

Graphical abstract
Explorations of the S1′ subsite of ACE2 via modifications of the P1′ methylene biphenyl moiety of thiol-based metalloprotease inhibitors led to improvements in ACE2 selectivity versus ACE and NEP, while maintaining potent ACE2 inhibition.

Keywords: Angiotensin-converting enzyme 2, Metalloproteases, Protease inhibitors, Thiols
Abstract
Explorations of the S1′ subsite of ACE2 via modifications of the P1′ methylene biphenyl moiety of thiol-based metalloprotease inhibitors led to improvements in ACE2 selectivity versus ACE and NEP, while maintaining potent ACE2 inhibition.
Dampening of the renin-angiotensin signaling cascade (RAS) has proven invaluable in the treatment of cardiovascular disease. Drugs that inhibit the M2 family dicarboxymetallopeptidase angiotensin-converting enzyme (ACE, EC 3.4.15.1) or the A1 family aspartic protease renin (EC 3.4.23.15), as well as those that antagonize the 7-transmembrane G protein-coupled angiotensin receptor II (AT2), improve hypertension. Dual ACE and M13 metalloprotease neutral endopeptidase (neprilysin, NEP, EC 3.4.24.11) inhibition has also been explored for the treatment of high blood pressure. Recently, a new member of the RAS, the M2 family monocarboxymetallopeptidase angiotensin-converting enzyme 2 (ACE2), was identified.1, 2 It is a membrane-associated and secreted metalloprotease, expressed in heart, kidney, testes, intestine, and lung, with highest homology to ACE. ACE2 has been implicated in cardiovascular disease, kidney disease, obesity, and lung disease.3, 4, 5
Angiotensin I and the AT1 & AT2 receptor agonist angiotensin II are substrates for ACE2, which converts them into angiotensin (1-9) and the mas receptor agonist angiotensin (1-7), respectively, revealing a role for ACE2 in RAS regulation. Moreover, ACE2 (−/−) mice studies have revealed roles for this protease in cardiac contractility,6 angiotensin II-induced hypertension,7 and heart failure.8 Furthermore, male ACE2 (−/Y) mice, but not female ACE2 (−/−) mice, also develop glomerulosclerosis of the kidneys,9 but ACE2’s exact role in the renin-angiotensin system (RAS) still needs to be clarified. In addition, ACE2 (−/−) mice are resistant to weight gain on a high fat diet.10 Finally, ACE2 is the primary receptor for the severe acute respiratory syndrome (SARS) corona virus’s entry into cells.11 ACE2 (−/−) mice resist SARS corona virus infection.12 Because of limited space, the authors refer the interested reader to a recent review by Hamming et al. for a more comprehensive summary of the physiology and pathology of ACE2.13
With the many putative roles of ACE2, small molecule modulators of this enzyme’s activity could be utilized to help further define the physiological functions of this enzyme. Although small molecule inducers of proteases are difficult to discover, ACE2 activators could provide further insight into the functions of ACE2 and the substrates it processes. Similarly, inhibitors could also shed more light on the roles of ACE2 and the proteins/peptides it activates/degrades.
Efforts to discover ACE2 inhibitors have recently been disclosed. Millennium Pharmaceutical researchers reported the discovery of a reversible, subnanomolar ACE2 inhibitor MLN-4760 (IC50 = 0.44 nM), derived from a micromolar lead.14 In addition, Dyax researchers identified micromolar peptide inhibitors of ACE2 using a phage display library approach, including the 29 amino acid cyclic peptide DX600 (IC50 = 10 nM).15 Furthermore, a pharmacophore-based virtual screening approach of commercial databases identified several micromolar ACE2 inhibitors. The best inhibitor was 4S-16659 (IC50 = 62,000 nM).16 Finally, researchers at the University of Florida also utilized a virtual screening exercise of the NCI database to identify N-(2-aminoethyl)-1 aziridine-ethanamine (NAAE) (IC50 = 57,000 nM) as an irreversible inhibitor of ACE2.17

Recently, employing a directed screen of a set of metalloprotease inhibitors from the GlaxoSmithKline compound collection, GSK researchers reported the identification of a known ACE/NEP inhibitor as a potent inhibitor of the zinc metalloprotease ACE2.18 Structure/activity studies of the S1 subsite of ACE2 led to the identification of P1 modified thiol acid 1a as a more potent ACE2 inhibitor (K i App = 1.5 nM) with improved selectivity versus ACE (K i App = 490 nM) and NEP (K i App = 27 nM). It also inhibits the M14 metalloprotease carboxypeptidase A1 (CpA, EC 3.4.17.1, K i App = 11,000 nM). Speculating that selectivity could be improved by exploiting potential differences in the S1′ subsites of ACE, ACE2, and NEP, the structure–activity relationships of the P1′ position of inhibitor 1a were explored, with the goal of maintaining potency and reducing ACE and NEP inhibitory activity.

The thiol analogs 1a–1t were prepared as depicted in Scheme 1 . The amine hydrochlorides 2a–2t were coupled to the acid 3, after its in situ activation to the aza-hydroxybenzotriazole ester via the carbodiimide, to produce the fully protected amides. Then, hydrolysis of the thioacetate and methyl ester with lithium hydroxide afforded the thiol acids 1a–1t.19
Scheme 1.

Reagents and conditions: (a) EDC, HOAt, i-Pr2NEt, CH2Cl2, rt, 39–76%; (b) LiOH·H2O, THF, H2O, rt, 56–94%.
Amine hydrochlorides 2a–2g, 2l, and 2o were commercially available. The remaining amine hydrochlorides 2h–2k, 2m–2n, and 2p–2t were prepared as depicted in Scheme 2 . The commercially available amino acids 4a–4b were converted to their corresponding methyl esters with thionyl chloride and methanol at reflux. Then, the resulting amine hydrochlorides were protected as the tert-butyl carbamates 5a–5b with di-tert-butyl dicarbonate. A Suzuki cross coupling of the o-bromide 5a (X = Br) with phenyl boronic acid afforded the o-biphenyl derivative 6a. Acid catalyzed cleavage of the carbamate gave the o-biphenyl amine hydrochloride 2h. The m-phenol 5b (X = OH) was converted into the triflate with trifluoromethanesulfonic anhydride, and then coupled via the Suzuki protocol to phenyl boronic acid, providing the fully protected m-biphenyl derivative 6b. Subsequent hydrolysis of the amine masking group gave the m-biphenyl amine hydrochloride 2i. The o-bromide 5a (X = Br) was converted into the o-phenol 5c (X = OH) via palladium-mediated transformation of the bromide into a boronate ester, and then oxidation of the boronate to the phenol with hydrogen peroxide. Employing a modified Ullmann ether synthesis protocol, the o-phenol 5c and m-phenol 5b were coupled with phenyl boronic acid to afford the o- and m-diaryl ethers 6c and 6d. Acidic uncloaking of the tert-butyl carbonyl protecting group, then produced the phenyl ethers 2j and 2k. Finally, utilizing the Williamson ether synthesis, the o-phenol 5c, the m-phenol 5b, and the commercially available p-phenol 5d were alkylated with various benzyl bromides to give the fully masked benzyl aryl ethers 6e–6k. Then, unmasking of the carbamates afforded the amine hydrochlorides 2m–2n and 2p–2t.
Scheme 2.

Reagents and conditions: (a) SOCl2, MeOH, ↑↓, 95–99%; (b) Boc2O, i-Pr2NEt, CH2Cl2, 0 °C to rt, 97–99%; (c) X = Br or OTf, PhB(OH)2, Pd(PPh3)4, K2CO3, PhMe, 90 °C, 41–57%; (d) X = OH, Tf2O, pyridine, CH2Cl2, 0 °C to rt, 88%; (e) X = Br, PdCl2(dppf), bis(pinacolato)diborane, KOAc, DMF, 80 °C; 30% H2O2 (aq), MeOH, 44%; (f) X = OH, Cu(OAc)2, PhB(OH)2, pyridine, powdered 4 Å molecular sieves, CH2Cl2, rt, 70–96%; (g) X = OH, ArCH2Br, K2CO3, acetone, rt, 71–99%; (h) HCl, MeOH, 91–98%.
The structure/activity relationships of P1′ analogs are depicted in Table 1 . Complete removal of the P1′ substituent as in analog 1b resulted in a large decrease in ACE2 (K i App = 90 nM) and NEP (K i App = 5100 nM) inhibitory activity. The potential reduction in van der Waals interactions from the absence of a P1′ side chain as well as the entropic gain from increased rotational freedom of the acid could account for this decrease in potency. In contrast, this change had little affect on ACE (K i App = 250 nM) or CpA (K i App = 35,000 nM). The alanine-derived analog 1c (ACE2 K i App = 14 nM, NEP K i App = 950 nM) decreases rotational freedom and recovers some of the ACE2 inhibitory activity lost upon removal of the P1′ group, while not dramatically changing the activity versus ACE (K i App = 180 nM) or CpA (K i App = 11,000 nM). The phenylalanine and tyrosine derivatives 1d (K i App = 1.5 nM) and 1e (K i App = 1.3 nM) were equipotent ACE2 inhibitors to the methylene p-biphenyl analog 1a with similar ACE (1d K i App = 570 nM, 1e K i App = 410 nM) and NEP (1d K i App = 35 nM, 1e K i App = 75 nM) activities as well. In contrast to 1a, the benzyl derivatives 1d and 1e were >10-fold more potent inhibitors of CpA (1d K i App = 530 nM, 1e K i App = 350 nM). Since many good CpA substrates contain P1′ phenylalanine residues, this increase in CpA inhibition for 1d and 1e is not surprising.
Table 1.

| # | R | ACE2 | ACE | NEP | CpA |
|---|---|---|---|---|---|
| Ki Appa (nM) | Ki Appb (nM) | Ki Appc (nM) | Ki Appd (nM) | ||
| 1a |  |
1.5 | 490 | 27 | 11,000 |
| 1b | H | 90 | 250 | 5100 | 35,000 |
| 1c | Me | 14 | 180 | 950 | 11,000 |
| 1d |  |
1.5 | 570 | 35 | 530 |
| 1e |  |
1.3 | 410 | 75 | 350 |
| 1f | 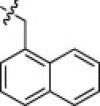 |
2.2 | 1800 | 12 | 3600 |
| 1g |  |
2.4 | 310 | 46 | 7200 |
| 1h | 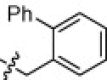 |
37 | 20,000 | 190 | 9900 |
| 1i |  |
2.2 | 1000 | 19 | 5000 |
| 1j |  |
0.90 | 5000 | 180 | 2300 |
| 1k | 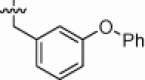 |
5.8 | 1100 | 79 | 14,000 |
| 1l | 2.2 | 250 | 10 | 8900 | |
| 1m |  |
6.0 | 770 | 120 | 1600 |
| 1n | 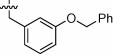 |
2.4 | 750 | 37 | 5000 |
| 1o | 2.7 | 570 | 300 | 4600 | |
| 1p | 2.5 | 4000 | 180 | 16,000 | |
| 1q | 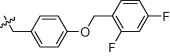 |
2.0 | 950 | 75 | 1900 |
| 1r |  |
0.85 | 1800 | 640 | 3300 |
| 1s | 35 | 2600 | 1500 | 4600 | |
| 1t |  |
84 | 25,000 | 11,000 | 11,000 |
Inhibition of recombinant human ACE2 activity in a fluorescence assay using 0.4 nM ACE2, 30 μM MCA-Tyr-Val-Ala-Asp-Ala-Pro-Lys(DNP)-OH as substrate in 1 μM Zn(OAc)2, 100 μM TCEP, 50 mM Hepes, 300 μM CHAPS, and 300 mM NaCl at pH = 7.5. The Ki App values are means of at least two inhibition assays.
Inhibition of recombinant human ACE activity in a fluorescence assay using 0.5 nM ACE, 10 μM MCA-Ala-Ser-Asp-Lys-Dap(DNP)-OH as substrate in 1 μM Zn(OAc)2, 100 μM TCEP, 50 mM Hepes, 300 μM CHAPS, and 300 mM NaCl at pH = 7.5. The Ki App values are means of at least two inhibition assays.
Inhibition of recombinant human NEP activity in a fluorescence assay using 0.15 nM NEP, 2 μM FAM-Gly-Pro-Leu-Gly-Leu-Phe-Ala-Arg-Lys(TAMRA)-NH2 as substrate in 1 μM Zn(OAc)2, 100 μM TCEP, 50 mM Hepes, 300 μM CHAPS, and 300 mM NaCl at pH = 7.5. The Ki App values are means of at least two inhibition assays.
Inhibition of recombinant human CpA activity in a fluorescence assay using 37 nM CpA, 30 μM Abz-Gly-Gly-Nph-OH as substrate in 1 μM Zn(OAc)2, 100 μM TCEP, 50 mM Hepes, 300 μM CHAPS, and 300 mM NaCl at pH = 7.5. The Ki App values are means of at least two inhibition assays.
It was decided to explore other bulky P1′ substituents with the goal of further improving on the potency and/or selectivity of analog 1a, since the bulky methylene biphenyl P1′ moiety is readily accommodated by the ACE2 enzyme, implying a large S1′ subsite. Although the α-methylene naphthyl and the β-methylene naphthyl derivatives 1f (K i App = 2.2 nM) and 1g (K i App = 2.4 nM) were also potent ACE2 inhibitors with >100-fold selectivity versus ACE (1f K i App = 1,800 nM, 1g K i App = 310 nM) and CpA (1f K i App = 3600 nM, 1g K i App = 7200 nM), they still retained substantial inhibitory activity against NEP (1f K i App = 12 nM, 1g K i App = 46 nM).
In contrast to the p-phenyl phenylalanine derivative 1a, the o-phenyl phenylalanine analog 1h (K i App = 37 nM) was an order of magnitude less potent ACE2 inhibitor. This substitution also reduced ACE (K i App = 20,000 nM) and NEP (K i App = 190 nM) inhibition, while not altering CpA potency (K i App = 9900 nM). In contrast to 1h, the methylene m-biphenyl derivative 1i (K i App = 2.2 nM) was an equipotent ACE2 inhibitor to the p-derivative 1a, despite branching from the phenyl moiety at a different angle, but it offered no clear selectivity advantages over ACE (K i App = 1000 nM), NEP (K i App = 19 nM), or CpA (K i App = 5000 nM) versus 1a.
Although the large biphenyl derivatives were fairly rigid, they were tolerated in the S1′ subsite of ACE2. Thus, bulkier P1′ groups were explored. The o-, m-, and p-phenoxy phenylalanine derivatives 1j (K i App = 0.90 nM), 1k (K i App = 5.8 nM), and 1l (K i App = 2.2 nM) retained good potency for ACE2 inhibition and >100-fold selectivity versus ACE (1j K i App = 5000 nM, 1k K i App = 1100 nM, 1l K i App = 250 nM) and CpA (1j K i App = 2300 nM, 1k K i App = 14,000 nM, 1l K i App = 8900 nM), despite altered branching termini. In contrast, their NEP selectivity differed. The o-phenyloxy analog 1j (K i App = 180 nM) was 200-fold selective versus NEP, while the m-phenoxy analog 1k (K i App = 79 nM) and the p-phenoxy analog 1l (K i App = 10 nM) were 14-fold and 5-fold NEP selective, respectively.
Surprisingly, the extended o-, m-, and p-benzyloxy phenylalanine derivatives 1m (K i App = 6.0 nM), 1n (K i App = 2.4 nM), and 1o (K i App = 2.7 nM) were also well tolerated in the S1′ subsite of ACE2. In addition, they maintained good selectivity versus ACE (1m K i App = 770 nM, 1n K i App = 750 nM, 1o K i App = 570 nM) and CpA (1m K i App = 1600 nM, 1n K i App = 5000 nM, 1o K i App = 4600 nM). In contrast to the phenyloxy analogs, the p-benzyl analog 1o (K i App = 300 nM) was quite selective versus NEP (110-fold), while the o-benzyloxy analog 1m (K i App = 120 nM) and the m-benzyloxy analog 1n (K i App = 37 nM) were less selective (20-fold and 15-fold, respectively).
Both the methylene o-phenoxyphenyl derivative 1j and the methylene p-benzyloxyphenyl analog 1o are potent and selective ACE2 inhibitors, but the latter inhibitor is readily derived from the cheap natural amino acid tyrosine. Therefore, fluorinated derivatives of inhibitor 1o were prepared with the goal of maintaining potency and selectivity, while blocking potential metabolic sites of the naked phenyl ring. It was hoped that electron withdrawing groups would not only sterically impede metabolism, but also electronically deactivate the aryl ring to oxidation. The mono- and difluoro analogs 1p (K i App = 2.5 nM), 1q (K i App = 2.0 nM), and 1r (K i App = 0.85 nM) were potent ACE2 inhibitors with good selectivity. The 3,4-difluorobenzyl tyrosine derivative 1r was the most selective analog with >750-fold separation in K i Apps between ACE2 and ACE (K i App = 1800 nM), NEP (K i App = 640 nM), and CpA (K i App = 3300 nM). In contrast to the ACE2 inhibition of the fluoro derivatives 1p–1r, the larger trifluoromethyl derivatives 1s (K i App = 35 nM) and 1t (K i App = 84 nM) were substantially less potent ACE2 inhibitors. Likely, these larger P1′ substituents clash with the residues that make up the S1′ subsite.
In vitro drug metabolism and pharmacokinetic assays were performed to help predict in vivo oral bioavailabilities and pharmacokinetics of the thiols. Although these inhibitors conform to Lipinski’s Rule of 5, the Madin-Darby canine kidney (MDCK) assay revealed that this inhibitor class had poor to moderate passive cell permeabilities (PAPP = 5–47 nm/s).20 Since many dipeptides are absorbed by active transport mechanisms, representative thiol-based inhibitors were dosed orally in the rat despite their poor predicted cell permeation, but they had limited absorption (F < 10%) in agreement with the MDCK assay.
A model21, 22 of the thiol 1r docked into the active site of ACE2 based on the recent Millennium X-ray crystal structure23 is shown in Figure 1 . It provides insight into the P1′ SAR of the thiol series. The terminal carboxylate of the inhibitor accepts hydrogen bonds from the imidazoles of 345His and 505His and forms a salt bridge to the guanidine of 273Arg. Also, the amide nitrogen of the inhibitor donates a hydrogen atom to the carbonyl of 346Pro, while the amide carbonyl accepts a hydrogen atom from 515Tyr. Moreover, in addition to its normal ACE2 ligands, the imidazole nitrogens of 374His and 378His and the carboxylate of 402Glu, the active site zinc coordinates the thiol of the inhibitor. Furthermore, the P1 sec-butyl group of the inhibitor forms van der Waals interactions with the S1 pocket composed of 347Thr, 504Phe, 510Tyr, and 514Arg. The greasy P1′ 3,4-difluorobenzyltyrosine side chain of 1r forms significant lipophilic interactions with the quite large S1′ channel composed of the lengthwise canal between the two subdomains, including residues 274Phe, 445Thr, 406Glu, 409Ser, 370Leu, 371Thr, 276Thr and extending over to 441Lys and 442Gln. Although several residues in this pocket are hydrophilic, most of the polar groups are either involved in hydrogen bonds with other enzyme residues or oriented away from the S1′ pocket, maintaining the lipophilic nature of this subsite. Based on this model, the 3,4-difluorobenzyl portion of the P1′ substituent is hypothesized to occupy a different part of this large pocket than the P1′ side chain of the carboxyl inhibitor MLN-4760 co-crystallized in 1R4L.
Figure 1.

A model of the thiol compound 1r bound to the active site of ACE2 based on the X-ray co-crystal structure of MLN-4760 bound to ACE2 (PDB code 1R4L). The ACE2 carbons are colored cyan with inhibitor 1r carbons colored green. The semi-transparent gray surface represents the molecular surface, while hydrogen bonds are depicted as yellow dashed lines. Several residues were removed for visual clarity. This figure was generated using PYMOL version 1.0 (Delano Scientific, www.pymol.org).
In contrast, as shown in Figure 2 , a model21, 22 of the thiol 1j docked into the active site of ACE2 revealed that the o-phenyloxy P1′ group occupies a different part of the S1′ subsite than the 3,4-difluorobenzyl substituent of analog 1r. The o-phenyloxy P1′ moiety fits into a portion of the S1′ subsite composed of 360Met, 346Pro, 362Thr, 271Trp, 368Asp, 371Thr, 127Tyr, 144Leu, 149Asn, 363Lys, and 269Asp, and the disulfide pair of 344Cys and 361Cys. Thus, the o-phenyloxy P1′ moiety of 1j binds similarly to the P1′ side chain of the Millennium carboxyl inhibitor MLN-4760 co-crystallized in 1R4L. The other interactions of inhibitor 1j are similar to those for analog 1r. Thus, these two models help explain the divergent P1′ SAR, since the very large, forked S1′ subsite can tolerate substituents with different branching points.
Figure 2.

A comparison of the models of the thiol compounds 1j (carbons colored in green) and 1r (carbons colored in magenta) bound to the active site of ACE2 (carbons colored in cyan) based on the X-ray co-crystal structure (PDB code 1R4L). The semi-transparent gray surface represents the molecular surface, while hydrogen bonds are depicted as yellow dashed lines. Several residues were removed for visual clarity. This figure was generated using PYMOL version 1.00.
In summary, variation of substituents at the P1′ position in a series of α-thiol amide-based inhibitors of ACE2 resulted in the discovery of potent inhibitors with good ACE and NEP selectivity. Inhibitors containing p-methylene aryl tyrosine P1′ moieties like 1o, 1p, and 1r were some of the more selective ACE2 inhibitors. In addition, o-phenyloxy phenylalanine analog 1j was also a potent and selective ACE2 inhibitor. These analogs may prove useful in further defining the roles ACE2 plays in the RAS cascade.
Acknowledgments
The authors thank Andrea Epperly, Chuck Poole, and Jo Salisbury for the pharmacokinetic studies.
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2008.01.046.
Supplementary data
References and notes
- 1.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R., Breitbart R.E., Acton S. Circ. Res. 2000;87:e1. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 2.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. J. Biol. Chem. 2000;275:33238. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 3.Danilczyk U., Penninger J.M. Circ. Res. 2006;98:463. doi: 10.1161/01.RES.0000205761.22353.5f. [DOI] [PubMed] [Google Scholar]
- 4.Kuba K., Imai Y., Penninger J.M. Curr. Opin. Pharm. 2006;6:271. doi: 10.1016/j.coph.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tallant E.A., Ferrario C.M., Gallagher P.E. Future Cardiol. 2006;2:335. doi: 10.2217/14796678.2.3.335. [DOI] [PubMed] [Google Scholar]
- 6.Crackower M.A., Sarao R., Oudit G.Y., Yagil C., Kozieradzki I., Scanga S.E., Oliveira-dos-Santos A.J., da Costa J., Zhang L., Pei Y., Scholey J., Ferrario C.M., Manoukian A.S., Chappell M.C., Backx P.H., Yagil Y., Penninger J.M. Nature. 2002;417:822. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 7.Gurley S.B., Allred A., Le T.H., Griffiths R., Mao L., Philip N., Haystead T.A., Donoghue M., Breitbart R.E., Acton S.L., Rockman H.A., Coffman T.M. J. Clin. Invest. 2006;116:2218. doi: 10.1172/JCI16980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto K., Ohishi M., Katsuya T., Ito N., Ikushima M., Kaibe M., Tatara Y., Shiota A., Sugano S., Takeda S., Rakugi H., Ogihara T. Hypertension. 2006;47:718. doi: 10.1161/01.HYP.0000205833.89478.5b. [DOI] [PubMed] [Google Scholar]
- 9.Oudit G.Y., Herzenberg A.M., Kassiri Z., Wong D., Reich H., Khokha R., Crackower M.A., Backx P.H., Penninger J.M., Scholey J.W. Am. J. Pathol. 2006;168:1808. doi: 10.2353/ajpath.2006.051091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acton, S. L.; Ocain, T. D.; Gould, A. E.; Dales, N. A.; Guan, B.; Brown, J. A.; Patane, M.; Kadambi, V. J.; Solomon, M.; Stricker-Krongrad, A. PCT Int. Appl. WO 039997, 2002; Chem. Abstr. 2002, 136, 402027.
- 11.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Nature. 2003;426:450. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuba K., Imai Y., Rao S., Gao H., Guo F., Guan B., Huan Y., Yang P., Zhang Y., Deng W., Bao L., Zhang B., Liu G., Wang Z., Chappell M., Liu Y., Zheng D., Leibbrandt A., Wada T., Slutsky A.S., Liu D., Qin C., Jiang C., Penninger J.M. Nat. Med. 2005;11:875. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamming I., Cooper M.E., Haagmans B.L., Hooper N.M., Korstanje R., Osterhaus A.D.M.E., Timens W., Tumer A.J., Navis G., van Goor H. J. Pathol. 2007;212:1. doi: 10.1002/path.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dales N.A., Gould A.E., Brown J.A., Calderwood E.F., Guan B., Minor C.A., Gavin J.M., Hales P., Kaushik V.K., Stewart M., Tummino P.J., Vickers C.S., Ocain T.D., Patane M.A. J. Am. Chem. Soc. 2002;124:11852. doi: 10.1021/ja0277226. [DOI] [PubMed] [Google Scholar]
- 15.Huang L., Sexton D.J., Skogerson K., Devlin M., Smith R., Sanyal I., Parry T., Kent R., Enright J., Wu Q., Conley G., DeOliveira D., Morganelli L., Ducar M., Wescott C.R., Ladner R.C. J. Biol. Chem. 2003;278:15532. doi: 10.1074/jbc.M212934200. [DOI] [PubMed] [Google Scholar]
- 16.Rella M., Rushworth C.A., Guy J.L., Turner A.J., Langer T., Jackson R.M. J. Chem. Inf. Model. 2006;46:708. doi: 10.1021/ci0503614. [DOI] [PubMed] [Google Scholar]
- 17.Huentelman M.J., Zubcevic J., Hernandez Prada J.A., Xiao X., Dimitrov D.S., Raizada M.K., Ostrov D.A. Hypertension. 2004;44:903. doi: 10.1161/01.HYP.0000146120.29648.36. [DOI] [PubMed] [Google Scholar]
- 18.Deaton D.N., Gao E.N., Graham K.P., Gross J.W., Miller A.B., Strelow J.M. Bioorg. Med. Chem. Lett. 2008;18:732. doi: 10.1016/j.bmcl.2007.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hydrolyses were performed under an argon atmosphere. When hydrolyses were carried out with reactions open to the atmosphere, varying amounts of disulfide products were isolated, likely from aerobic oxidation. The enzyme assays contain a reducing agent, Tris-(2-chloroethyl)-phosphate (TCEP), to prevent oxidation of the thiols to disulfides during the assays. Dilutions of 10 mM stock solutions of the thiols 1a–1t to final assay concentrations were done with 50% aqueous acetonitrile just prior to protease inhibition studies. Under these standard conditions, both the thiols and their corresponding disulfides showed enzyme inhibitory activity. Presumably, the disulfides were reduced to their corresponding thiols by TCEP during the pre-incubation period, before substrates were added. In contrast, if the assays were performed without TCEP, the disulfides were completely inactive, while the potencies of the thiols were attenuated, probably because of partial aerobic oxidation to their corresponding disulfides during the pre-incubation period.
- 20.Irvine J.D., Takahashi L., Lockhart K., Cheong J., Tolan J.W., Selick H.E., Grove J.R. J. Pharm. Sci. 1999;88:28. doi: 10.1021/js9803205. [DOI] [PubMed] [Google Scholar]
- 21.Lambert M.H. In: Practical Application of Computer-Aided Drug Design. Charifson P.S., editor. Marcel Dekker; New York, NY: 1997. p. 243. [Google Scholar]
- 22.The ‘grow’ algorithm within the MVP program was used to dock the inhibitor into the active site beginning from the carboxylic acid group.
- 23.Towler P., Staker B., Prasad S.G., Menon S., Tang J., Parsons T., Ryan D., Fisher M., Williams D., Dales N.A., Patane M.A., Pantoliano M.W. J. Biol. Chem. 2004;279:17996. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.