

Abstract
We previously showed that an intraperitoneal infection with mouse hepatitis virus (MHV) persists in interferon-γ (IFN-γ)-deficient C57BL/6 (B6-GKO) mice and results in subacute fatal peritonitis, which bears a resemblance to feline infectious peritonitis. To examine the role of other host factors in MHV infection in mice, IFN-γ-deficient mice with a BALB/c background (BALB-GKO) were infected intraperitoneally with MHV and compared with B6-GKO mice. In contrast to B6-GKO mice, BALB-GKO mice died within 1 week due to acute hepatic failure. The viral titer of the liver in BALB-GKO mice was significantly higher than that in B6-GKO mice. All hepatocytes in BALB-GKO mice were necrotic at 5 days post-infection, which was clearly distinct from large but limited lesion in the liver from infected B6-GKO mice. The serum alanine aminotransferase activity of infected BALB-GKO mice were higher than that of B6-GKO mice and was paralleled with the severity of the pathological changes and viral titers in infected mice. Administration of exogenous IFN-γ to BALB-GKO partially inhibited the acute death. These results indicate that BALB-GKO and B6-GKO mice clearly show different diseases following MHV infection, although wild type counterparts of both mice apparently showed the same clinical course after MHV infection.
Keywords: Acute hepatic failure, Genetic background, Interferon-γ-deficient mice, Mouse hepatitis virus
1. Introduction
Murine coronaviruses induce a variety of diseases, including hepatitis, enteritis and encephalitis in mice, depending on the virus strain, infection route, age, genetic background and immune status of the hosts (Kyuwa and Stohlman, 1990, Compton et al., 1993). Due to frequent outbreaks of murine coronavirus infection in laboratory animal facilities, the viruses are detested as the most detrimental infectious agent for laboratory mice. On the other hand, experimental murine coronavirus infections provide useful animal models of human diseases. In particular, murine coronavirus-induced demyelination is utilized in investigating the pathogenesis of demyelinating diseases, such as multiple sclerosis in humans (Liu et al., 2000, Marten et al., 2000, Wu et al., 2000). We previously reported a reduced viral clearance and a resultant systemic persistent infection associated with disseminated granulomatous serositis in IFN-γ-deficient C57BL/6 (B6-GKO) mice after intraperitoneal (i.p.) infection with murine coronavirus, strain JHM (JHMV) (Kyuwa et al., 1998). The disease bears a resemblance to feline infectious peritonitis, another coronavirus-induced fatal disease in cats. The same disease was also observed in naturally infected B6 mice deficient in IFN-γ (France et al., 1999) as well as IFN-γ, IL-10 double knockout mice (Boivin and Smith, 1998). This experimental infection may provide a unique model to examine the pathological mechanism of virus-induced peritonitis.
As mentioned above, the genetic background is a decisive factor that determines the fate of murine coronavirus infection in mice. For example, whereas SJL/J mice are resistant to intracerebral JHMV infection, other strains of laboratory mice are susceptible (Stohlman and Frelinger, 1978). Recently, it has been shown that the virus receptor genotype is correlated with susceptibility (Ohtsuka and Taguchi, 1997). The genetic profile of the mouse susceptible to MHV-3, another murine coronavirus, is distinct from that of JHMV (Levy et al., 1981), suggesting the complexity of susceptibility to murine coronavirus infection in vivo.
To see the role of the mouse genetic background in murine coronavirus infection in IFN-γ-deficient mice, we attempted an experimental i.p. MHV infection in IFN-γ-deficient mice with a BALB/c background (BALB-GKO) and compared with B6-GKO mice. Although previous studies demonstrated that wild type BALB/c mice suffered an acute hepatitis that was resolved within a few weeks after i.p. infection with JHMV similar to wild type B6 mice, BALB-GKO mice died within 1 week after JHMV infection, quite differently to B6-GKO mice.
2. Materials and methods
2.1. Mice
Production of mice deficient in IFN-γ was described previously (Tagawa et al., 1997). A 129/SvJ mouse with a disrupted IFN-γ gene was backcrossed with B6 and BALB/c mice more than eight times. The genotype of the mice was determined by PCR as described previously (Tagawa et al., 1997), except for a substitute primer (5′-GTGCTGTGCTCTGTGGATGAGAAA-3′) instead of primer 1 and 8- to 12-week-old female mice were used. Breeding mice were maintained in a laminar flow rack in an environmentally controlled area and routinely checked to ensure that they were serologically free of murine coronaviruses and other pathogenic agents. Infected mice were kept in a safety cabinet in a different area. The experiments were conducted according to institutional ethical guidelines for animal experiments and safety guidelines for gene manipulation experiments.
2.2. Virus
The DL variant of JHMV was propagated and plaque assayed on DBT cells, as described previously (Kyuwa et al., 1998). A single pool of virus was divided into aliquots and stored at −70 °C until use. The mice were infected i.p. with 1×106 PFU of JHMV in a volume of 0.2 ml. For viral isolation and titration, 10% tissue homogenates of samples were serially diluted and plaque assayed on DBT cells (Kyuwa et al., 1998).
2.3. Analysis of hepatocellular injury
For biochemical assessment of liver injury, the sera of JHMV-infected mice were collected and serum alanine aminotransferase (ALT) activity was determined by an enzymatic rate method with a commercial kit according to the manufacturer's instructions (Iatron Laboratories, Tokyo) (Kyuwa et al., 1998).
2.4. Interferon assay
Serum was collected and irradiated with ultraviolet rays to inactivate JHMV. Interferon activity was determined using vesicular stomatitis virus and L-929 cells in 96-well flat bottom plates. Inhibition of viral cytopathicity was detected by Naphthol blue-black staining and by taking a photometric reading at 610 nm.
2.5. Histopathology
Tissue samples were fixed in 10% phosphate-buffered formalin, embedded in paraffin, sectioned and stained with hematoxylin and eosin.
2.6. Administration of recombinant IFN-γ
Recombinant murine IFN-γ was kindly provided by Shionogi and Co. Ltd. (Osaka, Japan). BALB-GKO mice (n=7) were infected i.p. with 106 PFU of JHMV and injected with 1×104 U of IFN-γ 2 h before and 1 day after infection. Control BALB-GKO mice (n=7) were infected i.p. with 106 PFU of JHMV and injected with the same volume of phosphate-buffered saline (PBS).
2.7. Statistical analysis
Evaluation of statistical differences between data obtained from mutant and control mice were assessed for statistical significance by Student's t-test. A difference was considered statistically significant at P<0.05.
3. Results
3.1. Acute death of BALB-GKO mice after i.p. JHMV infection
A previous study showed that i.p. infection with JHMV induced a subacute granulomatous serositis in B6-GKO mice (Kyuwa et al., 1998). To see whether the same disease was induced in IFN-γ-deficient mice with other genetic backgrounds, IFN-γ−/−, IFN-γ+/− and IFN-γ+/+ with either B6 or BALB/c background were infected i.p. with 106 PFU of JHMV and observed for 50 days after infection. All the wild type and heterozygous B6 mice survived and appeared healthy throughout the experiment (Fig. 1 A). Although none of the B6-GKO mice died during the first week, their survival rate gradually decreased, as previously described (Kyuwa et al., 1998). All the wild type and heterozygous BALB/c mice survived and appeared healthy throughout the experiment, similar to mice with a B6 background (Fig. 1B). However, BALB-GKO mice died within 1 week after JHMV infection, in distinct contrast to B6-GKO mice. These results suggest that JHMV induces a different type of disease in IFN-γ-deficient mice of different genetic backgrounds.
Fig. 1.
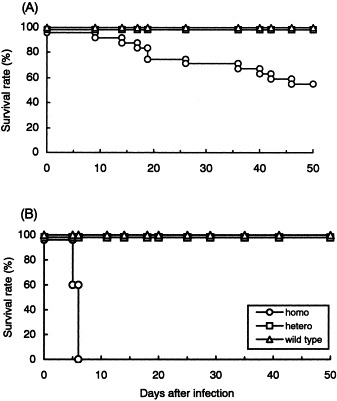
Mortality of IFN-γ-deficient B6 and BALB/c mice after i.p. JHMV infection. Twenty-four homozygous, ten heterozygous and ten wild-type B6 mice (A), and homozygous, heterozygous and wild-type BALB/c mice, ten of each (B), were infected i.p. with 106 PFU of JHMV and monitored for 50 days.
3.2. Histopathological changes after JHMV infection in IFN-γ-deficient mice
To understand JHMV infection in IFN-γ-deficient mice, histopathological changes of the liver in IFN-γ−/− and IFN-γ/+/− mice with either B6 or BALB/c background were compared. As reported previously (Kyuwa et al., 1998), JHMV-induced lesions at 5 days post-infection in B6-GKO mice were larger than those observed in heterozygous counterparts of the same background (Fig. 2 a,b). On the other hand, massive necrosis was observed in BALB-GKO mice at 5 days post-infection (Fig. 2c), which was clearly different from focal necrotic lesion observed in B6-GKO mice (Fig. 2a). In the liver of BALB-GKO mice, inflammatory cells were relatively few and lymphoid cells were scarcely infiltrated. JHMV-infected IFN-γ+/− mice with a BALB/c background had focal necrotic lesions the same as or slightly severer than those observed in IFN-γ+/− mice with a B6 background (Fig. 2d). Although a clear difference was observed between IFN-γ−/− and IFN-γ+/− mice with a BALB/c background in the histopathological changes of the liver at 5 days post-infection, it was not obvious at 3 days post-infection; both IFN-γ−/− and IFN-γ+/− mice showed focal inflammatory lesions in the liver (Fig. 2e,f).
Fig. 2.



Histopathological changes in the livers of IFN-γ-deficient BALB/c and B6 mice after i.p. JHMV infection. IFN-γ−/− and IFN-γ+/− mice with a B6 background (a, b) and those with a BALB/c background (c, d) at 5 days post-infection. IFN-γ−/− and IFN-γ+/− mice with a BALB/c background (e, f) at 3 days post-infection. (Magnification ×175).
3.3. Serum ALT activity in JHMV-infected IFN-γ-deficient mice
A slight rise in serum ATL activity was observed at 3 and 5 days post-infection in IFN-γ−/− and IFN-γ+/− mice with a B6 background, as reported previously (Table 1 ). In concord with severe histopathological changes in the liver of BALB-GKO mice, a remarkable increase in ALT activity was observed in BALB-GKO mice at 5 days post-infection. JHMV-infected IFN-γ+/− BALB/c mice also showed a moderate increase in ATL activity.
Table 1.
Serum ALT activity in IFN-γ−/− and IFN-γ+/− mice with either B6 or BALB/c background after i.p. JHMV infection
| Background of mice | Days after infection | ALT activity (KU/l)a |
|
|---|---|---|---|
| IFN-γ−/− | IFN-γ+/− | ||
| B6 | 3 | 74±37 | 80±100 |
| 5 | 99±72 | 76±6 | |
| BALB/c | 3 | 41±11 | 110±16 |
| 5 | 2700±260 | 860±4 | |
Samples were collected at the times indicated (n=3–5). Data are expressed as the mean±S.D.
Levels of ALT activity in uninfected mice were <30 KU/l.
3.4. Viral growth in the liver in IFN-γ-deficient mice
The viral titer in the liver of B6-GKO mice was slightly higher than that of heterozygous mice with a B6 background (Table 2 ), as reported previously (Kyuwa et al., 1998). The essentially same phenomenon was observed in mice with a BALB/c background. The viral titers of BALB-GKO mice were 20- and 6-fold higher than those of heterozygous mice at 3 and 5 days post-infection, respectively. However, it is worth noting that values of a BALB/c background were greater than those of a B6 background. For example, the viral titer of BALB-GKO mice at 5 days post-infection was significantly higher than that of B6-GKO mice.
Table 2.
Viral growth in the liver in IFN-γ−/− and IFN-γ+/− mice with either B6 or BALB/c background after i.p. JHMV infection
| Background of mice | Days after infection | Viral titer (log PFU/g) |
|
|---|---|---|---|
| IFN-γ−/− | IFN-γ+/− | ||
| B6 | 3 | 4.4±0.2 | 3.3±0.3 |
| 5 | 4.7±0.7 | 4.2±0.4 | |
| BALB/c | 3 | 5.1±0.2 | 3.8±0.4 |
| 5 | 6.5±0.2 | 5.7±0.3 | |
Samples were collected at the times indicated (n=4–5). Data are expressed as the mean±S.D.
3.5. Serum IFN activity in IFN-γ-deficient mice
A relatively high level of IFN activity was observed in IFN-γ-deficient mice with B6, as well as BALB/c background, at 24 h post-infection (Table 3 ). The same or higher level of IFN activity was detected in heterozygous mice with a BALB/c background, whereas IFN activity in heterozygous mice with a B6 background was noticeably low. We confirmed that no IFN-γ was detected in the samples from IFN-γ-deficient mice by enzyme-linked immunosorbent assay (data not shown).
Table 3.
Serum IFN activity in IFN-γ−/− and IFN-γ+/− mice with either B6 or BALB/c background after i.p. JHMV infection
| Background of mice | IFN activity |
|
|---|---|---|
| IFN-γ−/− | IFN-γ+/− | |
| B6 | 7400±1100 | 620±250 |
| BALB/c | 7400±2300 | 10,000±1100 |
Samples were collected at 24 h after infection (n=3–4). Data are expressed as the mean±S.D.
3.6. Effect of IFN-γ administration on JHMV infection in BALB-GKO mice
Since exogenous IFN-γ administration partially inhibited JHMV-induced subacute fatal disease in B6-GKO mice (Kyuwa et al., 1998), the same experiment was conducted using JHMV-induced acute disease in BALB-GKO mice. The administration of recombinant IFN-γ clearly increased the survival rate of BALB-GKO mice following JHMV infection compared with those treated with PBS (Fig. 3 ).
Fig. 3.

Effect of IFN-γ treatment on JHMV-induced acute disease in BALB-GKO mice. BALB-GKO mice (n=7) were infected i.p. with 106 PFU of JHMV and injected i.p. with 104 U of recombinant murine IFN-γ, 2 h before and 1 day after infection. Control BALB-GKO mice (n=7) were infected i.p. with 106 PFU and injected with the same volume of PBS. Both groups of mice were monitored for up to 50 days post-infection.
4. Discussion
In this study we demonstrated that i.p. infection with JHMV induced acute death of IFN-γ-deficient mice with a BALB/c background. High serum ALT activity and massive necrosis of the liver suggest that the mice died due to acute hepatic failure. The same disease was not observed in heterozygous or wild type mice with the same background and administration of exogenous IFN-γ into BALB-GKO mice, in part, protected the virus-induced acute death. These results suggest that IFN-γ play an essential role in protecting mice with a BALB/c background from JHMV infection. The disease, however, was noticeably distinct from subacute granulomatous peritonitis in IFN-γ-deficient mice with a B6 background that we reported previously (Kyuwa et al., 1998). Whereas hepatitis was observed in B6-GKO mice in the acute phase, it was not progressing and had been almost cured in the subacute phase when granulomatous peritonitis was evident (Kyuwa et al., 1998). This result was unexpected, because wild type mice with both genetic backgrounds are resistant to i.p. infection with JHMV (Kyuwa et al., 1988, Kyuwa et al., 1996, Stohlman et al., 1992) and both strains have the same genotype of MHV receptor (Ceacam1) that is believed to be important in determining mouse susceptibility (Ohtsuka and Taguchi, 1997). Taken together, the data presented here suggest that genetic factor(s) other than IFN-γ or MHV receptor affect MHV infection in mice.
We found that the viral titer of the liver from BALB-GKO mice was significantly higher than that of B6-GKO mice. We are not sure whether this is a critical factor in determining the profile of MHV infection in IFN-γ-deficient mice because heterozygous mice with a BALB/c background also had a relatively high viral burden in the liver. Although the viral titers in mice of a BALB/c background seem to be somewhat higher than those of a B6 background, it remains obscure whether the difference is dependent upon the viral replication rate in hepatocytes or extrinsic effects, such as immune responses.
It is interesting that the histopathological changes in the liver in IFN-γ−/− and IFN−γ+/− mice with a BALB/c background were drastically diverged between 3 and 5 days post-infection. It suggests that IFN-γ itself or together with other factor(s) would play a role in preventing the expansion of virus-induced lesions during the period. In a reconstitution experiment, we could protect only half of JHMV-infected BALB-GKO mice by administration of IFN-γ 2 h before and 1 day after infection. We should treat them once more with IFN-γ during the period, for example at 3 days post-infection, to protect them completely.
Although some papers reported on infectious diseases in cytokine-deficient C57BL/6 and BALB/c mice (Hu et al., 2000, Kamradt et al., 2000, Potter et al., 2000), there are few papers that report that the disruption of a single cytokine gene in mice generates notably different consequences among mice with different genetic backgrounds. Therefore, the results are unique and this experimental model may provide a distinctive opportunity to address the mechanism(s) underlying the protection against viral infections in mammals.
The importance of type I IFN in the defense against viral infections has been demonstrated by studies on mice with a targeted disruption of the INF-α/β receptor gene (Müller et al., 1994, Hwang et al., 1995). The effectiveness of type I IFN in protecting against murine coronavirus infection in mice was also reported (Uetsuka et al., 1996, Matsuyama et al., 2000). In the present study, we indicated that serum interferon activities in JHMV-infected B6-GKO and BALB-GKO were almost the same. The results suggest that type I IFN is not a key factor in the different profiles of JHMV infection in BALB-GKO and B6-GKO mice. It was of interest that heterozygous mice with a BALB/c background also showed a high level of serum interferon activity, which may play a role in concert with IFN-γ in the protection of the mice that have a relatively high viral burden in the liver.
Although we do not know the host genetic factor(s) which operate differentially in B6-GKO and BALB-GKO mice to induce the different form of disease between the two mice strains, we showed not only a disrupted-gene, but also other genetic factor(s) may affect the phenotype of the genetically engineered mice after virus infection. On the other hand, it has been reported that some gene-knockout mice show no typical phenotype due to being compensated by other genes, which have the same function to the gene disrupted (Doetschman, 1999). The gene-knockout technology thus, not only offers a good opportunity to see the function of the gene in vivo, it sometimes show us the existence of other gene(s) involved in the phenomenon of interest.
Acknowledgments
This work was supported in part by a Grant-in-Aid for scientific research from the Japan Society for the Promotion of Science.
References
- Boivin G.P, Smith F.N.L. Idiopathic granulomas in IFN-γ−/−, IL-10−/− double knockout mice. Lab. Anim. Sci. 1998;48:419. [Google Scholar]
- Compton S.R, Barthold S.W, Smith A.L. The cellular and molecular pathogenesis of coronaviruses. Lab. Anim. Sci. 1993;43:15–28. [PubMed] [Google Scholar]
- Doetschman T. Interpretation of phenotype in genetically engineered mice. Lab. Anim. Sci. 1999;49:137–143. [PubMed] [Google Scholar]
- France M.P, Smith A.L, Stevenson R, Barthold S.W. Granulomatous peritonitis and plueritis in interferon-γ gene knockout mice naturally infected with mouse hepatitis virus. Aust. Vet. J. 1999;77:600–604. doi: 10.1111/j.1751-0813.1999.tb13199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Mayadas-Norton T, Tanaka K, Chan J, Salgame P. Mycibacterium tuberculosis infection in complement receptor 3-deficient mice. J. Immunol. 2000;165:2596–2602. doi: 10.4049/jimmunol.165.5.2596. [DOI] [PubMed] [Google Scholar]
- Hwang S.Y, Hertzog P.J, Holland K.A, Sumarsono S.H, Tymms M.J, Hamilton J.A, Whitty G, Bertoncello I, Kola I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc. Natl. Acad. Sci. USA. 1995;92:11284–11288. doi: 10.1073/pnas.92.24.11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamradt A.E, Greiner M, Ghiare P, Kaufmann S.H. Helicobacter pylori infection in wild-type and cytokine-deficient C57BL/6 and BALB/c mouse strains. Microbes Infect. 2000;2:593–597. doi: 10.1016/s1286-4579(00)00367-1. [DOI] [PubMed] [Google Scholar]
- Kyuwa, S., Yamaguchi, K., Hayami, M., Hilgers, J., Fujiwara, K., 1988. Spontaneous production of interleukin-2 and interleukin-3 by spleen cells from mice infected with mouse hepatitis virus type 4. J. Virol. 62, 3506–3508. [DOI] [PMC free article] [PubMed]
- Kyuwa S, Stohlman S.A. Pathogenesis of a neurotropic murine coronavirus, strain JHM in the central nervous system of mice. Semin. Virol. 1990;1:273–280. [Google Scholar]
- Kyuwa, S., Machii, K., Okumura, A., Toyada, Y., 1996. Characterization of T cells expanded in vivo during primary mouse hepatitis virus infection in mice. J. Vet. Med. Sci. 58, 431–437. [DOI] [PubMed]
- Kyuwa S, Tagawa Y, Shibata S, Doi K, Machii K, Iwakura Y. Murine coronavirus-induced subacute fatal peritonitis in C57BL/6 mice deficient in gamma-interferon. J. Virol. 1998;72:9286–9290. doi: 10.1128/jvi.72.11.9286-9290.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy G.A, Leibowitz J.L, Edgington T.S. Induction of monocyte procoagulant activity by murine hepatitis virus type 3 parallels disease susceptibility in mice. J. Exp. Med. 1981;154:1150–1163. doi: 10.1084/jem.154.4.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.T, Chen B.P, Oertel P, Buchmeier M.J, Armstrong D, Hamilton T.A, Lane T.E. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J. Immunol. 2000;165:2327–2330. doi: 10.4049/jimmunol.165.5.2327. [DOI] [PubMed] [Google Scholar]
- Marten N.W, Stohlman S.A, Bergmann C.C. Role of viral persistence in retaining CD8+ T cells within the central nervous system. J. Virol. 2000;74:7903–7910. doi: 10.1128/jvi.74.17.7903-7910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Henmi S, Ichihara N, Sone S, Kikuchi T, Ariga T, Taguchi F. Protective effects of murine recombinant interferon-beta administered by intravenous, intramuscular or subcutaneous route on mouse hepatitis virus infection. Antivir. Res. 2000;47:131–137. doi: 10.1016/s0166-3542(00)00097-8. [DOI] [PubMed] [Google Scholar]
- Müller U, Steinhoff U, Reis L.F, Hemmi S, Pavlovic J, Zinkernagel R.M, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Ohtsuka N, Taguchi F. Mouse susceptibility to mouse hepatitis virus infection is linked to viral receptor genotype. J. Virol. 1997;71:8860–8863. doi: 10.1128/jvi.71.11.8860-8863.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter M.R, Noben-Trauth N, Weis J.H, Teuscher C, Weis J.J. Interleukin-4 (IL-4) and IL-13 signaling pathways do not regulate borrelia burgdorferi-induced arthritis in mice: IgG1 is not required for host control of tissue spirochetes. Infect. Immun. 2000;68:5603–5609. doi: 10.1128/iai.68.10.5603-5609.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S.A, Frelinger J.A. Resistance to fatal nervous system disease by mouse hepatitis virus, strain JHM. 1. Genetic analysis. Immunogenetics. 1978;6:277–281. [Google Scholar]
- Stohlman, S.A., Kyuwa, S., Cohen, M., Bergmann, C., Polo, J.M., Yeh, J., Anthony, R., Keck, J.G., 1992. Mouse hepatitis virus nucleocapsid protein-specific cytotoxic T lymphocytes are Ld restricted and specific for the carboxy terminus. Virology 189, 217–224. [DOI] [PMC free article] [PubMed]
- Tagawa Y, Sekikawa K, Iwakura Y. Suppression of Concanavalin A-induced hepatitis in IFN-g−/− mice, but not in TNF-a−/− mice. J. Immunol. 1997;159:1418–1428. [PubMed] [Google Scholar]
- Uetsuka K, Nakayama H, Goto N. Protective effect of recombinant interferon (IFN)-alpha/beta on MHV-2cc-induced chronic hepatitis in athymic nude mice. Exp. Anim. 1996;45:293–297. doi: 10.1538/expanim.45.293. [DOI] [PubMed] [Google Scholar]
- Wu G.F, Dandekar A.A, Pewe L, Perlman S. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J. Immunol. 2000;165:2278–2286. doi: 10.4049/jimmunol.165.4.2278. [DOI] [PubMed] [Google Scholar]