

Abstract
The interaction of human immunodeficiency virus type 1 (HIV-1) with CD4+ T lymphocytes is well studied and typically results in virally induced cytolysis. In contrast, relatively little is known concerning the interplay between HIV-1 and microglia. Recent findings suggest that, counter-intuitively, HIV-1 infection may extend the lifespan of microglia. We developed a novel cell line model system to confirm and mechanistically study this phenomenon. We found that transduction of a human microglial cell line with an HIV-1 vector results in a powerful cytoprotective effect following apoptotic challenge. This effect was reproduced by ectopic expression of a single virus-encoded protein, Tat. Subsequent studies showed that the pro-survival effects of intracellular Tat could be attributed to activation of the PI-3-kinase (PI3K)/Akt pathway in the microglial cell line. Furthermore, we found that expression of Tat led to decreased expression of PTEN, a negative regulator of the PI-3-K pathway. Consistent with this, decreased p53 activity and increased E2F activity were observed. Based on these findings, a model of possible regulatory circuits that intracellular Tat and HIV-1 infection engage during the cytoprotective event in microglia has been suggested. We propose that the expression of Tat may enable HIV-1 infected microglia to survive throughout the course of infection, leading to persistent HIV-1 production and infection in the central nervous system.
Abbreviations: HIV-1, human immunodeficiency virus type 1; PTEN, phosphatase and tensin homolog; CNS, central nervous system; HCV, hepatitis type C virus; HTLV-1, human T-cell leukemia virus type 1; PI-3-K, PI-3-kinase; CHX, cycloheximide; GFP, green fluorescent protein; CMV, cytomegalovirus; LPS, lipopolysaccharide; SNP, sodium nitroprusside; MOI, multiplicity of infection; PBMC, peripheral blood mononuclear cells
Keywords: HIV-1, microglia, viral reservoir, Tat, long-term survival
Introduction
Human immunodeficiency virus type-1 (HIV-1) infection induces apoptosis in several cell types of the infected host. CD4+ T cells infected by HIV-1 display a cell cycle arrest at the G2 phase, which eventually leads to cell death.1., 2. This is thought to contribute to the loss of CD4+ T cells and to the development of immunodeficiency.3., 4. A virally encoded accessory protein, Vpr, is a major player in the induction of G2 arrest and apoptosis in CD4+ T cells.5., 6., 7., 8. However, in the central nervous system (CNS), HIV-1 triggers the apoptosis of neurons via a different mechanism. Neurons undergo apoptosis as a result of exposure to extracellular viral proteins such as Tat and gp120, and this neurodegeneration can lead to HIV-1 associated dementia (HAD).9., 10., 11. These neurotoxic viral proteins12., 13., 14., 15. are believed to be secreted from HIV-1 infected microglia, the macrophage of the CNS.
Unlike CD4+ T cells, macrophage and microglia do not undergo apoptosis upon HIV-1 infection.16., 17. These two terminally differentiated cell types are infected by macrophage (M)-tropic/R5 viruses at the early stages of viral infection.18., 19., 20., 21. Since macrophage and microglia are non-dividing cells, the cell cycle arrest effect of virus-encoded Vpr, which is mechanistically linked to cell death, has been thought to be insignificant in these HIV-1 target cells. This is because Vpr cannot activate the pro-apoptotic pathway that it activates in T cells, since ATR, Rad17 and Chk1 are not expressed in macrophage.22 Interestingly, a recent study reported that HIV-1 infected microglia may have a survival advantage, as compared with uninfected microglia in the same patients.23 Since the phenotypic outcomes of HIV-1 infection in CD4+ T cells (apoptosis) and microglia (pro-survival) are opposite, it is of considerable interest to better understand how HIV-1 interacts with microglia. This may lead to insight into how HIV-1 infected microglia are able to contribute to the persistent production of HIV-1 within the central nervous system.17., 24., 25., 26.
Many cellular proteins and signaling pathways have been implicated in the regulation of cell fate. One of the most important, and best studied, is Akt.27 Upon cellular stimulation, PI-3-K recruits protein kinase B/Akt to the membrane through generation of PIP3 from PIP2, where it is catalyzed via specific phosphorylation. Once activated, Akt leaves the membrane to modulate the activity of its numerous targets, including GSK3β, p21, Mdm2 and Bad.28., 29., 30., 31. The overall outcome of these events is the induction of cellular proliferation and survival. In order to prevent excess cellular proliferation and survival, especially following damage and stress, this pathway must be regulated. One of the well-studied negative regulators, phosphatase and tensin homolog (PTEN), converts PIP3 to PIP2, inhibiting recruitment of Akt to the membrane and therefore preventing further activation of the PI-3-K pathway.32 PTEN also can modulate proliferation and survival through reciprocal positive regulation of p53. PTEN binds to the C-terminal end of p53, thereby stabilizing p53 while p53 transcriptionally induces PTEN.33 Both PTEN and p53 are often found mutated or deleted in cancers, alleviating the restraint on cellular survival pathways and allowing for long term survival and the subsequent establishment of a transformed phenotype.32
The activation of the PI-3-K/Akt cell survival signaling pathway has been observed in cells infected by several human viruses, including human T-cell leukemia virus type 1 (HTLV-1)34., 35. and hepatitis type C virus (HCV).36 For example, the expression of HTLV-1 Tax protein activates this survival pathway in infected cells and this event is required for its transformation activity.34 In the case of HCV, the activation of the PI-3-K/Akt pathway in HCV-infected Huh7 hepatoma cells was suggested to be involved in establishing and maintaining the latency of the virus and long-term cellular survival.31
Here, we report a new biological activity of intracellular Tat that renders microglia resistant to apoptotic challenge. We have established a human fetal microglial cell line model system to understand the cellular mechanism of the cytoprotective effect induced by expression of HIV-1 Tat. Indeed, our data demonstrate that the pro-survival effect of intracellular Tat protein is mediated by the activation of the cellular proliferation pathway and the PI-3-K/Akt pathway. This effect of HIV-1 Tat is consistent with the recent finding that infected microglia in HIV-1 patients display prolonged survival, and it suggests a previously unrecognized role for HIV-1 Tat in the establishment and maintenance of HIV-1 infected microglia as a long-lived viral reservoir within the CNS.
Results
A human fetal microglia cell line, CHME5, transduced with HIV-1 vector displays enhanced survival
Limited access of primary human microglia has been a major obstacle in studying the molecular and cellular mechanisms involved in the cell fate change of microglia induced by HIV-1 infection. In this study, we tested whether the human fetal microglial cell line, CHME5, displays the extended survival of microglia following HIV-1 infection as previously observed in an ex vivo setting with HIV-1 infected patient samples.23 First, we attempted to validate the CHME5 cell line as a target cell type of M-tropic HIV-1 by examining whether CHME5 cells can support the replication of an M-tropic HIV-1 strain, YU-2, which was cloned from HIV-1 infected human brain tissue.37 As shown in Figure 1 , the CHME5 cells were able to produce and amplify M-tropic virus during serial viral passages, and the efficiency of the HIV-1 production in this cell line was similar with that in primary human peripheral blood mononuclear cells (PBMCs). However, as expected, the CHME5 cells could not support the replication of a T-tropic HIV-1 strain, NL4-3, which is known to replicate in CD4+ T cells but not in macrophages and microglia. Indeed, the data shown in Figure 1 validate CHME5 cells as an M-tropic HIV-1 target cell.
Figure 1.
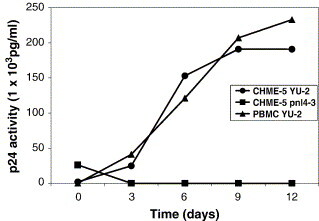
M-tropic HIV-1 replicates efficiently in the CHME5 microglial cell line. A total of 1 × 106 CHME5 cells and human PBMCs were infected with M-tropic YU2 HIV-1 (2.5 × 105 pg of p24) and cultured for 12 days. As a control, T tropic NL4-3 HIV-1 (2.5 × 106 pg of p24) was also inoculated in the CHME5 cells. Viral supernatants were collected at 3, 6, 9 and 12 days post-infection and the p24 level in each viral sample was measured as described in Experimental Procedures . The SD value of the p24 level in each time point was within 5%.
Next, we analyzed the survival capability of HIV-1 infected CHME5 cells in response to cellular insults. In order to visualize the infected CHME5 cells, we initially employed an HIV-1 pseudotyped vector expressing green fluorescent protein (GFP) as well as all HIV-1 proteins except Env and Nef.38 For a control, we used an HIV-1 transfer plasmid, pHR'-GFP, expressing eGFP under the control of the cytomegalovirus (CMV) promoter. Unlike the HIV-1 vector system, this plasmid does not express any HIV-1 proteins, since it contains only the viral LTR and packaging site.39 CHME5 cells were either transfected with the plasmid pHR'-GFP or transduced with the eGFP expressing HIV-1 vector (HIV-GFP) at a multiplicity of infection (MOI) of 1. Then 48 h later, cells were treated with lipopolysaccharide (LPS) to induce cell death. Since it has been reported that LPS-induced cell death in macrophage and microglia requires blockade of protein synthesis,40 the cells were also treated with cycloheximide (CHX;10 μg/ml). Cells were then analyzed by the cytotoxicity/viability assay, which uses a combination of intracellular esterase activity (a characteristic of live cells, green) and binding of the ethidium homodimer to DNA (a marker for dead cells, red). Following HIV-1 vector transduction or lipofectamine-mediated plasmid transfection (for pHR'GFP), approximately 75% of the cells expressed GFP (green). As shown in Figure 2(a), CHME5 cells that were either transfected with the GFP expressing plasmid (pHR'-GFP) or transduced with the HIV-1 GFP vector (HIV-GFP), did not undergo cell death in the absence of LPS/CHX (as shown by the lack of red or yellow cells in the merged fields). However, when cells transfected with pHR'GFP were treated with LPS/CHX, extensive cell death was induced (as shown by the red/yellow staining in the merged fields). In contrast, CHME5 cells transduced with pseudotyped HIV-1 (HIV-GFP) remained viable (no yellow staining in merged field) following treatment with LPS/CHX. Under these conditions, only the adjacent, untransduced cells lacking HIV-GFP expression underwent cell death (Figure 2(a)). These data demonstrate that the HIV-1 vector transduced CHME5 cells, which express HIV-1 Gag, Pol, Tat, Rev, Vpr and Vif, have an extended survival phenotype.
Figure 2.
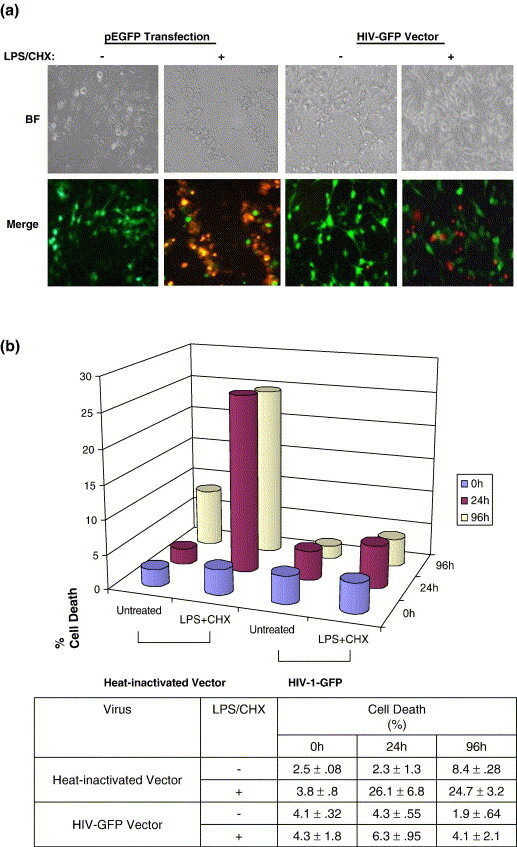
HIV-1 expression exerts a cytoprotective effect in the CHME5 microglial cell line. (a) CHME5 microglial cells were either transduced with pseudotyped HIV-GFP (MOI = 1) or directly transfected with the pHR-GFP plasmid (pEGFP). Treatments with LPS and CHX (both at 10 μg/ml) and the cytotoxicity assay were then performed as described using only the cell death-specific ethidium homodimer stain (red). Representative bright fields (BF) and merged fields (green + red) are shown. Green cells represent cells expressing GFP while red cells represent dead cells. (b) CHME-5 cells (1 × 105) were transduced with HIV-GFP (MOI 1) or incubated with heat-inactivated vector and then treated with LPS/CHX (at the same concentration as above) for a total of 96 h. Cell viability (shown as a percentage of cell death) was measured at 0 h (blue), 24 h (pink) and 96 h (yellow) using the trypan blue exclusion assay. The mean percentages cell death ± SEM induced in the CHME5 cell line following LPS/CHX treatment are summarized below. Experiments were performed in triplicate.
Next, we quantitated the extent of cell death in vector-transduced cultures following LPS/CHX treatment. CHME5 cells were transduced, resulting in greater than 95% transduction (as determined by GFP expression), then treated and analyzed for viability. The cultures were then treated with LPS/CHX at 48 h post-transduction. The number of viable cells was quantitated by trypan blue exclusion assay at 24 and 96 h post-treatment. As shown in Figure 2(b), the control CHME5 cells (incubated with heat-inactivated HIV GFP vector) showed significant cell death upon LPS/CHX treatment, whereas the CHME5 cells transduced with the HIV-1 vector (HIV-GFP) displayed greatly increased survival under similar conditions. Further quantification of the amount of cell death induced in CHME5 cells also confirmed that transduced cells (HIV-GFP vector) exhibited much less cell death (<4%) as compared with cells incubated with heat-inactivated vector (∼25%). Importantly, as expected, we also found that the CHME5 cells infected with infectious M-tropic HIV-1 also showed the elevated survival capability as observed in the CHME5 cells transduced with the HIV-GFP vector (data not shown).
These data demonstrate that HIV-1 infection in the microglial cell line induces a strong cytoprotective effect upon apoptotic challenge. Basically, due to difficulties and limited access in studying primary human microglia, the CHME5 system can serve as a model system, allowing us to investigate the cellular mechanism involved in the pro-survival effect of HIV-1 infection in primary human microglia.
HIV-1 Tat promotes survival of the microglial cell line
First, using the CHME5 model system, we attempted to identify the HIV-1 protein(s) responsible for the cytoprotective effect of HIV-1 infection. Although the HIV-1 vector used in Figure 1, Figure 2 expresses multiple viral proteins, we reasoned that the expressed viral accessory proteins (Tat, Vpr, Vif or Vpu) might contribute to microglial protection. In particular, we sought to determine whether the expression of Tat protein, the HIV-1 transcriptional activator, plays a role in the survival of microglia. To test this, we developed stable sublines of the human microglial cell line CHME5 containing either an empty vector (pCDNA3.1-hygromycin; control) or a vector encoding the 101 amino acid long full-length Tat (pTat101) previously cloned from the M-tropic YU-2 HIV-1 strain. The clonally selected sublines were analyzed for the presence of the Tat gene by performing PCR amplification of genomic DNA and functionally for the activation of the HIV LTR using the Magi assay (data not shown). In order to induce cell death in the CHME5 sublines, the cells were exposed to two different concentrations of LPS (10 μg and 100 μg/ml) and CHX (10 μg/ml) for 6 h and analyzed for viability as described above. As shown in Figure 3(a), the treatment of LPS at 10 μg and 100 μg/ml, together with CHX, induced 20% and 34.5% cell death, respectively, in the control CHME5 subline (pCDNA3.1), as determined by the trypan blue exclusion assay. In contrast, CHME5 cells expressing Tat displayed only 2–3% cell death at both concentrations of LPS (Figure 3(a)). In these assays, CHX treatment alone failed to induce cell death in the CHME5 sublines. Next, cellular viability was examined by performing the cytotoxicity assay (Figure 3(b)). The results confirmed the viable cell counts and revealed that cells expressing either Tat (pTat101) or the control plasmid (pCDNA3.1) remained viable (green) in the absence of LPS/CHX treatment. However, upon treatment, control cells (but not Tat-expressing cells) underwent extensive cell death (as shown by extensive red staining) and loss of viability (as reflected by the loss of the green esterase staining). Importantly, we tested a total of three independent clonal Tat expressing CHME5 sublines, and we observed that these three CHME5 sublines displayed similar elevated survival comparable to the subline data presented in Figure 3. Together, these results indicate that the intracellular expression of Tat may promote the survival of CHME5 microglial cells upon apoptotic challenge.
Figure 3.
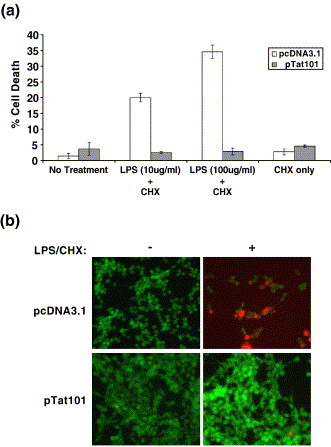
Expression of HIV-1 Tat in a microglial cell line promotes survival in response to apoptotic stimuli. (a) Control CHME5 subline cells (white bars, pCDNA3.1) and those expressing HIV-1 Tat (grey bars, pTat101) were either left untreated, or treated with LPS (10 or 100 μg/ml) plus 10 μg/ml of cycloheximide (CHX) or CHX alone for 6 h. The cells were harvested and viability was assessed by trypan blue exclusion (shown as a percentage of cell death). The data represent the mean ± SEM derived from three independent experiments. (b) Representative results from the cytotoxicity assay. Cells were treated with 10 μg/ml of LPS + CHX for 24 h before performing the cytotoxicity assay. Live cells are represented by fluorescent green staining (intracellular esterase activity) while dead cells appear fluorescent red (ethidium homodimer binding). The fields shown represent merged images. Three independent CHME5 subline clones were tested in this study.
Since Tat can be actively secreted by cells, it remained possible that the ability of Tat-expressing cells to survive in LPS-containing media might be due to the signaling effects triggered by secreted/extracellular Tat protein. To exclude this possibility, we treated CHME5 cells with full-length recombinant Tat protein (0.2 μM, Xeptagen), and then with CHX and LPS (at 10 and 100 μg/ml) for 24 h and observed that extracellular Tat protein was unable to protect CHME5 cells from cell death (data not shown). These findings suggested that intracellular expression of Tat is required to protect human microglial cells from apoptotic challenge.
Role of the PI-3-kinase/AKT pathway in mediating the cytoprotective effect of intracellular Tat in CHME5 cells
In order to investigate how intracellularly expressed Tat might protect microglial cells following apoptotic challenge, we tested whether the activation of the PI-3-K/Akt pathway is involved. This assumption was based on the fact that the expression of Tat mediates activation of this pathway in Kaposi's sarcoma cells, thereby protecting cells from apoptosis.41 To test this possibility, we exposed CHME5 sublines (containing either pTat101 or empty vector, pCDNA3.1) to LPS and CHX (at 10 μg/ml each) for 6 h. Prior to LPS treatment, cells were left alone or pretreated with the PI-3-K/Akt-specific pharmacologic inhibitors Wortmannin and LY294002 (at 100 nM and 50 μM, respectively). Following pretreatment and incubation with LPS and CHX, cell survival was analyzed by the trypan blue exclusion assay. As shown in Figure 4 , the Tat expressing CHME5 cells still exhibited the cytoprotective effect upon LPS/CHX treatment. However, when the Tat-expressing microglial cells were pre-treated with PI-3-K/Akt inhibitors, they showed restricted survival similar to that seen in the control CHME5 cells (pcDNA3.1). These data imply that the PI-3-K/Akt inhibitors abolished the protective effect of Tat.
Figure 4.
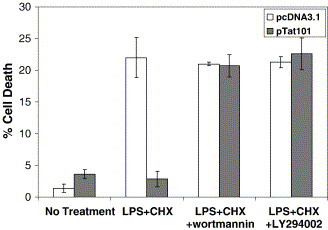
Inhibitors of PI-3-K reverse the pro-survival effect induced by expression of Tat. CHME5 subline cells harboring pcDNA3.1 (white bars) or pTat101 (gray bars) were pretreated with or without Wortmannin (100 nM) or LY294002 (50 μM) for 30 min. CHX (10 μg/ml) and LPS (10 μg/ml) were added to the cells and incubated for 6 h. Cell viability was then assessed by trypan blue staining (shown as a percentage of cell death). The data represent the mean ± SEM values in three separate sets of experiments.
Next, since pharmacologic agents may exert effects on additional cellular pathways, we sought to confirm these results through genetic approaches. To achieve this, we expressed a dominant negative mutant form of Akt in the CHME5 cells. This mutant molecule (Akt K179A, referred to as dn-Akt) contains a substitution of lysine residue 179 with alanine, and functions as a dominant negative inhibitor of signal-mediated Akt activation.41 In these experiments, CHME5 sublines (containing pTat101 or pCDNA3.1) were transiently transfected with an empty vector (representing endogenous Akt) or a vector encoding dn-Akt 24 h prior to treatment. Cells were then exposed to LPS and CHX (100 and 10 μg/ml, respectively) for 24 h and cellular viability was analyzed. As expected, we found that transfection of K179A resulted in an increase in the amount of cell death induced following LPS/CHX treatment (Figure 5 ). Transient transfection of the Akt mutant did not seem to influence the survival of untreated cells. This suggests that expression of dn-Akt was able to reverse the cytoprotective effect of Tat in CHME5 cells. The extent of susceptibility restored in the Tat-expressing cells reflects the transfection efficiency of the dominant negative Akt mutant (∼75% efficiency). Based on the results from the pharmacological (Figure 4) and genetic (Figure 5) interferences of the PI-3-K/Akt pathway, we concluded that the activation of the PI-3-K/Akt pathway by intracellular Tat might be required for Tat to promote microglial survival.
Figure 5.
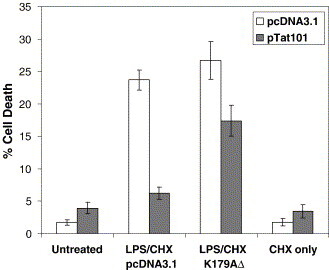
Akt is involved in the cytoprotective effect elicited by HIV-1 Tat. CHME5 cell lines (pcDNA3.1, white bars; pTat101, gray bars) were transfected with either an empty vector (pcDNA3.1) or Akt-K179A, a dominant negative Akt mutant. Twenty-four hours post-transfection, cells were treated with CHX (10 μg/ml) and LPS (10 μg/ml) for 24 h. The transfection efficiency was approximately 75% and the total cell population was analyzed for viability using trypan blue; results are presented as the percentage of cell death (mean ± SEM, derived from three experiments).
Regulation of PTEN and cell proliferative signals by expression of HIV-1 Tat
PI3-kinase activity is negatively regulated by PTEN, which dephosphorylates PIP3 to PIP2.32 In addition, PTEN and p53 positively regulate each other. More specifically, p53 upregulates the transcription of PTEN, and PTEN stabilizes p53 by directly binding to the C-T end of p53.33 Importantly, it has been demonstrated that HIV-1 Tat also directly interacts with the C-T region of p53.42 Therefore, we tested whether intracellular Tat may compete with PTEN for p53 binding, which may induce the destabilization of p53 and decrease of PTEN levels, ultimately facilitating activation of the PI-3-K/Akt survival pathway. To test this possibility, we examined the PTEN level of CHME5 cells in the presence and absence of intracellular Tat expression as well as HIV-1 transduction. As shown in Figure 6(a), the PTEN level significantly decreased upon transduction with the HIV-GFP vector. More interestingly, the expression of HIV-1 Tat in CHME5 microglial cells also led to a drastic decrease in PTEN levels. This suggests that the down-regulation of PTEN by intracellular Tat and HIV-1 infection may be a potential mechanism for stimulation of the PI-3-K/Akt survival pathway.
Figure 6.
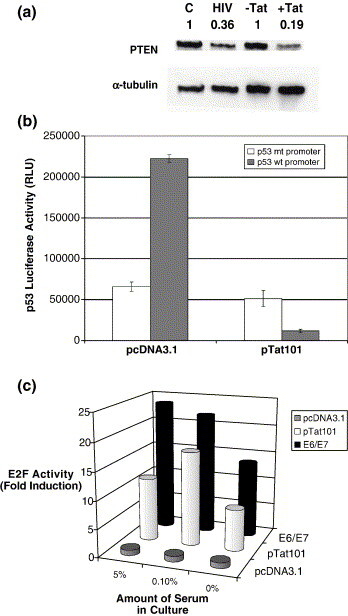
Tat expression stimulates cellular responses associated with proliferation. (a) PTEN levels in CHME5 sublines with (+Tat) and without (−Tat) intracellular Tat expression or CHME5 cells transduced with HIV-1 GFP vector (HIV) or control heat-inactivated vector (C) (48 h post-transduction) were analyzed by Western blot using PTEN antibody. α-Tubulin was used as a loading control. Ratios of PTEN level normalized by the α-tubulin level are marked. (b) CHME5 cells were transfected with pcDNA3.1 or pTat101, pcDNA-LacZ and a p53-dependent luciferase reporter construct. Activity from the wild-type p53 promoter construct is shown in gray while the activity from the mutated p53 promoter construct is shown in white. The cells were then harvested 24 h later, at which time lysates were prepared and luciferase assays were performed. The data represent p53 luciferase activity in relative light units (RLUs) and denote mean data values ± SEM from three independent experiments. (c) CHME5 cells were transiently transfected with pcDNA3.1 (gray), pTat101 (white) or E6/E7 (black) along with an E2F-dependent luciferase reporter construct and pcDNA3.1-LacZ. The cells were then incubated in DMEM containing 5%, 0.1% or 0% fetal bovine serum. 72 h later, the cells were lysed and luciferase activity was measured. All luciferase activities were normalized to equal amounts of β-galactose activity. Data were compared to the level of luciferase activity in control cells (pcDNA3.1, gray). Results shown represent fold induction; data denotes mean values ± SEM from three experiments.
Next, since the decrease of PTEN levels induces the destabilization of p53,43 we tested whether Tat expression may induce the decrease of p53 transcriptional activity in CHME5 cells. For this test, we transfected CHME5 cells with a p53-regulated luciferase reporter plasmid together with pcDNA-lacZ and pTat101 or pCDNA3.1. CHME5 cells were derived from human fetal microglia following transformation with the simian virus 40 (SV40) large T antigen. This molecule is known to directly interact with p53 and inhibit its activity.49 However, as shown in Figure 6(b), we observed that CHME5 cells exhibited a basal activity of p53 which was still detectable. This basal activity was comparable with the basal activity of p53 in other cell types that contain SV40 large T antigen.50 Importantly, as predicted, p53 activity was further drastically decreased (5.3% of control) following expression of Tat in CHME5 cells (Figure 6(b)). As an additional control, the cells were transfected with a control p53- luciferase plasmid that lacks functional p53 binding elements (p53 mt promoter) and the luciferase expression from this control mutant construct remained low following expression of HIV-1 Tat in CHME5 cells. These data suggest that expression of Tat in the microglial cell line system induces the inhibition of the transcription activator function of p53, which is consistent with the decrease of PTEN in the CHME5 Tat expression model system.
One of the cellular consequences of p53 dysfunction is the activation of cell proliferation,46 which is a well-characterized mechanism of cell survival. One of the key molecular events involved in the promotion of cell proliferation is the activation of the E2F transcription factor which is responsible for the expression of a number of genes essential for the cell cycle exit from G1 and entry into S phase.45 Therefore, we examined E2F activity in CHME5 cells expressing HIV-1 Tat. CHME5 cells were transiently transfected with an E2F-driven luciferase reporter plasmid together with either an empty expression vector (pCDNA3.1) or pTat101. As a positive control, cells were transfected with an expression plasmid (E6/E7) that contains the human papilloma virus type 16 (HPV-16)-encoded E6/E7 proteins, which are known to increase E2F activity.47 In all cases, pCDNA3.1-LacZ was also included as an internal control for normalization of luciferase assay results. After transfection, cells were incubated for 72 h in media containing either 5%, 0.1% or 0% fetal bovine serum (FBS) and luciferase activity was measured. As expected, high luciferase activity was observed following co-expression of the E6/E7 proteins, presumably due to the activation of E2F by E6/E7 (Figure 6(c)).48 Similarly, Tat-expressing cells also displayed significantly higher luciferase activity as compared to the control cells even following the stress of serum starvation. These data imply that the intracellular expression of Tat contributes to the activation of E2F in the microglial cell line. We also found that expression of Tat in our system stimulated enhanced proliferative activity and DNA synthesis capability compared with control cells (data not shown).
In summary, employing the CHME5 model system, we observed that HIV-1 Tat expression induces (1) a decrease in PTEN level, (2) a decrease of p53 function and (3) an increase of E2F function, which collectively facilitate cell survival by the activation of the Akt pathway and cell proliferation potential.
HIV-1 infection induces a cytoprotective effect in primary human monocyte-derived macrophages
Evidence suggests that HIV-1 infected microglia survive longer as compared to uninfected microglia in AIDS patients.23 Furthermore, since our CHME5 model system also displays an elevated survival capability upon HIV-1 infection,16., 17. we attempted to confirm whether HIV-1 infection influences the survival of primary human monocyte-derived macrophage upon cytotoxic challenge. Macrophages were infected with M-tropic HIV-1 Bal (1.1 × 107 pg, equivalent to vector MOI 40) and treated with LPS (15 μg/ml) and CHX (10 μg/ml) at 48 h post-infection. The live (green) and dead (red) cells were monitored at 24 h post-treatment. As shown in Figure 7(a), macrophage infected with HIV-1 Bal displayed a decreased number of dead cells (red), as compared to uninfected macrophage (incubated with heat-inactivated virus). This suggests that infection with HIV-1 Bal induces a protective effect in primary human macrophage following cytotoxic challenge. Next, we employed an HIV-1 vector pseudotyped with the VSV-G envelope protein. This HIV-1 vector system expresses eGFP and all HIV-1 viral proteins except the Env and Nef proteins.39 Following transduction with the pseudotyped HIV-1 vector (HIV-GFP), macrophages were treated with LPS (25 μg/ml) and CHX (10 μg/ml), and cell viability was examined. In this assay, we used only the cell death specific ethidium homodimer to detect dead cells (red). As shown in Figure 7(b), human macrophage transduced by the HIV-1 vector system (green) exhibited greatly diminished levels of cell death (red) than the control macrophage (incubated with heat-inactivated vector). It is important to point out that none of the transduced cells (green) showed characteristics of cell death (as shown by distinct red or green staining in the merged field). These data demonstrate that HIV-1 infection induces a cytoprotective effect in human monocyte-derived macrophage. Interestingly, the data with the HIV-1 vector system also suggest that Env and Nef, which are deleted in the pseudotyped vector, are not involved in this HIV-1 induced cell survival effect.
Figure 7.
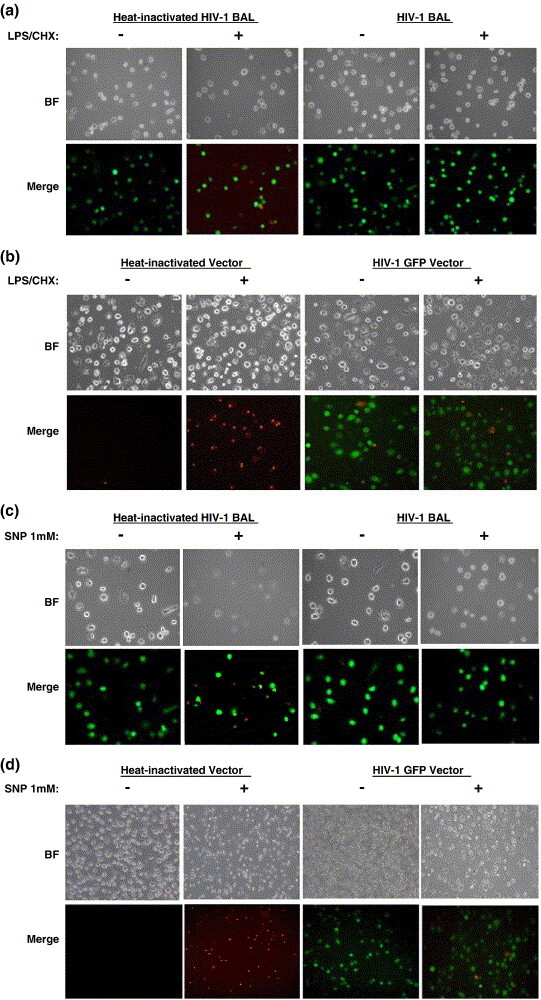
HIV-1 infection protects primary human macrophage from cell death. Primary human macrophage were either infected with 1.1 × 107 pg M-tropic HIV-1 BAL ((a) and (c)) or transduced with pseudotyped HIV-GFP vector ((b) and (d)) at an MOI of 40 for two days prior to treatment. Heat-inactivated virus or vector was used as a control. Following 24 h exposure to LPS/CHX or SNP, cells were analyzed for viability using the cytotoxicity assay. LPS was used at a concentration of 15 μg/ml (BAL infections) or 25 μg/ml (viral transductions) with a constant CHX concentration of 10 μg/ml while SNP was used at 1 mM. Representative bright fields (BF) and merged fields (green + red) are shown. Green cells represent either viable cells ((a) and (c)) or cells expressing GFP ((b) and (d)). In (a)–(d), red cells represent dead cells stained with ethidium homodimer.
Since microglial cells are long-lived cells known for their role in immune surveillance of the CNS, there is little known about natural mechanisms involved in apoptosis of these cells. One of the known ways to induce microglial apoptosis is through increased nitric oxide (NO) production.44 In fact, HIV-1 infection is known to activate microglia, which leads to the production of nitric oxide.45 Another source of NO during HIV-1 infection is through the secretion of gp120, the viral envelope glycoprotein, which can induce production from bystander CNS cell types.46 This additive effect on NO production by HIV-1 infection induces apoptosis in neighboring cells such as neurons. Normally, the overactivation of microglia sensitizes them to undergo apoptosis through a feedback mechanism.47 However, despite the large amounts of NO produced during HIV-1 infection, microglia still exhibit an extended survival phenotype. Therefore, we next tested whether primary human macrophages infected with HIV-1 exhibited this cytoprotective effect following exposure to sodium nitroprusside, an NO donor.48 As shown in Figure 7(c), macrophage exposed to heat-inactivated virus exhibited significant cell death (red cells) upon exposure to sodium nitroprusside (SNP). However, macrophage infected with HIV-1 BAL did not display any significant induction in cell death even under the conditions of increased NO production. Consistent with this, primary macrophage transduced using our pseudotyped vector system (green cells) also exhibited a survival effect following SNP treatment (Figure 7(d)), whereas macrophages incubated with heat-inactivated vector displayed significant cell death upon SNP treatment. It is also important to emphasize that although SNP treatment resulted in a greater stress than LPS, inducing higher amounts of cell death, HIV-1 infected macrophages still exhibited a potent cytoprotective effect (the absence of red cells in Figure 7(c) and (d)).
To measure the extent of cell death induced in primary human macrophage, the mean percentages of cell death were obtained by counting the number of dead cells (red) out of the number of total cells from various fields. In the case of pseudotyped vector transduced cells, the number of dead cells (red) out of GFP-expressing cells was determined. As shown in Figure 8 , primary human macrophage infected with M-tropic HIV-1 Bal or transduced with pseudotyped GFP vector showed significantly less cell death than the uninfected macrophage (incubated with heat-inactivated virus or heat-inactivated vector) following treatment with either the LPS or SNP apoptotic stimulus. These data demonstrate that as observed with HIV-1 infected primary microglia23 and our CHME5 model system, HIV-1 infection of primary human macrophage also leads to increased cell survival upon apoptotic challenge.
Figure 8.
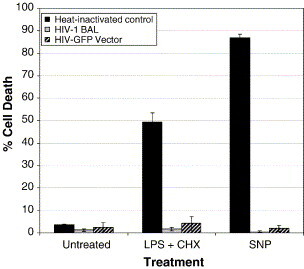
Quantitation of the percentage of cell death in primary human macrophage following LPS/CHX or SNP treatment. Primary human macrophage (2 × 105) were either infected with HIV-1 Bal (1.1 × 107 pg, equivalent to vector MOI = 40) or transduced with HIV-GFP (85% GFP positive) and treated with LPS (15 μg/ml, Bal infections; 25 μg/ml, vector transductions) and CHX (10 μg/ml) or 1 mM SNP at 48 h post-infection. Heat-inactivated virus or vector was used as a control. The mean percentages cell death ± SEM were obtained by counting the number of dead cells (red) out of the number of total cells from various fields following staining with ethidium homodimer (i.e. Figure 7). In the case of pseudotyped vector transduced cells, the number of dead cells out of GFP-expressing cells was determined. These numbers reflect the mean percentage of cell death from five fields (∼75 cells per field).
Discussion
The interaction of HIV-1 with CD4+ T cells has been well studied, and typically results in cell cycle arrest and cytolysis. In contrast, the cellular effects following HIV-1 infection in macrophage and microglia are much less clearly understood. Hence, we set out to conduct experiments that might shed light on this interaction, based on an intriguing recent report demonstrating that HIV-1 infection may promote the survival of primary human microglia.23 However, a major obstacle for approaching this question is the limited access to primary human microglia. In addition, it is technically challenging to study primary human microglia because many experimental approaches often require large quantities of cells. Here, we attempted to develop a new cell line model that mimics the interplay between HIV-1 and microglia in terms of cell survival.
We initially demonstrated that this cell line model system using the CHME5 microglial cell line can support the replication and amplification of M-tropic HIV-1, and displays a potent cytoprotective effect upon HIV-1 infection. Importantly, we demonstrated that the intracellular expression of the HIV-1 Tat protein was able to induce a strong cytoprotective effect in CHME5 cells. The possibility that secreted Tat protein from the CHME5 cells expressing Tat may also induce the same effect was eliminated when we observed that CHME5 cells treated with recombinant Tat protein (0.2 μM) did not show increased cell survival (data not shown). Furthermore, we later confirmed that primary human monocyte-derived macrophages infected with HIV-1 also exhibited prolonged survival upon cytotoxic challenges, further validating the CHME5 microglial cells as a model system to study the molecular mechanisms involved with the cytoprotective effect.
One of the most extensively studied survival pathways is the PI-3-K/Akt pathway.32., 49. We therefore tested the possible involvement of the PI-3-K/Akt pathway using Wortmannin and LY294002, two unrelated inhibitors specific for PI-3-K. Both of these inhibitors reversed the survival effect induced by the expression of Tat, suggesting that the PI-3-K pathway is indeed involved in the pro-survival effect of Tat in CHME5 cells. This interpretation was confirmed using an independent genetic test in which cells were transfected with a dominant negative (kinase inactive) Akt mutant;41 this led to the restoration of cell death in Tat-expressing cells. This mutant, however, did not affect the sensitivity of the control cells to cell death in response to LPS and CHX. These data strongly suggest that Tat promotes survival in microglia via activation of the PI-3-K/Akt pathway.
The same phenotype has been observed in cells infected by oncogenic viruses or cells that express viral oncogenes such as the E6/E7 gene products of human papilloma virus type 16 (HPV-16).50., 51. Consistent with the activation of the PI-3-K/Akt pathway, expression of HIV-1 Tat protein in CHME5 cells also repressed the transcription activation activity of p53, while stimulating that of E2F. These changes are reflective of enhanced proliferative activity likely without actual cell division since primary microglia are terminally differentiated/non-dividing cells.
Based on the data presented in Figure 6, the expression of HIV-1 Tat induced the (1) decrease of PTEN, (2) the decrease of p53 function and (3) increase of E2F activity. We therefore modeled the mechanism of intracellular HIV-1 Tat and HIV-1 infection-induced cell survival in the CHME5 human microglia system (Figure 9 ). It is known that both Tat and PTEN bind to the C-terminal region of p53, and therefore these two proteins may compete with each other for p53. Importantly, PTEN binding to p53 stabilizes p53, and p53 upregulates PTEN expression transcriptionally.43 Therefore, expression of Tat and HIV-1 infection possibly induce the destabilization of p53 by competing with PTEN, and this ultimately leads to the decrease of PTEN levels, which we observed in Figure 6(a). The decreased PTEN level is known to facilitate the activation of the PI-3-K/Akt pathway, which directly promotes cell proliferation and cell survival (pathway A in Figure 9). In addition, the decrease of p53 function by HIV-1 Tat expression may also lead to the activation of cell proliferation signals such as the E2F transcription factor, following release from Rb (pathway B in Figure 9). The free E2F can then up-regulate a number of genes essential for G1/S transition and cell proliferation, which is a key mechanism in cellular survival. E2F also transcriptionally induces expression of an adaptor protein involved in the activation of the Akt pathway, an upstream mechanism of regulating this survival pathway.52 Future studies on downstream cellular factors involved in Akt cellular survival (i.e. GSK3β, Bad) and cell cycle control pathways (p53, MDM2, p21Cip1, CyclinD and Rb) may reveal more precisely the point at which Tat influences cell signaling cascades in microglia, in order to elicit its protective effect.29., 30., 53., 54., 55.
Figure 9.
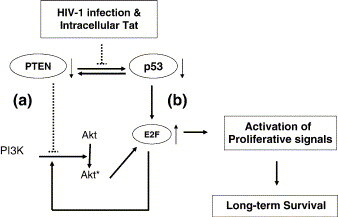
Model for the cytoprotective mechanism exerted by HIV-1 infection and intracellular HIV-1 Tat expression in microglial cells. HIV-1 infection or expression of intracellular HIV-1 Tat results in decreased PTEN expression which alleviates the restraint on activation of the PI-3-K/Akt pathway. The increased activation of PI-3-K and Akt leads to long-term cellular survival.85 Intracellular Tat can also decrease activity of p53, which in turn leads to progression through the cell cycle, activating the E2F transcription factor and proliferation-specific signals.86 E2F also regulates survival by transcriptionally activating an upstream adaptor protein involved in the activation of the PI-3-K/Akt pathway.52 The combined modulation of PTEN and p53 by expression of Tat leads to the increased activation of proliferation and long-term cellular survival exhibited in microglia during HIV-1 infection: dotted lines, negative regulation; thick arrows, positive regulation; thin arrows, effects of intracellular Tat expression and HIV-1 infection.
In addition to HIV-1, many viruses have developed mechanisms of hijacking the PI-3-K/Akt pathway to benefit their particular needs, whether it is for enhancement of viral replication, cellular proliferation or aiding in the establishment of a persistent infection. For example, tumor-causing viruses such as human papilloma virus (HPV),56 Epstein Barr virus (EBV)57 and polyoma virus58 employ viral genes to activate proliferation through this pathway, resulting in cellular transformation. Other viruses such as human cytomegalovirus (HCMV),59 herpes simplex virus type 1(HSV-1)60 and EBV61 utilize viral proteins and downstream effectors of Akt to increase viral replication. However, as is the case with our HIV-1 Tat results, most viruses hijack this particular pathway to promote cellular survival and therefore establish a persistent infection. Hepatitis C virus (HCV),36 SARS CoV62 and EBV63 activate Akt to establish persistence and latency.
Evidence suggests that PI-3-K/Akt activation prevents cellular death by modulating functions of pro-apoptotic molecules such as GSK-3β,64 Bad and the BH3-interacting domain death agonist BID.65 This leads to the inhibition of caspase 1, 3, and 9-like activity and the modulation of mitochondrial membrane potential and cytochrome c release.66 , 67 The cytoprotective actions of PI-3-K/Akt also involve the upregulated expression of survival-promoting genes such as Bcl-2, Bcl-xL, and c-IAP.65., 68., 69. Akt may regulate the expression of these genes via the activation of transcription factors including NF-κB, CREB and Forkhead (FKHR).70., 71., 72., 73. Indeed, intracellular expression of HIV-1 Tat is known to activate NF-κB and CREB transcription factors in other cell types,74., 75., 76., 77. suggesting that intracellular Tat might employ these signaling events in microglial cells to promote survival.
It has been reported that the secretion of NO is one of the major in vivo toxic effects induced during HIV-1 infection, which contributes to AIDS-induced neuronal cell death. Interestingly, we have also observed that primary human macrophages infected with HIV-1 display a strong cytoprotective effect upon treatment with SNP, an NO donor (Figure 7(c) and (d); Figure 8). Another well characterized cytotoxic extracellular protein secreted from HIV-1 infected macrophages is TRAIL (TNF related apoptosis inducing ligand), which can directly induce cell death in not only neighboring neuron cells but also the infected microglia in CNS.78., 79., 80. It has been demonstrated that HIV-1 infected macrophages become sensitive to TRAIL, whereas uninfected macrophages are resistant to TRAIL.79 In addition, it was reported that the TRAIL levels in HIV-1 infected patients are elevated, compared to uninfected healthy individuals.79 Indeed, this finding led to the testing of TRAIL as an agent to specifically kill infected macrophages. Unexpectedly, however, the treatment with recombinant TRAIL did not reduce viral production and cell death.80 One possible explanation for this failure was that the local concentration of TRAIL near infected microglia may not reach a high enough concentration to induce the apoptosis of infected macrophage and microglia.80., 81. Alternatively, HIV-1 might have already evolved to escape efficiently from the cytotoxic effect of TRAIL in microglia and macrophages as implied by the observation of persistent HIV-1 infection in the CNS.
In summary, using the CHME5 cell line model, we demonstrated that intracellular expression of HIV-1 Tat has an unexpected pro-survival effect in microglia. This may have important implications for the pathogenesis of HIV-associated dementia, since virally infected microglia represent an important source of soluble effector molecules that can lead to the dysfunction and death of neurons. Finally, our studies show that Tat manipulates Akt signaling and cell cycle regulatory pathways in such a way as to extend cell survival. This in turn may lead to the establishment of long-lived cellular reservoirs of HIV-1 infection in the CNS. Furthermore, our cell line model can serve as a useful tool for elucidating the molecular, cellular and virological elements that contribute to persistent HIV-1 infection in the CNS.
Experimental Procedures
Plasmids and cells
The p53 and E2F luciferase reporter plasmids (wild-type and mutant), the HPV-16 E6/E7 expression plasmid and the K179A dominant-negative Akt, were gifts from Drs Dennis McCance and Robert Freeman (University of Rochester).82 The HIV-1 Tat cDNA derived from the YU-2 M-tropic HIV-1 strain,83 was sub-cloned into pcDNA3.1+/Hygromycin (Invitrogen). To do this, the full-length Tat cDNA (101 amino acid residues) was amplified by polymerase chain reaction and a hemagglutinin (HA) epitope-tag sequence (2×) was cloned at its N terminus using specific primers. The purified Tat-HA-tag segment was then inserted into a pcDNA3.1 vector using the KpnI and XbaI restriction sites, placing it under control of the CMV promoter. The generated plasmid was designated as pTat101. An HIV-1 transfer plasmid, pHR-GFP, which expresses green fluorescent protein (eGFP) from the CMV promoter was obtained from Dr Vicente Planelles (University of Utah).38
The CHME5 cell line,84 a human fetal microglial cell line, was obtained from Dr David Mock (University of Rochester). To establish a CHME5 cell line that stably expresses full-length Tat, cells were transfected with the plasmid pTat101 using Lipofectamine 2000 (Invitrogen) and Hygromycin-resistant cells were clonally selected in 1 mg/ml of Hygromycin (Invitrogen) for 15 days. The control cells were transfected with an empty vector (pCDNA3.1-Hygro, Invitrogen) and subjected to drug selection as indicated above. The parental CHME5 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; CellGro) containing 5% fetal bovine serum (FBS; HyClone) and maintained as described,84 whereas the stable subline media was supplemented with 500 μg/ml of Hygromycin. The presence of Tat was confirmed by PCR amplification of Tat DNA sequences using gene-specific primers and functionally by activation of the viral LTRs.
HIV-1 replication assay
CHME5 microglial cells were infected with M-tropic HIV-1 YU-2 (2.5 × 105pg of p24) or T tropic HIV-1 NL4-3 (2.5 × 106pg of p24). Viral supernatants were collected every three days for a period of 12 days before they were subjected to the p24 HIV-1 capsid-enzyme linked immunosorbent assay as per the manufacturer's protocol (Beckman-Coulter).
Isolation of primary human macrophage
Primary human monocyte-derived macrophage were isolated and differentiated as described.39 Macrophage were maintained in culture in RPMI 1640 (CellGro) supplemented with 10% fetal bovine serum (HyClone).
Pseudotyped HIV-1 vector production
Pseudotyped HIV-1 expressing EGFP was prepared as described.39 Primary human macrophage were transduced with the HIV-1 vector at a MOI of 40 (1.1 × 107 pg) while an MOI of 1 was used to transduce the CHME5 microglial cell line.
Preparation of M-tropic HIV-1
M-tropic HIV-1 BAL (NIH Aids Reagent Program) was propagated in human PBMCs for 14 days. Viral supernatants were pooled and the p24 activity of the virus was determined using the p24 enzyme-linked immunosorbent assay (Beckman-Coulter) according to the manufacturer's protocol. The p24 levels were normalized to p24 levels equivalent to a vector MOI of 40 and primary human macrophage were infected with HIV-1 BAL (1.1 × 107 pg) for 48 h prior to treatment. CHME5 cells were infected with HIV-1 YU-2 (1.38 × 106 pg) 48 h prior to treatment.
Cell survival assays
CHME5 cells or primary human macrophage were exposed to CHX (10 μg/ml) and Escherichia coli serotype O26:B6 LPS (Sigma) at the concentrations indicated in the Figure legends for the specified times. Sodium nitroprusside (Sigma) was used at 1 mM for treatment of cells. Cells were then trypsinized and mixed with 0.4% (w/v) trypan blue solution (Gibco) at a 1:1 dilution and the number of blue (dead) cells was calculated by cell counts. For inhibition of the PI-3-K pathway, cells were pretreated with either Wortmannin (100 nM) or LY294002 (50 μM) (Sigma) for 30 min prior to LPS/CHX treatment. For immunofluorescent detection of viable and non-viable cells, the Live/Dead-Viability/Cytotoxicity kit (Molecular Probes) was used to stain cells as per the manufacturer's protocol. Images were taken at a magnification of 200× 24 h post-treatment using a fluorescence microscope (Leica). Each assay was performed in triplicate.
In order to determine the survival of HIV-1-infected cells, approximately 1 × 105 cells/well were transduced with pseudotyped HIV-1 vector eGFP at various MOI values as described in the Figure legends. For CHME5 cells, transductions were performed in the presence of Polybrene (10 μg/ml). All infections and transductions were performed 48 h prior to treatments. Following treatment, cellular viability was analyzed by the cytotoxicity assay. Similarly, the viability of uninfected, as well as transiently transfected cells was analyzed using identical methods. For trypan blue analysis of transduced cells, cells were treated for a total of 96 h, changing media every 24 h and replacing with fresh media and LPS/CHX. Viability was measured by trypan blue exclusion every 24 h. Each assay was performed in triplicate.
For quantitation of the percentage of cell death induced in primary human macrophage following LPS/CHX treatment, cells were infected or transduced 48 h prior to LPS/CHX treatment. The mean percentages of cell death were obtained by counting the number of dead cells out of the number of total cells from various fields following staining with ethidium homodimer (described above). In the case of pseudotyped vector transduced cells, the number of dead cells out of GFP-expressing cells was determined. These numbers reflect the mean percentage of cell death from five fields (∼75 cells per field).
Luciferase assay
For measuring p53 activity, CHME5 cells were co-transfected with pcDNA3.1/LacZ (Invitrogen), a luciferase reporter plasmid (containing either a wild-type or mutated responsive element) and either pcDNA3.1-Hygro (Invitrogen) or pTat101 using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. At 24 h post-transfection, cells were washed with DPBS (CellGro), lysed in 50 μl Galacto-Light Plus lysis buffer (Tropix) for 10 min and the β-galactosidase assay was performed using the Galacto-Light Plus kit (Tropix). Lysates were normalized according to β-galactosidase activity and the luciferase assay was performed (Promega). Luciferase activity was then immediately measured with the Lumicount Microplate Luminometer (Packard). To detect E2F activity, the transfected CHME5 cells were incubated in DMEM (CellGro) with either 5%, 0.1% or 0% FBS for 72 h post-transfection before lysing cells for the β-galactosidase and luciferase assays as described above. As an additional control, a plasmid expressing the E6/E7 genes of human papilloma virus type 16 (HPV-16) was also used. Each assay was performed in triplicate.
Western blot analysis
Cell lysates were prepared in RIPA buffer supplemented with protease inhibitors (Roche) and phosphatase inhibitor cocktail (Sigma) and samples (20 μg) were run on an SDS 8% (w/v) polyacrylamide gel. PTEN expression was detected by probing with the PTEN (138G6) rabbit monoclonal antibody (Cell Signaling) at a dilution of 1:1000. Donkey anti-rabbit Ig (Amersham Biosciences) was used at 1:5000 for secondary antibody followed by ECL detection using the SuperSignal West Femto kit (Pierce). For a loading control, blots were probed for α-tubulin (Cell Signaling) followed by sheep anti-mouse IgG (Amersham Biosciences). ECL detection was performed as described above. Expression of PTEN in each sample was normalized to alpha-tubulin levels for analysis.
Acknowledgements
We thank Drs Dennis McCance, Robert S. Freeman and David Mock for providing reagents used in this study. This work was supported by research grants: AI058774 (to B.K.), MH64570 (to S. D. and S. M.) and training grant AI07362 and F31 AI64136-01 (to P.C.) from the National Institutes of Health.
Edited by J. Karn
References
- 1.Selliah N., Finkel T.H. Biochemical mechanisms of HIV induced T cell apoptosis. Cell Death Differ. 2001;8:127–136. doi: 10.1038/sj.cdd.4400822. [DOI] [PubMed] [Google Scholar]
- 2.Zhao R.Y., Elder R.T. Viral infections and cell cycle G2/M regulation. Cell Res. 2005;15:143–149. doi: 10.1038/sj.cr.7290279. [DOI] [PubMed] [Google Scholar]
- 3.Douek D.C. Disrupting T-cell homeostasis: how HIV-1 infection causes disease. AIDS Rev. 2003;5:172–177. [PubMed] [Google Scholar]
- 4.Yue F.Y., Kovacs C.M., Dimayuga R.C., Gu X.X., Parks P., Kaul R., Ostrowski M.A. Preferential apoptosis of HIV-1-specific CD4+ T cells. J. Immunol. 2005;174:2196–2204. doi: 10.4049/jimmunol.174.4.2196. [DOI] [PubMed] [Google Scholar]
- 5.Fukumori T., Akari H., Yoshida A., Fujita M., Koyama A.H., Kagawa S., Adachi A. Regulation of cell cycle and apoptosis by human immunodeficiency virus type 1 Vpr. Microbes Infect. 2000;2:1011–1017. doi: 10.1016/s1286-4579(00)01255-7. [DOI] [PubMed] [Google Scholar]
- 6.Muthumani K., Choo A.Y., Hwang D.S., Chattergoon M.A., Dayes N.N., Zhang D., et al. Mechanism of HIV-1 viral protein R-induced apoptosis. Biochem. Biophys. Res. Commun. 2003;304:583–592. doi: 10.1016/s0006-291x(03)00631-4. [DOI] [PubMed] [Google Scholar]
- 7.Muthumani K., Choo A.Y., Premkumar A., Hwang D.S., Thieu K.P., Desai B.M., Weiner D.B. Human immunodeficiency virus type 1 (HIV-1) Vpr-regulated cell death: insights into mechanism. Cell Death Differ. 2005;12(Suppl. 1):962–970. doi: 10.1038/sj.cdd.4401583. [DOI] [PubMed] [Google Scholar]
- 8.Andersen J.L., Planelles V. The role of Vpr in HIV-1 pathogenesis. Curr. HIV Res. 2005;3:43–51. doi: 10.2174/1570162052772988. [DOI] [PubMed] [Google Scholar]
- 9.Mattson M.P., Haughey N.J., Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12(Suppl. 1):893–904. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- 10.Shi B., Raina J., Lorenzo A., Busciglio J., Gabuzda D. Neuronal apoptosis induced by HIV-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV-1 dementia. J. Neurovirol. 1998;4:281–290. doi: 10.3109/13550289809114529. [DOI] [PubMed] [Google Scholar]
- 11.Singh I.N., Goody R.J., Dean C., Ahmad N.M., Lutz S.E., Knapp P.E., et al. Apoptotic death of striatal neurons induced by human immunodeficiency virus-1 Tat and gp120: differential involvement of caspase-3 and endonuclease G. J. Neurovirol. 2004;10:141–151. doi: 10.1080/13550280490441103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kruman I.I., Nath A., Mattson M.P. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- 13.Garden G.A., Budd S.L., Tsai E., Hanson L., Kaul M., D'Emilia D.M., et al. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J. Neurosci. 2002;22:4015–4024. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garden G.A., Guo W., Jayadev S., Tun C., Balcaitis S., Choi J., et al. HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 2004;18:1141–1143. doi: 10.1096/fj.04-1676fje. [DOI] [PubMed] [Google Scholar]
- 15.Aksenov M.Y., Hasselrot U., Wu G., Nath A., Anderson C., Mactutus C.F., Booze R.M. Temporal relationships between HIV-1 Tat-induced neuronal degeneration, OX-42 immunoreactivity, reactive astrocytosis, and protein oxidation in the rat striatum. Brain Res. 2003;987:1–9. doi: 10.1016/s0006-8993(03)03194-9. [DOI] [PubMed] [Google Scholar]
- 16.Aquaro S., Bagnarelli P., Guenci T., De Luca A., Clementi M., Balestra E., et al. Long-term survival and virus production in human primary macrophages infected by human immunodeficiency virus. J. Med. Virol. 2002;68:479–488. doi: 10.1002/jmv.10245. [DOI] [PubMed] [Google Scholar]
- 17.Lambotte O., Deiva K., Tardieu M. HIV-1 persistence, viral reservoir, and the central nervous system in the HAART era. Brain Pathol. 2003;13:95–103. doi: 10.1111/j.1750-3639.2003.tb00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herbein G., Coaquette A., Perez-Bercoff D., Pancino G. Macrophage activation and HIV infection: can the Trojan horse turn into a fortress? Curr. Mol. Med. 2002;2:723–738. doi: 10.2174/1566524023361844. [DOI] [PubMed] [Google Scholar]
- 19.Khati M., James W., Gordon S. HIV-macrophage interactions at the cellular and molecular level. Arch. Immunol. Ther. Exp. (Warsz) 2001;49:367–378. [PubMed] [Google Scholar]
- 20.Gorry P.R., Churchill M., Crowe S.M., Cunningham A.L., Gabuzda D. Pathogenesis of macrophage tropic HIV-1. Curr. HIV Res. 2005;3:53–60. doi: 10.2174/1570162052772951. [DOI] [PubMed] [Google Scholar]
- 21.Verani A., Gras G., Pancino G. Macrophages and HIV-1: dangerous liaisons. Mol. Immunol. 2005;42:195–212. doi: 10.1016/j.molimm.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 22.Zimmerman E.S., Sherman M.P., Blackett J.L., Neidleman J.A., Kreis C., Mundt P., et al. Human immunodeficiency virus type 1 vpr induces DNA replication stress in vitro and in vivo. J. Virol. 2006;80:10407–10418. doi: 10.1128/JVI.01212-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cosenza M.A., Zhao M.L., Lee S.C. HIV-1 expression protects macrophages and microglia from apoptotic death. Neuropathol. Appl. Neurobiol. 2004;30:478–490. doi: 10.1111/j.1365-2990.2004.00563.x. [DOI] [PubMed] [Google Scholar]
- 24.Persidsky Y., Gendelman H.E. Mononuclear phagocyte immunity and the neuropathogenesis of HIV-1 infection. J. Leukoc Biol. 2003;74:691–701. doi: 10.1189/jlb.0503205. [DOI] [PubMed] [Google Scholar]
- 25.Cosenza M.A., Zhao M.L., Si Q., Lee S.C. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002;12:442–455. doi: 10.1111/j.1750-3639.2002.tb00461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kramer-Hammerle S., Rothenaigner I., Wolff H., Bell J.E., Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 27.Franke T.F., Yang S.I., Chan T.O., Datta K., Kazlauskas A., Morrison D.K., et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 28.van Weeren P.C., de Bruyn K.M., de Vries-Smits A.M., van Lint J., Burgering B.M. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB. J. Biol. Chem. 1998;273:13150–13156. doi: 10.1074/jbc.273.21.13150. [DOI] [PubMed] [Google Scholar]
- 29.del Peso L., Gonzalez-Garcia M., Page C., Herrera R., Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 30.Zhou B.P., Liao Y., Xia W., Spohn B., Lee M.H., Hung M.C. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nature Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 31.Zhou B.P., Liao Y., Xia W., Zou Y., Spohn B., Hung M.C. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nature Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 32.Osaki M., Oshimura M., Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667–676. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 33.Freeman D.J., Li A.G., Wei G., Li H.H., Kertesz N., Lesche R., et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–130. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y., Wang Y., Yamakuchi M., Masuda S., Tokioka T., Yamaoka S., et al. Phosphoinositide-3 kinase-PKB/Akt pathway activation is involved in fibroblast Rat-1 transformation by human T-cell leukemia virus type I tax. Oncogene. 2001;20:2514–2526. doi: 10.1038/sj.onc.1204364. [DOI] [PubMed] [Google Scholar]
- 35.Jeong S.J., Pise-Masison C.A., Radonovich M.F., Park H.U., Brady J.N. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene. 2005;24:6719–6728. doi: 10.1038/sj.onc.1208825. [DOI] [PubMed] [Google Scholar]
- 36.Mannova P., Beretta L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J. Virol. 2005;79:8742–8749. doi: 10.1128/JVI.79.14.8742-8749.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y., Kappes J.C., Conway J.A., Price R.W., Shaw G.M., Hahn B.H. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J. Virol. 1991;65:3973–3985. doi: 10.1128/jvi.65.8.3973-3985.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu Y., Feuer G., Day S.L., Wrzesinski S., Planelles V. Multigene lentiviral vectors based on differential splicing and translational control. Mol. Ther. 2001;4:375–382. doi: 10.1006/mthe.2001.0469. [DOI] [PubMed] [Google Scholar]
- 39.Diamond T.L., Roshal M., Jamburuthugoda V.K., Reynolds H.M., Merriam A.R., Lee K.Y., et al. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 2004;279:51545–51553. doi: 10.1074/jbc.M408573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki T., Kobayashi M., Isatsu K., Nishihara T., Aiuchi T., Nakaya K., Hasegawa K. Mechanisms involved in apoptosis of human macrophages induced by lipopolysaccharide from Actinobacillus actinomycetemcomitans in the presence of cycloheximide. Infect. Immun. 2004;72:1856–1865. doi: 10.1128/IAI.72.4.1856-1865.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deregibus M.C., Cantaluppi V., Doublier S., Brizzi M.F., Deambrosis I., Albini A., Camussi G. HIV-1-Tat protein activates phosphatidylinositol 3-kinase/ AKT-dependent survival pathways in Kaposi's sarcoma cells. J. Biol. Chem. 2002;277:25195–25202. doi: 10.1074/jbc.M200921200. [DOI] [PubMed] [Google Scholar]
- 42.Longo F., Marchetti M.A., Castagnoli L., Battaglia P.A., Gigliani F. A novel approach to protein-protein interaction: complex formation between the p53 tumor suppressor and the HIV Tat proteins. Biochem. Biophys. Res. Commun. 1995;206:326–334. doi: 10.1006/bbrc.1995.1045. [DOI] [PubMed] [Google Scholar]
- 43.Tang Y., Eng C. PTEN autoregulates its expression by stabilization of p53 in a phosphatase-independent manner. Cancer Res. 2006;66:736–742. doi: 10.1158/0008-5472.CAN-05-1557. [DOI] [PubMed] [Google Scholar]
- 44.Kawahara K., Mori M., Nakayama H. NO-induced apoptosis and ER stress in microglia. Nippon Yakurigaku Zasshi. 2004;124:399–406. doi: 10.1254/fpj.124.399. [DOI] [PubMed] [Google Scholar]
- 45.Zhao M.L., Kim M.O., Morgello S., Lee S.C. Expression of inducible nitric oxide synthase, interleukin-1 and caspase-1 in HIV-1 encephalitis. J. Neuroimmunol. 2001;115:182–191. doi: 10.1016/s0165-5728(00)00463-x. [DOI] [PubMed] [Google Scholar]
- 46.Kan H., Xie Z., Finkel M.S. HIV gp120 enhances NO production by cardiac myocytes through p38 MAP kinase-mediated NF-kappaB activation. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H3138–H3143. doi: 10.1152/ajpheart.2000.279.6.H3138. [DOI] [PubMed] [Google Scholar]
- 47.Liu B., Wang K., Gao H.M., Mandavilli B., Wang J.Y., Hong J.S. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J. Neurochem. 2001;77:182–189. doi: 10.1046/j.1471-4159.2001.t01-1-00216.x. [DOI] [PubMed] [Google Scholar]
- 48.Lee P., Lee J., Kim S., Lee M.S., Yagita H., Kim S.Y., et al. NO as an autocrine mediator in the apoptosis of activated microglial cells: correlation between activation and apoptosis of microglial cells. Brain Res. 2001;892:380–385. doi: 10.1016/s0006-8993(00)03257-1. [DOI] [PubMed] [Google Scholar]
- 49.Suh H.S., Kim M.O., Lee S.C. Inhibition of granulocyte-macrophage colony-stimulating factor signaling and microglial proliferation by anti-CD45RO: role of Hck tyrosine kinase and phosphatidylinositol 3-kinase/Akt. J. Immunol. 2005;174:2712–2719. doi: 10.4049/jimmunol.174.5.2712. [DOI] [PubMed] [Google Scholar]
- 50.Nair P., Nair K.M., Jayaprakash P.G., Pillai M.R. Decreased programmed cell death in the uterine cervix associated with high risk human papillomavirus infection. Pathol. Oncol. Res. 1999;5:95–103. doi: 10.1053/paor.1999.0161. [DOI] [PubMed] [Google Scholar]
- 51.Basile J.R., Zacny V., Munger K. The cytokines tumor necrosis factor-alpha (TNF-alpha) and TNF-related apoptosis-inducing ligand differentially modulate proliferation and apoptotic pathways in human keratinocytes expressing the human papillomavirus-16 E7 oncoprotein. J. Biol. Chem. 2001;276:22522–22528. doi: 10.1074/jbc.M010505200. [DOI] [PubMed] [Google Scholar]
- 52.Chaussepied M., Ginsberg D. Transcriptional regulation of AKT activation by E2F. Mol. Cell. 2004;16:831–837. doi: 10.1016/j.molcel.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 53.Diehl J.A., Cheng M., Roussel M.F., Sherr C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ogawara Y., Kishishita S., Obata T., Isazawa Y., Suzuki T., Tanaka K., et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002;277:21843–21850. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- 55.El-Deiry W.S. Akt takes centre stage in cell-cycle deregulation. Nature Cell Biol. 2001;3:E71–E73. doi: 10.1038/35060148. [DOI] [PubMed] [Google Scholar]
- 56.Westbrook T.F., Nguyen D.X., Thrash B.R., McCance D.J. E7 abolishes raf-induced arrest via mislocalization of p21(Cip1) Mol. Cell Biol. 2002;22:7041–7052. doi: 10.1128/MCB.22.20.7041-7052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moody C.A., Scott R.S., Amirghahari N., Nathan C.A., Young L.S., Dawson C.W., Sixbey J.W. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J. Virol. 2005;79:5499–5506. doi: 10.1128/JVI.79.9.5499-5506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peace B.E., Toney-Earley K., Collins M.H., Waltz S.E. Ron receptor signaling augments mammary tumor formation and metastasis in a murine model of breast cancer. Cancer Res. 2005;65:1285–1293. doi: 10.1158/0008-5472.CAN-03-3580. [DOI] [PubMed] [Google Scholar]
- 59.Basha W., Kitagawa R., Uhara M., Imazu H., Uechi K., Tanaka J. Geldanamycin, a potent and specific inhibitor of Hsp90, inhibits gene expression and replication of human cytomegalovirus. Antivir. Chem. Chemother. 2005;16:135–146. doi: 10.1177/095632020501600206. [DOI] [PubMed] [Google Scholar]
- 60.Prejean C., Sarma T., Kurnasov O., Usacheva A., Hemmings B., Cantley L., et al. Phosphatidylinositol 3-kinase confers resistance to encephalomyocarditis and herpes simplex virus-induced cell death through the activation of distinct downstream effectors. J. Immunol. 2001;167:4553–4559. doi: 10.4049/jimmunol.167.8.4553. [DOI] [PubMed] [Google Scholar]
- 61.Mainou B.A., Everly D.N., Raab-Traub N. Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene. 2005;24(46):6917–6924. doi: 10.1038/sj.onc.1208846. [DOI] [PubMed] [Google Scholar]
- 62.Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. JNK and PI3k/Akt signaling pathways are required for establishing persistent SARS-CoV infection in Vero E6 cells. Biochim. Biophys. Acta. 2005;1741:4–10. doi: 10.1016/j.bbadis.2005.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dawson C.W., Tramountanis G., Eliopoulos A.G., Young L.S. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J. Biol. Chem. 2003;278:3694–6704. doi: 10.1074/jbc.M209840200. [DOI] [PubMed] [Google Scholar]
- 64.Enguita M., DeGregorio-Rocasolano N., Abad A., Trullas R. Glycogen synthase kinase 3 activity mediates neuronal pentraxin 1 expression and cell death induced by potassium deprivation in cerebellar granule cells. Mol. Pharmacol. 2005;67:1237–1246. doi: 10.1124/mol.104.007062. [DOI] [PubMed] [Google Scholar]
- 65.Boehme S.A., Lio F.M., Maciejewski-Lenoir D., Bacon K.B., Conlon P.J. The chemokine fractalkine inhibits Fas-mediated cell death of brain microglia. J. Immunol. 2000;165:397–403. doi: 10.4049/jimmunol.165.1.397. [DOI] [PubMed] [Google Scholar]
- 66.Chong Z.Z., Kang J.Q., Maiese K. AKT1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-xL and caspase 1, 3, and 9. Exp. Cell Res. 2004;296:196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 67.Chong Z.Z., Maiese K. W.N.T. Targeting, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol. Histopathol. 2004;19:495–504. doi: 10.14670/hh-19.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dhandapani K.M., Wade F.M., Wakade C., Mahesh V.B., Brann D.W. Neuroprotection by stem cell factor in rat cortical neurons involves AKT and NFkappaB. J. Neurochem. 2005;95:9–19. doi: 10.1111/j.1471-4159.2005.03319.x. [DOI] [PubMed] [Google Scholar]
- 69.Vyas S., Juin P., Hancock D., Suzuki Y., Takahashi R., Triller A., Evan G. Differentiation-dependent sensitivity to apoptogenic factors in PC12 cells. J. Biol. Chem. 2004;279:30983–30993. doi: 10.1074/jbc.M400692200. [DOI] [PubMed] [Google Scholar]
- 70.Li W., He H., Kawakita T., Espana E.M., Tseng S.C. Amniotic membrane induces apoptosis of interferon-gamma activated macrophages in vitro. Exp. Eye Res. 2005 doi: 10.1016/j.exer.2005.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Das S., Cordis G.A., Maulik N., Das D.K. Pharmacological preconditioning with resveratrol: role of CREB-dependent Bcl-2 signaling via adenosine A3 receptor activation. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H328–H335. doi: 10.1152/ajpheart.00453.2004. [DOI] [PubMed] [Google Scholar]
- 72.Vandermoere F., El Yazidi-Belkoura I., Adriaenssens E., Lemoine J., Hondermarck H. The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-kappaB activation induced via interaction between Akt and IkappaB kinase-beta in breast cancer cells. Oncogene. 2005;24:5482–5491. doi: 10.1038/sj.onc.1208713. [DOI] [PubMed] [Google Scholar]
- 73.Hwang S.Y., Jung J.S., Lim S.J., Kim J.Y., Kim T.H., Cho K.H., Han I.O. LY294002 inhibits interferon-gamma-stimulated inducible nitric oxide synthase expression in BV2 microglial cells. Biochem. Biophys. Res. Commun. 2004;318:691–697. doi: 10.1016/j.bbrc.2004.04.082. [DOI] [PubMed] [Google Scholar]
- 74.Harhaj E., Blaney J., Millhouse S., Sun S.C. Differential effects of I kappa B molecules on Tat-mediated transactivation of HIV-1 LTR. Virology. 1996;216:284–287. doi: 10.1006/viro.1996.0062. [DOI] [PubMed] [Google Scholar]
- 75.Fortin J.F., Barat C., Beausejour Y., Barbeau B., Tremblay M.J. Hyper-responsiveness to stimulation of human immunodeficiency virus-infected CD4+ T cells requires Nef and Tat virus gene products and results from higher NFAT, NF-kappaB, and AP-1 induction. J. Biol. Chem. 2004;279:39520–39531. doi: 10.1074/jbc.M407477200. [DOI] [PubMed] [Google Scholar]
- 76.Harrod R., Nacsa J., Van Lint C., Hansen J., Karpova T., McNally J., Franchini G. Human immunodeficiency virus type-1 Tat/co-activator acetyltransferase interactions inhibit p53Lys-320 acetylation and p53-responsive transcription. J. Biol. Chem. 2003;278:12310–12318. doi: 10.1074/jbc.M211167200. [DOI] [PubMed] [Google Scholar]
- 77.Hottiger M.O., Nabel G.J. Interaction of human immunodeficiency virus type 1 Tat with the transcriptional co-activators p300 and CREB binding protein. J. Virol. 1998;72:8252–8256. doi: 10.1128/jvi.72.10.8252-8256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lum J.J., Pilon A.A., Sanchez-Dardon J., Phenix B.N., Kim J.E., Mihowich J., et al. Induction of cell death in human immunodeficiency virus-infected macrophages and resting memory CD4 T cells by TRAIL/Apo2l. J. Virol. 2001;75:11128–11136. doi: 10.1128/JVI.75.22.11128-11136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miura Y., Misawa N., Kawano Y., Okada H., Inagaki Y., Yamamoto N., et al. Tumor necrosis factor-related apoptosis-inducing ligand induces neuronal death in a murine model of HIV central nervous system infection. Proc. Natl Acad. Sci. USA. 2003;100:2777–2782. doi: 10.1073/pnas.2628048100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang Y., Erdmann N., Peng H., Herek S., Davis J.S., Luo X., et al. TRAIL-mediated apoptosis in HIV-1-infected macrophages is dependent on the inhibition of Akt-1 phosphorylation. J. Immunol. 2006;177:2304–2313. doi: 10.4049/jimmunol.177.4.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dorr J., Bechmann I., Waiczies S., Aktas O., Walczak H., Krammer P.H., et al. Lack of tumor necrosis factor-related apoptosis-inducing ligand but presence of its receptors in the human brain. J .Neurosci. 2002;22:RC209. doi: 10.1523/JNEUROSCI.22-04-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crowder R.J., Freeman R.S. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J. Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maggirwar S.B., Tong N., Ramirez S., Gelbard H.A., Dewhurst S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J. Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- 84.Janabi N., Peudenier S., Heron B., Ng K.H., Tardieu M. Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neurosci. Letters. 1995;195:105–110. doi: 10.1016/0304-3940(94)11792-h. [DOI] [PubMed] [Google Scholar]
- 85.Franke T.F., Hornik C.P., Segev L., Shostak G.A., Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 86.Wunderlich M., Berberich S.J. Mdm2 inhibition of p53 induces E2F1 transactivation via p21. Oncogene. 2002;21:4414–4421. doi: 10.1038/sj.onc.1205541. [DOI] [PubMed] [Google Scholar]