11.7095-7103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Z., Huang C., Fan K., Wei P., Chen H., Liu S., Pei J., Shi L., Li B., Yang K., Liu Y., Lai L. J. Chem. Inf. Comput. Sci. 2005;45:10–17. doi: 10.1021/ci049809b. [DOI] [PubMed] [Google Scholar]
- 41.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirayama K., Aoki S., Nishikawa K., Matsumoto T., Wada K. Bioorg. Med. Chem. 2007;15:6810–6818. doi: 10.1016/j.bmc.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 43.Luzhkov V.B., Selisko B., Nordqvist A., Peyrane F., Decroly E., Alvarez K., Karlen A., Canard B., Qvist J. Bioorg. Med. Chem. 2007;15:7795–7802. doi: 10.1016/j.bmc.2007.08.049. [DOI] [PubMed] [Google Scholar]
- 44.Hegyi A., Ziebuhr J. J. Gen. Virol. 2002;83:595–599. doi: 10.1099/0022-1317-83-3-595. [DOI] [PubMed] [Google Scholar]
- 45.Jain R.P., Pettersson H.I., Zhang J., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James M.N., Wishart D.S., Vederas J.C. J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 46.Desai P.V., Patny A., Sabnis Y., Tekwani B., Gut J., Rosenthal P., Srivastava A., Avery M. J. Med. Chem. 2004;47:6609–6615. doi: 10.1021/jm0493717. [DOI] [PubMed] [Google Scholar]
- 47.Desai P.V., Patny A., Gut J., Rosenthal P.J., Tekwani B., Srivastava A., Avery M. J. Med. Chem. 2006;49:1576–1584. doi: 10.1021/jm0505765. [DOI] [PubMed] [Google Scholar]
- 48.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.