

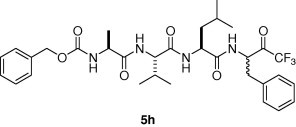
Graphical abstract
Keywords: SARS, Protease inhibitor, Trifluoromethyl ketone, Time-dependent inhibition
Abstract
A series of trifluoromethyl ketones as SARS-CoV 3CL protease inhibitors was developed. The inhibitors were synthesized in four steps from commercially available compounds. Three different amino acids were explored in the P1-position and in the P2–P4 positions varying amino acids and long alkyl chain were incorporated. All inhibitors were evaluated in an in vitro assay using purified enzyme and fluorogenic substrate peptide. One of the inhibitors showed a time-dependent inhibition, with a Ki value of 0.3 μM after 4 h incubation.
1. Introduction
Severe acute respiratory syndrome-associated coronavirus (SARS-CoV), identified to be the causative agent of this life-threatening epidemic,1, 2, 3, 4, 5 leads to a respiratory disease with the symptoms including cough, high fever, chills, rigor, myalgia, headache, dizziness, and progressive radiographic changes of the chest and lymphopenia. The spread of this contagious disease in 2003 infected more than 8000 people with a high mortality. In total, there were 774 deaths reported around the world. During the life cycle of SARS-CoV, 3CL protease cleaves the polyprotein into individual polypeptides to provide all the essential proteins for viral replication and transcription.6, 7 This enzyme is thus recognized as a primary target for the therapeutic intervention.
In contrast to the common serine proteases containing a Ser-His-Asp catalytic triad, SARS-CoV 3CL protease has a Cys-His catalytic dyad (Cys-145 and His-41), which is similar to porcine transmissible gastroenteritis virus main protease (Cys-144 and His-41) and human coronavirus 229E main protease (Cys-144 and His-41).8 In addition, it cleaves the replicase polyprotein at no less than 11 conserved sites with canonical Leu-Gln↓(Ser, Ala, Gly) sequences.9 Taken together, this information provides good understanding to the design of potent inhibitors.
To date, a number of 3CL protease inhibitors have been prepared, including C 2-symmetric diols,10 bifunctional aryl boronic acids,11 keto-glutamine analogs,12 isatin derivatives,13 α,β-unsaturated esters,14 anilide,15 and benzotriazole.16 Here, we report the synthesis of trifluoromethyl ketones as inhibitors against SARS-CoV 3CL protease, and provide kinetic analysis and computer modeling to address the issue of covalent binding.
Trifluoromethyl ketones (TFMKs) are well known as the inhibitors of serine17 and cysteine18 proteases. Owing to the high electronegativity of fluorine, the carbonyl carbon of TFMK is a highly active electrophile. It is generally believed that hemiketal or hemithioketal is formed by the nucleophilic attack of the hydroxyl or thiol group at the active site when TFMKs are employed as the inhibitors against serine or cysteine proteases, respectively. Previous studies19 indicated that TFMKs demonstrate a competitive slow, tight-binding inhibition against human leukocyte elastase. Recently, Zhang et al.20a described N,N-dimethyl glutaminyl fluoromethyl ketones as 3CL protease inhibitors. One of these compounds was found to have low toxicity in mice, and another one was found to have an EC50 value of 2.5 μM based on the cytopathic effect (CPE) inhibition assay. However, the in vitro inhibition has not been characterized in detail. Sydnes et al.20b also reported the synthesis of glutamic acid and glutamine peptides with a CF3-ketone unit as 3CL protease inhibitors.
2. Results and discussion
In order for the synthetic simplicity, we assumed that the benzyl group as the P1 site can mimic the Gln residue of the substrate. Scheme 1 shows the four-step synthesis of various N-protected trifluoromethyl ketones. The preparation of nitro alcohols 3 was carried out by C–C bond formation between nitroalkanes 2 and trifluoroacetaldehyde ethyl hemiacetal under the basic condition of catalytic potassium carbonate. The choice of nitroalkanes defines the P1 group of the final inhibitor. For instance, 1-nitro-2-phenylethane 2a introduces a benzyl group at the P1 site. Subsequent reduction to amine alcohols was performed either by PtO2- or Raney nickel-catalyzed hydrogenation. The use of PtO2 was avoided in the reduction of 3a because undesired saturation of the phenyl ring was observed. At this stage, the trifluoroamine alcohols were coupled with N-protected amino acids or long-chain acids by using HBTU and DIEA (or Et3N) to afford 4a–g. Final oxidation using Dess–Martin reagent generated the desired trifluoromethyl ketones 5a–h.
Scheme 1.
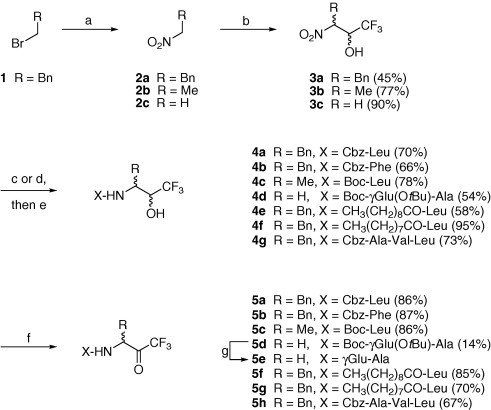
Synthesis of trifluoromethyl ketones 5a–5h. Reagents and conditions: (a) NaNO2 (1.3 equiv), DMF, −78 → 23 °C, 15 h, 68%; (b) trifluoroacetaldehyde ethyl hemiacetal (1.27 equiv), K2CO3 (cat.), neat, 50–60 °C, 3 h, then 23 °C, 25.5 h, 45–90%; (c) H2 (1 atm), cat. PtO2·×H2O (79–84% Pt), MeOH/CHCl3 (16:1), 23 °C, 43 h; (d) H2 (1 atm), Ra-Ni, H2O, EtOH, 23 °C, 14 h; (e) N-protected amino acids or long-chain acids, HBTU, DIEA (or Et3N), DMF, 23 °C, 36 h, 54–95%; (f) Dess-Martin reagent (3 equiv), TFA (3 equiv), CH2Cl2, 22 °C, 3 h, 14–87%; (g) TFA, 40.5 h. DMF = N, N-dimethylformamide; HBTU = (1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; DIEA = diisopropylethylamine; TFA = trifluoroacetic acid.
TFMKs 5a–h were evaluated to interfere with SARS-CoV 3CL protease activity according to the reported procedure21 (Table 1 ). The activity of 5a, 5b, 5f, 5g, and 5h, having benzyl group as the side chain at the P1 site, supports the idea that the P2–P4 sites still have a significant contribution to the binding affinity though they are far from the active site. The best inhibitor 5h, containing the same residues as the reported substrate sequence at the P2, P3, and P4 sites, displayed a competitive inhibition against 3CL protease (Fig. 1 ). Moreover, in consistence with the previous reports of cathepsin B and human leukocyte elastase,(b), 19 prolonged incubation of 3CL protease with 5h exhibited a time-dependent decrease in enzyme activity as a function of the inhibitor concentration. The inhibitor was found to produce progressive tightening of inhibition, as shown by a 30-fold decrease in the K i value (from 8.8 to 0.3 μM) in 4 hr (Table 2 and Fig. 2 ). As indicated by the NMR studies, the trifluoromethyl ketone moiety exists as an equilibrium mixture of ketone and hydrate forms. The time-dependent tightening of inhibition is likely due to the slow formation of a covalent adduct through the nucleophilic attack of the thiol group on the carbonyl carbon.
Table 1.
Inhibition of trifluoromethyl ketones against SARS-CoV 3CL protease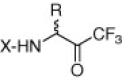
| No. | R | X | IC50 (μM) |
|---|---|---|---|
| 5a | Bn | Cbz-Leu | 15 |
| 5b | Bn | Cbz-Phe | 20 |
| 5c | Me | Boc-Leu | 40 |
| 5d | H | Boc-γGlu(OtBu)-Ala | 40 |
| 5e | H | γGlu-Ala | 50 |
| 5f | Bn | CH3(CH2)8CO-Leu | 50 |
| 5g | Bn | CH3(CH2)7CO-Leu | >50 |
| 5h | Bn | Cbz-Ala-Val-Leu | 10 |
Figure 1.
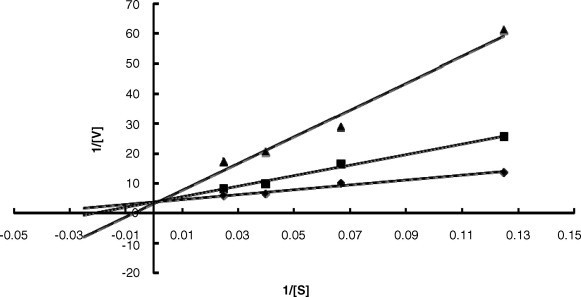
Lineweaver-Burk plots of compound 5h incubated with 3CL protease for 4 h. The enzyme activities were measured using 8–40 μM fluorogenic substrate in the absence (♦) or presence of 1 × IC50 (■) and 2× IC50 (▴) inhibitor. The pattern of these plots displayed competitive inhibition.
Table 2.
Time-dependent inhibition of 5h against SARS-CoV 3CL protease
| Incubation time | IC50 (μM) | Ki (μM) |
|---|---|---|
| 10 min | 10 | 8.76 ± 1.61 |
| 30 min | 7 | 2.69 ± 0.47 |
| 1 h | 4 | 1.30 ± 0.19 |
| 2 h | 2 | 0.73 ± 0.07 |
| 4 h | 0.8 | 0.29 ± 0.09 |
Figure 2.
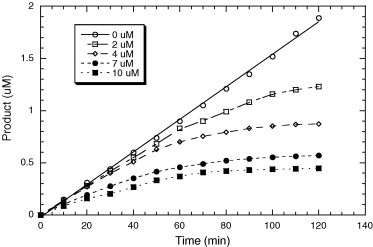
The progress curves in the presence of 2–10 μM inhibitor for reactions initiated by adding enzyme (final concentration of 0.005 μM) into a mixture of substrate (6 μM) and inhibitor 5h. Over the entire 120 min time window, the uninhibited enzyme displayed a linear progress curve, whereas the inhibited enzyme with a different concentration of inhibitor showed a time-dependent reduction of activity.
Compound 5h and 3CL protease complex have been crystallized in our laboratory, but the X-ray crystallography experiments were nevertheless unsuccessful in structural refinement due to fragmented electron density maps. Alternatively, computational molecular modeling was used to construct a model for the acyl–enzyme complex. On the basis of the crystal structure of 3CL protease with a chloromethyl ketone (CMK) inhibitor, the analog of trifluoromethyl ketone, determined by Yang et al.,7 we first constructed the models for the four possible stereoisomers of the covalent adducts between the protein and compound 5h. All the models were constrained with a covalent link between the thiol group of Cys-145 and compound 5h, in consistent with the analog experimental complex structure by Yang et al.7 In comparison with the analog experimental structure, only the (S,S,S,S) isomer of compound 5h with the R configuration of carbonyl carbon adjacent to CF3 group agreed with the binding mode of the CMK inhibitor, in particular all four amino acid side chains of compound 5h fitted into the bind pockets of the 3CL protease active site. All the other three stereoisomers were ruled out because all these molecules were unable to bind to the active site under the covalent constraint. The computational model of the (S,S,S,S) isomer is different from the binding mode of the CMK-3CL protease complex structure in that the P1, P2, P4 side chains in compound 5h occupied S2, S1, and S4 sites, respectively, in 3CL protease (Fig. 3 ). The binding mode discrepancies were expected consequences due to the difference between the amino acid side chains of the two inhibitor analogs. The proposed detailed covalent attacking mechanism was shown in Figure 4 .
Figure 3.
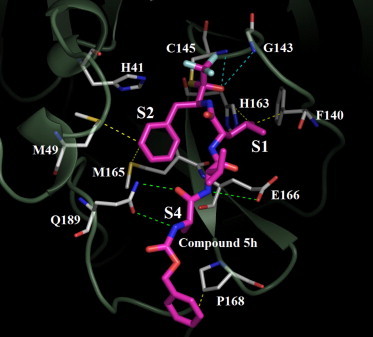
The model of compound 5h and SARS-CoV 3CL protease. The hydrogen bondings are shown in the green and blue (oxyanion hole) dotted lines, and the hydrophobic interactions are shown in yellow dotted lines. The thiol group on Cys-145 forms a covalent bonding to compound 5h.
Figure 4.
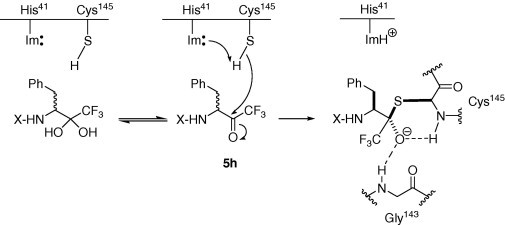
Proposed mechanism of inhibition of 3CL protease with compound 5h.
3. Conclusion
The substrate-based design and synthesis of trifluoromethyl ketones as SARS-CoV 3CL protease inhibitors have been reported. The most potent inhibitor 5h, which possesses the same moiety as the substrate on P1-P4 site, supported the covalent binding. Also, the time-dependent inhibition displayed by inhibitor 5h advanced our understanding of the interactions between the cysteine protease and the electrophilic compound, thereby furthering the discovery of cysteine protease inhibitors.
4. Experimental
4.1. General methods
All reactions with air- and moisture-sensitive materials were performed in oven-dried glassware fitted with rubber septa or three-way T taps under a positive pressure of argon or nitrogen. Air- and moisture-sensitive liquids and solutions were transferred via syringe. Organic solutions were concentrated by rotary evaporation at 23–80 °C (water-bath temperature). Column chromatography was performed employing Merck silica gel (60 Å pore size, 70–230 mesh ASTM). Analytical thin-layer chromatography (TLC) was performed using glass plates pre-coated with Merck silica gel (60 Å pore size) impregnated with a fluorescent indicator (254 nm). TLC plates were visualized by I2 vapors, UV lamp, phosphomolybdic acid solution in ethanol, or 0.5% ninhydrin in ethanol followed by brief heating on a hot plate. Commercial solvents and reagents were used as received without further purification. They were purchased from Aldrich, ACROS, BACHEM, or other commercial sources. Compounds are characterized by nuclear magnetic resonance spectroscopy and high resolution mass spectroscopy. Proton nuclear magnetic resonance (1H NMR) spectra and carbon nuclear magnetic resonance (13C NMR) spectra were recorded with Bruker Avance 600 (600 MHz/ 150 MHz), Bruker DRX 500 (500 MHz/ 125 MHz), and Bruker Avance 400 (400 MHz/100 MHz) NMR spectrometers. Chemical shifts for protons are reported in parts per million (ppm; δ scale) and are referenced to residual protium in the NMR solvents (CHCl3: δ 7.26, D2HCOD: δ 3.31, C2D5HSO: δ 2.50, C2D5HCO: δ 2.05). Chemical shifts for carbon are reported in parts per million (ppm; δ scale) and are referenced to the carbon resonances of the solvent (CDCl3: δ 77.23, CD3OD: δ 49.15, DMSO-d 6: δ 39.50, acetone-d 6: δ 29.84). Data are represented as follows: chemical shift, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad), coupling constant in Hz, integration, and assignment. High resolution mass spectra were obtained using Bruker Daltonics BioTOF III.
4.2. SARS-CoV 3CL protease inhibition assay
As described,21, 22 the inhibitory effects of each compound on the enzymatic activities of 3CL protease were evaluated using purified enzyme and fluorogenic substrate peptide. The kinetic measurements were performed in 20 mM Bis–Tris (pH 7.0) at 25 °C. The initial velocities of the inhibited reactions of 50 nM 3CL protease and 6 μM fluorogenic substrate were plotted against the different inhibitor concentrations to obtain the IC50 by fitting with Eq. 1. K i measurement was performed at two fixed inhibitor concentrations of 1 × IC50 and 2× IC50. Substrate concentrations ranged from 8 to 40 μM in a reaction mixture containing 50 nM 3CL protease. Lineweaver–Burk plots of kinetic data were fitted with the computer program KinetAsyst II (IntelliKinetics, State College, PA) by nonlinear regression to obtain the K i values of competitive inhibitors using Eq. 2
| (1) |
| (2) |
In Eq. 1, A(I) is the enzyme activity with inhibitor concentration [I], A(0) is the enzyme activity without inhibitor, and [I] is the inhibitor concentration. In Eq. 2, K m is the Michaelis constant of the substrate, V m is the maximal velocity, K i is the inhibition constant, and [I] and [S] represent the inhibitor and substrate concentrations in the reaction mixture, respectively.
4.3. Synthesis of compounds 2–5
4.3.1. 1-Nitro-2-phenylethane (2a)
In a 25 mL round-bottom flask fitted with a stirrer were placed sodium nitrite (156 mg, 2.26 mmol) and anhydrous DMF (10 mL). The clear solution was cooled to −78 °C and stirred under N2 (in the absence of light) for 10 min, after which (2-bromoethyl)benzene (238 μL, 1.74 mmol) was added. The mixture was stirred for 15 h, during which the temperature was gradually returned to 23 °C. DMF was removed under reduced pressure, and the residue was extracted with EtOAc. The organic layers were washed with H2O, dried over Na2SO4, filtered, and concentrated under reduced pressure to afford compound 2a as a yellow oil (178.9 mg, 68%). 1H NMR (400 MHz, CDCl3): δ = 7.39–7.23 (m, 5H; ArH), 4.62 (t, J = 7.34 Hz, 2H; O2NCH 2), 3.33 (t, J = 7.35 Hz, 2H; O2NCH2CH 2); 13C NMR (100 MHz, CDCl3): δ = 135.6, 128.8, 128.4, 127.2, 76.1, 33.2.
4.3.2. 3-Nitro-4-phenyl-1,1,1-trifluorobutan-2-ol (3a)
To compound 2a (4161 mg, 27.5 mmol) at 23 °C were added trifluoroacetaldehyde ethyl hemiacetal (90%, 4519 μL, 35 mmol) and K2CO3 (255 mg, 1.84 mmol). The mixture was stirred at 50–60 °C for 3 h, and then at 23 °C for 25.5 h. 1 N HCl (20 mL) and Et2O (20 mL) were added and the water layer was separated. After extraction with Et2O (twice), the combined organic layers were washed with H2O, dried over Na2SO4, filtered, and concentrated. The residue was purified by silica gel chromatography (9% → 20% → 100% EtOAc–hexanes) to give 3a as a yellow oil (3117 mg, 45%). R f = 0.44 (hexanes/EtOAc 2:1); 1H NMR (500 MHz, CDCl3): δ = 7.41–7.20 (m, 5H; ArH), 5.02–4.96 (m, 1H; O2NCH), 4.67–4.27 (m, 1H; CHOHCF3), 3.90–3.65 (br s, 1H; OH), 3.44–3.35 (m, 2H; CH 2Ph); 13C NMR (125 MHz, CDCl3): δ = 134.4, 129.1, 128.8, 128.1, 123.3 (q, J = 281 Hz), 87.2, 70.4 (q, J = 32 Hz), 36.3; HRMS (ESI): calcd for C10H9F3NO3 [M−H]−: 248.0535, found: 248.0555.
4.3.3. 3-Nitro-1,1,1-trifluorobutan-2-ol (3b)
Compound 3b was prepared in a similar way to compound 3a, except nitroethane was used here in place of 2-nitrophenyl ethane (77% yield). R f = 0.47 (hexanes/EtOAc 2:1); 1H NMR (400 MHz, CDCl3): δ = 4.87–4.39 (m, 2H; O2NCH + CHOHCF3), 3.68–3.47 (br s, 1H; OH), 1.69 (d, J = 6.9 Hz, 3H; CH 3); 13C NMR (100 MHz, CDCl3): δ = 123.5 (q, J = 280.5 Hz), 82.1, 71.1, 14.4; HRMS (ESI): calcd for C4H5F3NO3 [M−H]−: 172.0222, found: 172.0237.
4.3.4. 3-Nitro-1,1,1-trifluoropropan-2-ol (3c)
Compound 3c was prepared in a similar way to compound 3a, except nitromethane was used here in place of 2-nitrophenyl ethane (90% yield). R f = 0.72 (hexanes/EtOAc 1:1); 1H NMR (400 MHz, CDCl3): δ = 4.83 (br s, 1H; CHOHCF3), 4.67 (dd, J = 14.2, 2.6 Hz, 1H; O2NCHH′), 4.58 (dd, J = 14.0, 9.5 Hz, 1H; O2NCHH′), 4.13 (br s, 1H; OH); 13C NMR (100 MHz, CDCl3): δ 123.2 (q, J = 280.3 Hz; CF3), 74.3 (CNO2), 67.6 (q, J = 32.8 Hz; CH(OH)CF3).
4.3.5. 3-[N-(N-tert-Butoxycarbonyl-l-Leu)]-1,1,1-trifluorobutan-2-ol (4a)
To a stirred Raney-nickel (aqueous suspension) solution was added compound 3a (472.8 mg, 1.90 mmol) in EtOH (8 mL), and the mixture was hydrogenated under H2 bubbling at 23 °C for 14 h. The catalyst was filtered over Celite, and ethanol and water were evaporated under reduced pressure to afford the amine as a white solid (396.7 mg, 95%). R f = 0.13 (hexanes/EtOAc 2:1); 1H NMR (500 MHz, CD3OD): δ = 7.33–7.21 (m, 5H; ArH), 3.91–3.68 (m, 1H; CHOHCF3), 3.30–3.20 (m, 1H; H2NCH), 3.11–2.56 (dd, J = 13.4, 7.8 Hz, 2H; CH 2Ph); 13C NMR (125 MHz, CD3OD): δ = 139.6, 130.5, 129.8, 127.8, 73.5, 53.5, 40.1; HRMS (ESI): calcd for C10H11F3NO [M−H]−: 218.0793, found: 218.0855.
To a stirred solution of the above amine (385.9 mg, 1.76 mmol) and Cbz–Leu–OH (492 mg, 1.76 mmol) in dry DMF (15 mL) were added HBTU (1720 mg, 4.40 mmol) and Et3N (1227 μL, 8.80 mmol). The reaction mixture was stirred under N2 at 23 °C for 36 h. DMF was evaporated under reduced pressure, and the resulting brown oil was diluted with CH2Cl2 and washed with 1 N HCl. The water layer was separated and extracted with CH2Cl2 for three times. The organic layers were combined and washed with H2O for two times, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was then purified by SiO2 column chromatography (20% → 25% EtOAc–hexanes) to give 4a as a yellow solid (574 mg, 70%). R f = 0.26 (hexanes/EtOAc 2:1); 1H NMR (500 MHz, CD3OD): δ = 7.34–7.18 (m, 10H; ArH), 5.14–5.05 (m, 2H; PhCH 2O), 4.42–4.31 (m, 1H; CH(OH)CF3), 4.14–3.89 (m, 2H; 2× CH α), 3.12–2.77 (m, 2H; CH 2β(Phe)), 1.63–1.00 (m, 3H; CH 2β(Leu)+ CH γ(Leu)), 0.91–0.76 (m, 6H; 2× CH 3δ(Leu)); 13C NMR (125 MHz, CD3OD): δ = 174.8, 158.3, 139.1, 138.5, 130.6, 130.4, 129.6, 129.5, 129.4, 129.3, 129.0, 128.9, 128.8, 128.7, 127.7, 127.5, 127.4, 55.0, 51.3, 42.0, 37.6, 25.7, 23.3, 21.9; 19F NMR (376 MHz, CDCl3): δ = −77.88, −77.90, −77.97, −77.99; HRMS (ESI): calcd for C24H28F3N2O4 [M−H]−: 465.2001, found: 465.2044.
4.3.6. 3-[N-(N-Benzyloxycarbonyl-l-Leu)]-4-phenyl-1,1,1-trifluorobutan-2-ol (4b)
Compound 4b was prepared in a similar way to compound 4a, except Cbz–Phe–OH was used here in place of Cbz–Leu–OH. Compound 4b was isolated as a white solid (1240.7 mg, 66%). R f = 0.21 (hexanes/EtOAc 7:3); 1H NMR (400 MHz, CD3OD): δ = 7.33–7.06 (m, 15H; ArH), 5.05–4.94 (m, 2H; PhCH 2O), 4.48–3.93 (m, 3H; 2× CH α + CHOHCF3), 3.15–2.50 (m, 4H; 2 × CH 2β(Phe)); 13C NMR (100 MHz, CD3OD): δ = 173.7, 158.3, 139.3–138.3 (C(Ar)), 130.7–127.4 (CH(Ar) + CF3), 72.3–69.5 (CHOHCF3), 67.6, 57.8, 51.4, 39.1, 36.1.
4.3.7. 3-[N-(N-Benzyloxycarbonyl-l-Phe)]-4-phenyl-1,1,1-trifluorobutan-2-ol (4c)
Compound 3b was hydrogenated under H2 using PtO2 as catalyst and MeOH/CHCl3 (16:1) as solvent. After the work-up, the corresponding amine hydrochloride salt was obtained in a satisfactory yield (3482.8 mg, 84%). 1H NMR (400 MHz, DMSO-d 6): δ = 8.38 (br s, 3H; NH 3), 4.29 (m, 1H; CHOHCF3), 3.35 (m, 1H; H3NCH), 1.21 (d, J = 6.6 Hz, 3H; CH 3); 13C NMR (100 MHz, DMSO-d 6): δ = 124.9 (q, J = 281.9 Hz), 68.9 (q, J = 29.7 Hz), 46.8, 13.7; HRMS (ESI): calcd for C4H8ClF3NO [M−H]−: 178.0247, found: 178.0268. The subsequent coupling reaction is similar to that for compound 4a, except Boc–Leu–OH was used here in place of Cbz–Leu–OH. Compound 4c was isolated as a white solid (855.7 mg, 78%). R f = 0.53 (hexanes/EtOAc 1:1); 1H NMR (400 MHz, CDCl3): δ = 7.06 (d, J = 6.7 Hz, 1H; NH), 6.06 (br s, 1H; OH), 5.22 (d, J = 7.9 Hz, 1H; NH), 4.25–3.92 (m, 3H; 2× CH α + CHOHCF3), 1.57 (m, 2H; CH 2β(Leu)), 1.41 (s, 10H; OC(CH 3)3 + CH γ(Leu)), 1.31 (d, J = 6.5 Hz, 3H; CH 3β(Ala)), 0.90 (s, 6H; 2 × CH 3δ(Leu)); 13C NMR (100 MHz, CD3OD): δ = 172.8, 156.2, 124.5 (q, J = 281.4 Hz), 80.6, 72.1 (q, J = 29.9 Hz), 53.1, 45.1, 40.7, 28.2, 24.5, 22.5, 22.1, 17.5; HRMS (ESI): calcd for C15H27F3N2NaO4 [M + Na]+: 379.1821, found: 379.1865.
4.3.8. 3-{N-[N-tert-Butoxycarbonyl-l-γGlu(OtBu)-l-Ala]}-1,1,1-trifluoropropan-2-ol (4d)
Compound 3c was hydrogenated under H2 using Ra-Ni as catalyst to give the corresponding amine, which was used directly for coupling to Cbz-Ala-OSu to produce dipeptide adduct. R f = 0.19 (hexanes/EtOAc 1:1); 1H NMR (400 MHz, CDCl3): δ = 7.32 (m, 5H; ArH), 7.05 (br s, 1H; NH), 5.70 (br d, J = 6.3 Hz, 1H; NH), 5.06 (AB, J = 12.1 Hz, ν ab = 25.0 Hz, 2H; PhCH 2O), 4.89 (br s, 1H; OH), 4.23 (t, J = 6.6 Hz, 1H; CHOHCF3), 4.04 (br s, 1H; CH α), 3.68 (br s, 1H; CH α), 3.28 (m, 1H; CH α), 1.34 (d, J = 7.0 Hz, 3H; CH 3β(Ala)); 13C NMR (100 MHz, CDCl3): δ = 174.8, 156.4, 135.7, 128.3, 128.0, 127.9, 124.3 (q, 1 J(C,F) = 280.4 Hz; CF3), 68.7 (q, 2 J(C,F) = 15.1 Hz; CH(OH)CF3), 67.2, 50.8, 39.4, 17.8; HRMS (ESI): calcd for C14H17F3N2NaO4 [M + Na]+: 357.1038, found: 357.1001.
The Cbz group of the above dipeptide adduct was deprotected followed by amino acid coupling using Boc–Glu–OtBu as acid. Compound 4d was isolated as a white solid (1238.7 mg, 54%). R f = 0.48 (EtOAc); 1H NMR (600 MHz, CDCl3): δ = 7.56 (d, J = 22.0 Hz, 1H; NH), 7.20 (br s, 1H; NH), 5.48 (br s, 1H; NH), 4.51 (br s; CHOHCF3), 4.09–3.36 (br, 4H; 4 × CH α), 2.35–1.93 (br, 4H; CH 2β(Glu) + CH 2γ(Glu)), 1.46–1.43 (2s, 21H; 2× OC(CH 3)3 + CH 3β(Ala)); 13C NMR (150 MHz, CDCl3): δ = 173.9, 172.7, 171.5, 155.9, 124.4 (q, J = 280.5 Hz; CF3), 82.4, 80.2, 69.1 (q, J = 30 Hz; CHOHCF3), 53.5, 49.3, 49.1, 39.6, 31.9, 28.4, 27.8, 17.7.
4.3.9. 3-{N-[N-CH3(CH2)8(C O)-l-Leu]}-4-phenyl-1,1,1-trifluorobutan-2-ol (4e)
To a stirred solution of compound 4a (107.4 mg, 0.23 mmol) in MeOH (10 mL) was added Pd(OH)2 (20% Pd, 81 mg), and the whole mixture was stirred under H2. When tlc analysis indicated that the starting material has reacted completely, the catalyst was removed by filtration through Celite. Solvent was evaporated, and the crude amine (78.5 mg) was used without further purification. Subsequent coupling was performed using decanoic acid (40.7 mg, 0.24 mmol), HBTU (98.7 mg, 0.26 mmol), and DIEA (117 μL, 0.71 mmol) by the routine procedure. After SiO2 column chromatography (10% → 15% → 25% EtOAc–hexanes), compound 4e was obtained as a white solid (65.2 mg, 58%). R f = 0.45 (hexanes/EtOAc 2:1); 1H NMR (400 MHz, CDCl3): δ = 7.23–7.09 (m, 5H; ArH), 7.60–7.30, 7.00–5.30 (m, 2H; 2× NH), 4.60–3.80 (m, 3H; 2× CH α + CHOHCF3), 3.00–2.83 (m, 2H; CH 2β(Phe)), 2.13–2.03 (m, 2H; CH 2β(Leu)), 1.54–1.09 (br m, 16H; CH3 (CH 2)8C( O)NH), 0.86–0.73 (m, 7H; CH γ(Leu) + 2× CH 3δ(Leu)), 0.65 (d, J = 5.9 Hz, 3H; CH 3(CH2)8C( O)NH); 13C NMR (100 MHz, CDCl3): δ = 174.3, 172.3, 137.1, 129.2, 128.5, 126.7, 124.6 (q, J = 281.4 Hz), 70.3 (q, J = 30.1 Hz), 51.8, 50.4, 40.8, 36.3, 31.8, 29.4, 29.3, 29.2, 25.7, 24.5, 22.6, 22.3, 22.1, 14.0; HRMS (ESI): calcd for C26H42F3N2O3 [M + H]+: 487.3148, found: 487.3161.
4.3.10. 3-{N-[N-CH3(CH2)7(C O)-l-Leu]}-4-phenyl-1,1,1-trifluorobutan-2-ol (4f)
Compound 4f was prepared in a similar way to compound 4e, except nonanoic anhydride was used here in place of decanoic acid (95% yield). R f = 0.42 (hexanes/EtOAc 2:1); 1H NMR (400 MHz, CDCl3): δ = 7.60–6.10 (m, 7H; ArH+2× NH), 5.10–3.15 (m, 3H; 2× CH α + CHOHCF3), 3.15–2.80 (m, 2H; CH 2β(Phe)), 2.25–2.05 (m, 2H; CH 2β(Leu)), 1.70–0.65 (m, 24H; CH 3(CH 2)7C( O)NH + CH γ(Leu) + 2 × CH 3δ(Leu)); 13C NMR (100 MHz, CDCl3): δ = 178.1, 173.4, 137.0, 129.1, 128.7, 126.9, 71.8, 51.8, 51.5, 40.6, 36.4, 34.0, 31.8, 29.2, 25.6, 24.8, 22.6, 15.0; HRMS (ESI): calcd for C25H39F3N2NaO3 [M+Na]+: 495.2810, found: 495.2797.
4.3.11. 3-[N-(N-Benzyloxycarbonyl-l-Ala-l-Val-l-Leu)]-4-phenyl-1,1,1-trifluorobutan-2-ol (4g)
Compound 4g was prepared in a similar way to compound 4e, except Cbz–Ala–Val–OH was used here in place of decanoic acid (73% yield). R f = 0.41 (hexanes/EtOAc 1:1); 1H NMR (400 MHz, CD3OD): δ = 7.34–7.14 (m, 10H; ArH), 5.13–4.92 (m, 2H; OCH 2Ph), 4.47–3.83 (m, 5H; 4 × CH α + CHOHCF3), 3.15–2.74 (m, 2H; CH 2β(Phe)), 2.06 (m, 1H; CH β(Val)), 1.50–0.75 (m, 18H; 5 × CH 3 + CH 2β(Leu) + CH γ(Leu)); 13C NMR (100 MHz, CD3OD): δ = 175.8, 174.0, 173.2, 158.5, 139.0, 138.1, 130.5, 129.6, 129.5, 129.1, 128.9, 127.9, 67.8, 60.3, 53.1, 51.9, 41.9, 39.0, 36.4, 32.1, 31.5, 25.8, 23.6, 22.0, 20.0, 18.9, 18.2; HRMS (ESI): calcd for C32H43F3N4NaO6 [M + Na]+: 659.3032, found: 659.3000.
4.3.12. 3-[N-(N-Benzyloxycarbonyl-l-Leu)]-4-phenyl-1,1,1-trifluorobutan-2-one (5a)
To a solution of 4a (57.6 mg, 0.12 mmol) in dry CH2Cl2 (5 mL) was added the Dess–Martin reagent (15 wt% soln. in CH2Cl2, 769 μL, 0.37 mmol). TFA (28 μL, 0.37 mmol) was added and then the reaction mixture was stirred at 22 °C for 3 h. The reaction was concentrated under reduced pressure and the remaining residue was treated with a mixture of EtOAc and saturated aqueous solutions of NaHCO3. The water layer was extracted with EtOAc, washed with brine, dried (Na2SO4), filtered, and concentrated under vacuum. Purification (twice) by SiO2 column chromatography (1st: 25% EtOAc–hexanes; 2nd: 10% EtOAc–CH2Cl2) afforded 5a as a white solid (49.6 mg, 86%). R f = 0.17 (CH2Cl2/EtOAc 5:1); 1H NMR (400 MHz, CDCl3): δ = 7.35–7.13 (m, 10H; ArH), 6.68–6.45 (m, 1H; NH), 5.14–4.88 (m, 3H; PhCH 2O + NH), 4.29–3.93 (m, 2H; 2× CH α), 3.31–3.23 (m, 1H; CH βH′β(Phe)), 3.02–2.87 (m, 1H; CHβ H′β(Phe)), 1.60–1.36 (m, 3H; CH 2β(Leu) + CH γ(Leu)), 0.90–0.75 (m, 6H; 2× CH 3δ(Leu)); 13C NMR (100 MHz, CDCl3): δ = 189.7 (m; (C=O)CF3), 171.9, 156.3, 144.0, 135.7, 134.1, 129.2, 129.0, 128.9, 128.8, 128.6, 128.4, 128.1, 127.7, 126.9, 67.4, 54.9, 53.0, 40.4, 36.0, 24.6, 22.7, 21.8; 19F NMR (376 MHz, CDCl3): δ = − 77.0, −77.1 (ketones), −82.8, − 83.1 (hydrates); HRMS (ESI): calcd for C24H28F3N2O4 [M + H]+: 465.2001, found: 465.2001. Notes: In this and many other TFMK related compounds described, the NMR data are rather complex due to the presence of diastereomers and ketone/hydrate mixtures, which are not routinely separated.
4.3.13. 3-[N-(N-Benzyloxycarbonyl-l-Phe)]-4-phenyl-1,1,1-trifluorobutan-2-one (5b)
Compound 5b was prepared in a similar way to compound 5a, except the starting material used was 4b (87% yield). R f = 0.56 (hexanes/EtOAc 1:1; Notes: This compound has very similar mobility to the precursor alcohol 4b, but stains very differently with phosphomolybdic acid on silica TLC plates.); 1H NMR (400 MHz, acetone-d 6): δ = 7.27–7.07 (m, 15H; ArH), 6.61–6.40 (m, 2H; 2× NH), 4.98–4.92 (m, 2H; PhCH 2O), 4.43–4.34 (m, 2H; 2× CH α), 3.29–2.50 (m, 4H; 2× CH 2β(Phe)); 13C NMR (100 MHz, acetone-d 6): δ = 190.9–189.7 (C(=O)CF3), 175.1–171.7, 157.3–155.9, 139.5–137.1 (C(Ar)), 130.5–127.3 (CH(Ar)), 116.8 (q, J = 291.6 Hz; CF3), 98.0–94.8 (q, J = 29.8 Hz; C(OH)2CF3), 67.0, 57.0, 50.9, 38.7, 35.3; 19F NMR (376 MHz, acetone-d 6): δ = − 71.5, −72.3 (ketones), −76.8, −77.1 (hydrates); HRMS (ESI): calcd for C27H26F3N2O4 [M + H]+: 499.1845, found: 499.1890.
4.3.14. 3-[N-(N-tert-Butoxycarbonyl)-l-Leu]-1,1,1-trifluorobutan-2-one (5c)
Compound 5c was prepared in a similar way to compound 5a, except the starting material used was 4c (86% yield). R f = 0.24 (hexanes/EtOAc 2:1); 1H NMR (400 MHz, CDCl3): δ = 7.30–7.07 (m, 1H; NH), 5.23–4.88 (m, 1H; NH), 4.25–4.00 (m, 2H; 2× CH α), 1.66–1.49 (m, 3H; CH 2β(Leu)+ CH γ(Leu)), 1.42 (s, 9H; OC(CH 3)3), 1.34 (d, J = 6.9 Hz, 3H; CH 3β(Ala)), 0.95–0.85 (m, 6H; 2× CH 3δ(Leu)); 13C NMR (100 MHz, CDCl3): δ = 175.4, 156.1, 123.2 (q, J = 287.0 Hz), 94.6 (q, J = 30.5 Hz), 80.9, 53.2, 50.9, 40.6, 28.2, 24.6, 22.8, 21.8, 14.5; HRMS (ESI): calcd for C15H24F3N2O4 [M − H]−: 353.1688, found: 353.1705.
4.3.15. 3-{N-[N-tert-Butoxycarbonyl-l-γGlu(OtBu)-l-Ala]}-1,1,1-trifluoropropan-2-one (5d)
Compound 5d was prepared in a similar way to compound 5a, except the starting material used was 4d (14% yield). R f = 0.55 (EtOAc); 1H NMR (400 MHz, acetone-d 6): δ = 7.86 (m, 1H; NH), 7.60 (d, J = 6.3 Hz, 1H; NH), 6.52 (br d, J = 30.2 Hz, 1H; OH), 6.26 (d, J = 7.9 Hz, 1H; NH), 4.50–4.41 (m, 1H; CH α), 4.04–3.99 (m, 1H; CH α), 3.57 (d, J = 5.8 Hz, 2H; CH 2C(OH)2CF3), 3.10 (br s, 1H; OH), 2.38–1.85 (m, 4H; CH 2β(Glu)+ CH 2γ(Glu)), 1.44 (s, 9H; OC(CH 3)3), 1.41 (s, 9H; OC(CH 3)3), 1.34 (d, J = 7.1 Hz, 3H; CH 3β(Ala)); 13C NMR (100 MHz, acetone-d 6): δ = 177.5, 173.8, 173.1, 157.3, 125.2 (q, J = 285.7 Hz; CF3), 94.7 (q, J = 30.5 Hz; C(OH)2CF3), 82.3, 80.0, 55.6, 50.6, 45.7, 33.2, 31.4, 29.3, 28.9, 18.6; HRMS (ESI): calcd for C20H34F3N3NaO8 [M+H2O+Na]+: 524.2196, found: 524.2222.
4.3.16. 3-(N-l-γGlu-l-Ala)-1,1,1-trifluoropropan-2-one (5e)
Compound 5d (7.2 mg) was dissolved in TFA (5 mL) and stirred for 40.5 h. TFA was evaporated under reduced pressure to give 5e (TFA salt) as a white solid. 1H NMR (400 MHz, CD3OD): δ = 4.30 (m, 1H; CH α), 3.97 (m, 1H; CH α), 3.62 (m, 1H; CH α), 3.44 (m, 1H; CH α), 2.48 (t, J = 7.3 Hz, 2H; CH 2γ(Glu)), 2.20–2.06 (m, 2H; CH 2β(Glu)), 1.28 (d, J = 7.1 Hz, 3H; CH 3β(Ala)); 13C NMR (100 MHz, CD3OD): δ = 176.8, 174.3, 171.6, 95.9, 53.7, 50.7, 42.4, 32.4, 27.2, 18.0; 19F NMR (376 MHz, CD3OD): δ = − 74.7, −75.0, −80.3, −80.6; HRMS (ESI): calcd for C11H19F3N3O6 [M + H2O + H]+: 346.1226, found: 346.1066.
4.3.17. 3-{N-[N-CH3(CH2)8(C O)-l-Leu]}-4-phenyl-1,1,1-trifluorobutan-2-one (5f)
Compound 5f was prepared in a similar way to compound 5a, except the starting material used was 4e (85% yield). R f = 0.41 (hexanes/EtOAc 2:1); 1H NMR (400 MHz, CDCl3): δ = 7.33–7.14 (m, 5H; ArH), 6.38–5.27 (m, 2H; 2× NH), 5.10–4.13 (m, 2H; 2× CH α), 3.51–2.70 (m, 2H; CH 2β(Phe)), 2.18–2.07 (m, 2H; CH 2β(Leu)), 1.97–0.69 (m, 26H; CH 3(CH 2)8C( O)NH + CH γ(Leu) + 2× CH 3δ(Leu)); 13C NMR (100 MHz, CDCl3): δ = 189.4 (q, J = 34.4 Hz), 174.8, 173.7, 136.1, 129.1, 128.5, 126.6, 123.2 (q, J = 287.5 Hz), 94.5 (q, J = 30.9 Hz), 55.3, 51.6, 40.4, 36.4, 34.2, 33.3, 31.8, 29.4, 29.3, 29.2, 29.1, 25.6, 24.6, 22.6, 22.1, 14.1; HRMS (ESI): calcd for C26H41F3N2NaO4 [M + H2O + Na]+: 525.2916, found: 525.2906.
4.3.18. 3-{N-[N-CH3(CH2)7(C O)-l-Leu]}-4-phenyl-1,1,1-trifluorobutan-2-one (5g)
Compound 5g was prepared in a similar way to compound 5a, except the starting material used was 4f (70% yield). R f = 0.26 (hexanes/EtOAc 2:1); 1H NMR (400 MHz, CDCl3): δ = 7.34–7.14 (m, 5H; ArH), 7.10–5.65 (m, 2H; 2× NH), 5.36–4.95 (m, 1H; CH α), 4.48–4.11 (m, 1H; CH α), 3.30–2.79 (m, 2H; CH 2β(Phe)), 2.20–2.07 (m, 2H; CH 2β(Leu)), 1.74–0.71 (m, 24H; CH 3(CH 2)7C( O)NH + CH γ(Leu) + 2× CH 3δ(Leu)); 13C NMR (100 MHz, CDCl3): δ = 189.6, 174.6, 172.1, 136.0, 129.1, 128.5, 126.7, 117.0, 94.6, 55.7, 51.5, 40.4, 36.5, 31.8, 29.7, 29.2, 29.1, 25.6, 24.6, 22.6, 14.0; HRMS (ESI): calcd for C25H39F3N2NaO4 [M + H2O + Na]+: 511.2760, found: 511.2763.
4.3.19. 3-{N-[N-Benzyloxycarbonyl-l-Ala-l-Val-l-Leu]}-4-phenyl-1,1,1-trifluorobutan-2-one (5h)
Compound 5h was prepared in a similar way to compound 5a, except the starting material used was 4g (67% yield). R f = 0.32 (hexanes/EtOAc 1:1); 1H NMR (400 MHz, CD3OD): δ = 7.34–7.12 (m, 10H; ArH), 5.09 (m, 2H; OCH 2Ph), 4.61 (br s, 2H; C(OH)2CF3), 4.57–4.00 (m, 4H; 4 × CH α), 3.27–3.04 (m, 1H; CH βH′β(Phe)), 2.80–2.68 (m, 1H; CHβ H′β(Phe)), 2.02 (m, 1H; CH β(Val)), 1.38–0.70 (m, 18H; 5 × CH 3+ CH 2β(Leu) + CH γ(Leu)); 13C NMR (100 MHz, CD3OD): δ = 175.9, 174.5, 173.9, 158.5, 139.3, 138.1, 130.7, 129.6, 129.5, 129.2, 129.0, 127.5, 97.3 (q, J = 29.0 Hz), 67.9, 60.2, 55.6, 53.0, 52.2, 41.6, 35.4, 31.9, 25.7, 23.6, 21.9, 19.8, 18.7, 18.3; HRMS (ESI): calcd for C32H43F3N4NaO7 [M + H2O+Na]+: 675.2982, found: 675.3005.
4.4. Computer modeling
The crystal structure of SARS-CoV 3CL protease in complex with a substrate-analog inhibitor (coded 1uk4) was obtained from The Protein Data Bank (PDB; http://www.rcsb.org/pdb/). We constructed four stereomeric compound 5h complex as hemithioketal (DISCOVERY STUDIO 1.7) to determine which isomer can form the protein-inhibitor adduct. GOLD 3.223, 24 was used for the flexible docking of compound 5h into the enzyme to explore the wide range of its conformational flexibility. The atoms of the enzyme and compound 5h were assigned with Kollmann all-atom charges25 with SYBYL 7.3 program.26 To distinguish the four possible stereoisomers of enzyme-inhibitor complex, the carbonyl carbon adjacent to the CF3 group of compound 5h was constrained to form a covalent bonding with the sulfur atom of Cys-145. Initial 1000 independent genetic algorithm running cycles were carried out with inhibitor torsion angles varying between −180 and 180 degree. The search efficiency was set up at 200% to ensure the most exhaustive search for docking conformation space. The docking processes were carried out in a 40-CPU (Intel Xeon(TM) CPU 3.00 GHz) Linux cluster. For each stereoisomer conformation, the resultant enzyme-inhibitor complex structures were ranked with the CHEMSCORE scoring27 function to determine the top 10 hits.
Acknowledgments
This work is supported by National Science Council, Taiwan and Genomics Research Center, Academia Sinica. The SYBYL computation was conducted at the National Center for High Performance Computing, Taiwan. The DISCOVERY STUDIO 1.7 computation was conducted at the computational center of Academia Sinica.
References and notes
- 1.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.-E., Humphrey C.D., Shieh W.-J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.-Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J., the SARS working group N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.Peiris J.S.M., Lai S.-T., Poon L.L.-M., Guan Y., Yam L.Y.-C., Lim W., Nicholls J., Yee W.K.-S., Yan W.W., Cheung M.-T., Cheng V.C.-C., Chan K.-H., Tsang D.N.-C., Yung R.W.-H., Ng T.K., Yuen K.-Y., members of the SARS study group Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Peñaranda S., Bankamp B., Maher K., Chen M.-H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C.T., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Günther S., Osterhaus A.D.M.E., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 4.Marra M.A., Jones S.J.M., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S.N., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 5.Drosten C., Günther S., Preiser W., van der Werf S., Brodt H.-R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A.M., Berger A., Burguière A.-M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.-C., Müller S., Rickerts V., Stürmer M., Vieth S., Klenk H.-D., Osterhaus A., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 6.Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.-D., Rappuoli R. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 9.Xiong B., Gui C.-S., Xu X.-Y., Luo C., Chen J., Luo H.-B., Chen L.-L., Li G.-W., Sun T., Yu C.-Y., Yue L.-D., Duan W.-H., Shen J.-K., Qin L., Shi T.-L., Li Y.-X., Chen K.-X., Luo X.-M., Shen X., Shen J.-H., Jiang H.-L. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 10.(a) Wu C.-Y., Jan J.-T., Ma S.-H., Kuo C.-J., Juan H.-F., Cheng E.Y.-S., Hsu H.-H., Huang H.-C., Wu D., Brik A., Liang F.-S., Liu R.-S., Fang J.-M., Chen S.-T., Liang P.-H., Wong C.-H. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shao Y.-M., Yang W.-B., Peng H.-P., Hsu M.-F., Tsai K.-C., Kuo T.-H., Wang A. H.-J., Liang P.-H., Lin C.-H., Yang A.-S., Wong C.-H. ChemBioChem. 2007;8:1654–1657. doi: 10.1002/cbic.200700254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 12.Jain R.P., Pettersson H.I., Zhang J., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James Michael N.G., Wishart D.S., Vederas J.C. J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 13.Chen L.-R., Wang Y.-C., Lin Y.-W., Chou S.-Y., Chen S.-F., Liu L.-T., Wu Y.-T., Kuo C.-J., Chen Tom S.-S., Juang S.-H. Bioorg. Med. Chem. Lett. 2005;15:3058–3062. doi: 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shie J.-J., Fang J.-M., Kuo T.-H., Kuo C.-J., Liang P.-H., Huang H.-J., Wu Y.-T., Jan J.-T., Cheng E.Y.-S., Wong C.-H. Bioorg. Med. Chem. 2005;13:5240–5252. doi: 10.1016/j.bmc.2005.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shie J.-J., Fang J.-M., Kuo C.-J., Kuo T.-H., Liang P.-H., Huang H.-J., Yang W.-B., Lin C.-H., Chen J.-L., Wu Y.-T., Wong C.-H. J. Med. Chem. 2005;48:4469–4473. doi: 10.1021/jm050184y. [DOI] [PubMed] [Google Scholar]
- 16.Wu C.-Y., King K.-Y., Kuo C.-J., Fang J.-M., Wu Y.-T., Ho M.-Y., Liao C.-L., Shie J.-J., Liang P.-H., Wong C.-H. Chem. Biol. 2006;13:261–268. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Imperiali B., Abeles R.H. Biochemistry. 1986;25:3760–3767. doi: 10.1021/bi00361a005. [DOI] [PubMed] [Google Scholar]; (b) LaPlante S.R., Bonneau P.R., Aubry N., Cameron D.R., Déziel R., Grand-Maître C., Plouffe C., Tong L., Kawai S.H. J. Am. Chem. Soc. 1999;121:2974–2986. [Google Scholar]; (c) Ogilvie W., Bailey M., Poupart M.-A., Abraham A., Bhavsar A., Bonneau P., Bordeleau J., Bousquet Y., Chabot C., Duceppe J.-S., Fazal G., Goulet S., Grand-Maître C., Guse I., Halmos T., Lavallée P., Leach M., Malenfant E., O’Meara J., Plante R., Plouffe C., Poirier M., Soucy F., Yoakim C., Déziel R. J. Med. Chem. 1997;40:4113–4135. doi: 10.1021/jm970104t. [DOI] [PubMed] [Google Scholar]
- 18.(a) Peet N.P., Burkhart J.P., Angelastro M.R., Giroux E.L., Mehdi S., Bey P., Kolb M., Neises B., Schirlin D. J. Med. Chem. 1990;33:394–407. doi: 10.1021/jm00163a063. [DOI] [PubMed] [Google Scholar]; (b) Smith R.A., Copp L.J., Donnelly S.L., Spencer R.W., Krantz A. Biochemistry. 1988;27:6568–6573. doi: 10.1021/bi00417a056. [DOI] [PubMed] [Google Scholar]
- 19.Stein R.L., Strimpler A.M., Edwards P.D., Lewis J.J., Mauger R.C., Schwartz J.A., Stein M.M., Trainor D.A., Wildonger R.A., Zottola M.A. Biochemistry. 1987;26:2682–2689. doi: 10.1021/bi00384a005. [DOI] [PubMed] [Google Scholar]
- 20.(a) Zhang H.-Z., Zhang H., Kemnitzer W., Tseng B., Cinatl J., Jr., Michaelis M., Doerr H.W., Cai S.X. J. Med. Chem. 2006;49:1198–1201. doi: 10.1021/jm0507678. [DOI] [PubMed] [Google Scholar]; (b) Sydnes M.O., Hayashi Y., Sharma V.K., Hamada T., Bacha U., Barrila J., Freire E., Kiso Y. Tetrahedron. 2006;62:8601–8609. doi: 10.1016/j.tet.2006.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuo C.-J., Chi Y.-H., Hsu John T.-A., Liang P.-H. Biochem. Biophys. Res. Commun. 2004;318:862–867. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu John T.-A., Kuo C.-J., Hsieh H.-P., Wang Y.-C., Huang K.-K., Lin Coney P.-C., Huang P.-F., Chen X., Liang P.-H. FEBS Lett. 2004;574:116–120. doi: 10.1016/j.febslet.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones G., Willett P., Glen R.C. J. Mol. Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 24.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 25.Cornell W.D.C.P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 26.SYBYL 7.3; The Tripos Associates: St. Louis, MO.
- 27.Eldridge M.D., Murray C.W., Auton T.R., Paolini G.V., Mee R.P. J. Comput.-Aided Mol. Design. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]