

Abstract
Prohibitin 2 (PHB2) is an evolutionarily conserved and ubiquitously expressed multifunctional protein which is present in various cellular compartments including the nucleus. However, mechanisms underlying various functions of PHB2 are not fully explored yet. Previously we showed that PHB2 interacts with Akt and inhibits muscle differentiation by repressing the transcriptional activity of both MyoD and MEF2. Here we show that Calcium/Calmodulin-dependent kinase IV (CaMK IV) specifically binds to the C terminus of PHB2 and phosphorylates PHB2 at serine 91. Ectopic expression of CaMK IV and PHB2 in C2C12 cells results effectively in decreased PHB2-mediated repression of MEF2-dependent gene expression. Conversely, PHB2 mutant (S91A) resistant to CaMK IV phosphorylation has less effective in relieving the inhibition of MEF2 transcription by PHB2. Our findings suggest that CaMK IV interacts with and regulates PHB2 through phosphorylation, which could be one of the mechanisms underlying the CaMK-mediated activation of MEF2.
Abbreviations: BSA, bovine serum albumin; FBS, fetal bovine serum; PHB1, Prohibitin 1; PHB2, Prohibitin 2;CaMKs, calcium/calmodulin-dependent kinases; CaMKK, Calcium/Calmodulin-dependent kinase kinase; CaMK IV, Calcium/Calmodulin-dependent kinase IV; MEF2, myocyte enhancer factor2; bHLH, basic helix-loop-helix; HDAC, histone deacetylases; CREB, cyclic-AMP response element binding protein; AP-1, activator protein 1; ROR, retinoid orphan receptor; GM, growth medium; DM, differentiation medium; WCEs, Whole cell extracts; Co-Ip, Co–immunoprecipitation; ER, estrogen receptor; RNF2, ring finger 2; S1P, spningosine − 1- phosphate; Big 3, brefeldin A-inhibited guanine nucleotide-exchange protein 3; SARS-CoV, severe acute respiratory syndrome coronavirus; wt, wild type; mut, mutant; ca, constitutively active
Keywords: CaMKIV, PHB2, MEF2, Interaction, Phosphorylation
Graphical abstract

Research highlights
► CaMK IV specifically binds the C terminus of PHB2. ► PHB2 serine 91 serves as the major phosphorylation site for CaMK IV. ► CaMK IV effectively decreased PHB2-mediated repression of MEF2 activity through phosphorylation. ► Serine phosphorylation on PHB2 by CaMK IV relieves its inhibition on MEF2. ► Regulation of PHB2 by CaMK IV may contribute a mechanism underlying the CaMK-mediated activation of MEF2.
1. Introduction
Prohibitin 2 (PHB2), also designated as B-cell receptor-associated protein 37 (BAP 37) and repressor of estrogen receptor activity (REA), is an ubiquitous, abundant and evolutionarily highly conserved protein with homologues in bacteria, yeast, Drosophila and mammals [1]. In yeasts, it has been well defined that PHB2, in a complex with highly homologous prohibitin 1 (PHB1), is localized in mitochondria and plays an important role in maintaining mitochondrial inheritance and morphology [2]. PHB2 is involved in mitochondria-related cell processes like life span, cell growth, and apoptosis [3], [4]. In mammals, in addition to its mitochondrial functions, PHB2 has been found in multiple cellular compartments and possess diverse functions ranging from acting as scaffolding proteins at the plasma membrane to transcriptional regulators in the nucleus. PHB2 has been found at the plasma membrane of B lymphocytes in association with the IgM receptor [5]. PHB2 was shown to repress the transcriptional activity of various transcription factors, including steroid hormone receptors, either directly or through interactions with chromatin remodeling proteins [6], [7], [8], [9]. Interestingly, nuclear PHB2 was shown to protect sister-chromatid cohesion during mitosis [10]. In our previous work, we showed that PHB2, as a transcriptional repressor, interacts with both MyoD and myocyte enhancer factor2 (MEF2) and represses both MyoD- and MEF2-dependent gene transcription [6]. MyoD and MEF2 transcription factors are well-defined, critical factors in myogenesis [11]. Myoblast differentiation is regulated by cooperative interactions of myogeneic basic helix-loop-helix (bHLH) transcription factors (e.g. MyoD) with MEF2 and by signaling pathways that regulate MyoD and MEF2 activity [11], [12]. When stably expressed in C2C12 myogenic cells, PHB2 inhibits myogenic induction and phenotypic muscle differentiation [6]. Furthermore, PHB2 was found to mediate transcriptional repression by recruiting histone deacetylases HDAC1 and HDAC5 [6], [13]. However, the molecular mechanisms of regulating PHB2 function remained largely elusive.
Phosphorylation is a primary protein regulatory mechanism for controlling activity, stability, localization, and cofactor interaction. Evidence has emerged that suggests PHBs can be regulated by phosphorylation. Indeed, several studies have noted the presence of multiple potential serine/threonine and tyrosine phosphorylation sites in PHB2. Ross et al. have identified PHB2 as a serine and tyrosine phosphorylated protein using orthophosphate labeling coupled with phosphoamino acid analysis [14]. They mapped the tyrosine phosphorylation site of PHB2 to Tyr248 through mass spectrometry. However, the serine/threonine phosphorylation of these sites of PHB2 in vitro and in vivo and their upstream kinases has not been validated. Akt, as a serine/threonine kinase, was shown to bind to PHB2 directly in our previous study. Although PHB2 contains a consensus Akt phosphorylation site (86-RPRKIS-91), we failed to prove that Akt could phosphorylate PHB2 [6]. The consensus Akt phosphorylation site in PHB2 also fits the expected consensus calcium/calmodulin-dependent kinases (CaMKs) phosphorylation sites (RXXS/T). Therefore, it is possible that CaMKIV could phosphorylate PHB2 at this consensus site.
CaMKIV is a serine-threonine protein kinase and a member of CaMKs that functions as potent mediator of calcium-induced gene expression, primarily through its ability to phosphorylate and activate a variety of transcription factors, such as cyclic-AMP response element binding protein (CREB), activator protein 1 (AP-1), MEF2 and retinoid orphan receptor (ROR) [15]. CaMKIV is a potent activator of MEF2 activity [16]. Ectopic expression of activated CaMKIV both in skeletal muscle cells and cardiomyocytes led to dramatic increase in MEF2 transcriptional activity [17], [18].
In this work, we demonstrate that CaMK IV specifically interacts with and phosphorylates PHB2 and the C-terminus of PHB2 is responsible for CaMK IV binding. The CaMK IV phosphorylation site is mapped to Ser91 in PHB2. Co-expression of CaMK IV and PHB2 effectively relieves the PHB2-mediated repression of MEF2-dependent gene transcription, but it was less effective for the phosphorylation-defective PHB2. From these results, we concluded that PHB2 is a novel substrate of CaMK IV, and that CaMK IV phosphorylation of PHB2 could be one of the mechanisms underlying the CaMK IV-mediated activation of MEF2.
2. Materials and methods
2.1. Cell lines, antibodies and other reagents
C2C12 cells were grown in DMEM supplemented with 20% fetal bovine serum(FBS) and antibiotics (growth medium, or GM) and were induced to differentiate in DMEM containing 2% horse serum and antibiotics (differentiation medium or DM) when cells were near confluent. Anti-Flag was from Sigma; anti-Xpress (Omni-probe, M21) and anti-HA was from Santa Cruz Biotechnology. D(−)-Luciferin was purchased from Roche Applied Science. ATP was purchased from Sigma.
2.2. Plasmids
3 × MEF2-luc, CaMKKca, His-CaMKIVwt, CaMKIVca, Flag-CaMKIV, Xp-PHB2 and Flag-Six1 have been described previously [6], [17], [19]. Full-length mutant PHB2 in which serine 91 was substituted with alanine (S91A) was generated by PCR-mutagenesis using Xp-PHB2 as the template and then subcloned into pcDNA 3.1 C, termed as Xp-PHB2-mut. Mutant PHB2 cDNA was verified by DNA sequencing.
2.3. Yeast two-hybrid analysis
MATCHMAKER GAL4 Two-Hybrid System 3(BD Bioscience) was used to examine the interaction between CaMK IV and PHB2 and to identify the minimal region of PHB2 required for CaMK IV binding as previously described [5]. Briefly, CaMK IV cDNA was inserted into pGBKT7 and expressed as a fusion protein to the GAL4 -DNA binding domain. The cDNAs of PHB2(57-299) which was found in the yeast-two-hybrid screen with Akt as a bait and its various truncated forms were cloned into pGADT7 vector and expressed as fusion proteins to the GAL4 activation domain. CaMKIV-pGBKT7 and individual PHB2-pGADT7 were sequentially transformed into the host strain AH109. Positive clones were selected on synthetic dropout plates lacking leucine, tryptophan, histidine and adenine (SD/-his/-leu/-trp/-ade). Both pGBKT7 and pGADT7 without inserts were used as negative controls.
2.4. Transfection and reporter assays
Cells were first transfected with various plasmids using LipofectAMINE Plus reagents (Invitrogen) following the manufacturer's instructions and cultured in GM for 24–36 h with or without being followed by culturing in DM for another 24 h before lysis. Whole cell extracts (WCEs) were prepared by lysing cells in lysis buffers (50 mM HEPES, pH 7.6, 1% Triton X-100, 150 mM NaCl, 1 mM EGTA, 1.5 mM MgCl2, 100 mM NaF, 20 mM p-nitrophenylphosphate, 20 mM β-glycerol phosphate, 50 μM sodium vanadate, 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, 0.5 μg/ml leupeptin, and 0.7 μg/ml pepstatin), followed by removal of insoluble debris with centrifugation at 16,000 g for 2 min. Protein concentration in WCEs was determined by protein assay reagent from Bio-Rad (Hercules, CA). Reporter assay was performed as previously described: 20 μl of WCE were added to 150 μl of freshly made luciferase reaction buffer and the luciferase activity was determined with a LB9507 luminometer (EG&G Berthold). Luciferase units were normalized against total protein amount present in each sample.
2.5. Co-immunoprecipitation assay
Co-immunoprecipitation assay was performed as described in previous study [6]. Briefly, C2C12 cells were co-transfected with Xp-PHB2 together with either Flag-CaMKIV or Flag-Six1 (negative control). 24 h later, the cells were cross-linked with 200 μg ml− 1 DSP (Pierce) for 5 min followed by lysis in RIPA buffer. Protein-A/Sepharose beads were incubated with 200 μg of extracts and 2 μg of anti-Flag antibodies for 2 h at 4 °C. After extensive washing with the RIPA buffer, bound proteins were eluted by boiling and subjected to SDS-PAGE and western blot with anti-Xpress antibody.
2.6. Recombinant protein expression
Recombinant GST-PHB2wt and GST-PHB2mut vectors were generated by inserting truncated wild type and S91A mutant PHB2 (residues 57 to 120), which were obtained by PCR using Xp-PHB2 and Xp-PHB2mut as template respectively, into pGEX6p-1 plasmid. The corresponding recombinant protein GST-PHB2wt and GST-PHB2mut were expressed in BL21 following IPTG-induction overnight at 30 °C. Cells were lysed by French press and proteins were purified by incubation with glutathione-sepharose beads followed by extensive wash and then eluted with elution buffer containing glutathione. Eluted proteins were dialyzed and analyzed with SDS-PAGE and coommassie blue staining.
2.7. Kinase Assay
His-CaMK IV co-transfected with or without CaMKKca from 50 μg of WCE were pulled down with Talon beads (CLONTECH, palo Alto, CA) and washed. The pulled down His-CaMK IV was used as the kinase and incubated with 10 μl of recombinant GST-PHB2wt or GST-PHB2mut in 25 μl of kinase buffer (20 mM HEPES, pH 7.6, 10 mM MgCl2, 20 mM β-glycerol phosphate, 10 μM ATP, 1 mM DTT, 10 mM p–nitrophenylphosphate, 50 μM sodium vanadate, and 10 μCi of [γ-32P]-ATP at 30 °C for 30 min. The kinase mixtures were resolved by SDS-PAGE and labeled protein bands were visualized by autoradiography.
2.8. Western blot
20–30 μg of WCEs or eluted proteins was resolved by SDS-PAGE, transferred to a polyvinylidene fluoride membrane (Immobilon-P; Millipore), and probed with various antibodies. Protein bands were visualized using ECL kit (Amersham Biosciences).
2.9. Statistics
Data are expressed as mean ± SD from at least three experiments. Student's ttest was used to evaluate statistical significance of differences between two groups. p values of < 0.05 were considered statistically significant.
3. Results
3.1. Identification of PHB2 as a CaMK IV-interacting Protein
Our previous study showed that PHB2 interacts with Akt [6]. To gain more insight into the role of PHB2 in signaling transmission, we first examined the interaction between CaMK IV and PHB2. A positive clone encoding truncated PHB2 (amino acids 57–299) was identified from the yeast-two-hybrid screen of the cDNAs of PHB2 (57–299) using CaMK IV as a bait. This truncated PHB2 which has similar domain region found to interact with Akt in a yeast two-hybrid screening with the full-length Akt as bait [6], interacts specifically with CaMK IV but does not with the empty vector pGBKT7 (Fig. 1A left). As expected, the Akt, as positive control was also found to directly interact with PHB2 in yeast. To map the minimal region of PHB2 that interacts with CaMK IV, GAL4-AD constructs containing various regions of truncated PHB2 were generated and tested for their ability to interact with CaMK IV in yeast. As shown in Fig. 1B, the C-terminus of region of PHB2 (amino acid 120 to 299), but not the N-terminal region of PHB2 (amino acid 57–120), interacted with CaMK IV. As a control, we showed that all truncated PHB2 constructs were expressed in yeast (Fig. 1C).
Fig. 1.

PHB2 interacts with CaMKIV through C-terminus in yeast cells. (A) CaMKIVca cDNA was cloned into pGBKT7 and different deletion fragments of PHB2 were separately cloned into pGADT7. The mutual interaction between CaMKIV and individual PHB2 variants were tested by yeast two-hybrid assays. Positive clones were checked by being streaked on synthetic dropout plates lacking leucine, tryptophan, histidine and adenine (SD/-his/-leu/-trp/-ade). Akt-pGBKT7 and pGBKT7 were used as PHB2-binding positive and negative control respectively. The numbers marked on the plate represent the position of amino acids in various truncated PHB2. (B) Schematic representation of yeast two-hybrid assays of (A). The black bar represents the CaMKIV-interacting region on PHB2. The plus and minus signs denote growth and no growth on SD/-his/-leu/-trp/-ade plates. (C). The expression levels of different PHB2 fragments in AH109 yeast cells were detected by immunoblotting.
3.2. PHB2 interacts with CaMK IV in mammalian cells
To further confirm the PHB2-CaMK IV interaction in mammalian cells, C2C12 cells were transiently transfected with xp-PHB2 together with either Flag-CaMK IV or Flag-Six1 (a negative control). CaMK IV was immunoprecipitated from the cell lysate using anti-Flag antibody and the presence of PHB2 in the complex was detected with Xp-antibody. We found that PHB2 was able to interact with CaMK IV but not Six1 when co-transfected in C2C12 cells (Fig. 2 ), suggesting that PHB2 specifically interacts with CaMK IV in vivo.
Fig. 2.

PHB2 specifically interacts with CaMKIV in mammalian cells. C2C12 cells were co-transfected with Xp-PHB2 together with either Flag-CaMKIV or Flag-Six1 and WCE were harvested 24 h later. Immunoprecipitation and immunoblot were carried out as indicated. Abbreviations: CoIP, co-immunoprecipitation; IP, immunoprecipitation; IB, immunoblot; Xp, Xpress tag. Input represents 10% of total lysates used for immunoprecipitation. IgH, immunoglobin heavy chain; IgL, immunoglobin light chain.
3.3. PHB2 is phosphorylated by active CaMK IV
Examining the protein sequence of PHB2 revealed the presence of a putative CaMK IV phosphorylation consensus motif, suggesting that PHB2 may be phosphorylated directly by CaMK IV. We sought to determine whether PHB2 is a substrate for active CaMK IV using in vitro kinase assay. The recombinant PHB2 proteins encompassing the consensus phosphorylation site were expressed and purified from E. coli (Fig. 3A). 293T cells were co-transfected with wild type His-CaMK IV along with either CaMKKca or an empty vector (a negative control) and then His-CaMK IV was pulled down with talon beads for the kinase assay. As shown in Fig. 3B, recombinant PHB2 was efficiently phosphorylated by constantutively active Calcium/Calmodulin-dependent kinase kinase (CaMKKca) activated CaMK IV but not by non-activated CaMK IV, an observation in line with the presence of serine 91 in the context of preferential phosphorylation consensus sequence RXXS/T (88-RKIS-91). Consistent with the capacity of CaMK IV to phosphorylate PHB2 at this site, we demonstrated that a PHB2 S91A mutant is refractory to CaMK IV-directed phosphorylation (Fig. 3B, lane 3), suggesting that PHB2 serine 91 serves as the major phosphorylation site for CaMK IV.
Fig. 3.

CaMKIV could phosphorylate PHB2 on consensus phosphorylation site. (A) Wild type or CaMKIV consensus phosphorylation site mutant PHB2 were expressed as GST fusion protein in BL21 under the induction of IPTG. GST fusion proteins were purified on glutathione-sepharose beads. 5 μl and 10 μl of purified recombinant proteins were separated on SDS-PAGE and stained by coommassie blue. (B) 293T cells were cotransfected with His-CaMKIV along with either CaMKKca or an empty vectors and then His-CaMKIV was pulled down with talon beads and incubated with 10 μl of recombinant wild type or mutant PHB2 in kinase buffer plus [γ-32P]ATP. Abbreviations: wt, wild type; mut, mutant; ca, constitutively active.
3.4. Inhibition of MEF2 transactivation activity by CaMK IV-mediated PHB2 phosphorylation
Since PHB2 has been shown to interact with transcription factors including MyoD and MEF2, and repress MyoD and MEF2 dependent gene transcription [6], we determined whether CaMK IV-mediated phosphorylation of PHB2 has effect on MEF2 activity. To this end, we employed a MEF2 activity-dependent reporter assay in which the expression of the reporter gene luciferase is controlled by three MEF2 DNA binding elements (3 × MEF2-luc). C2C12 cells were transfected with 3 × MEF2-luc along with constructs for wild type PHB2 (Xp-PHB2) or phosphorylation-defective PHB2 (Xp-PHB2mut) in combination with or without constitutively active CaMK IV (CaMKIVca). As shown in Fig. 4A, both Xp-PHB2 and Xp-PHB2-mut significantly reduced MEF2-dependent luciferase activity. Since active CaMK IV strongly activates the MEF2 transactivation activity as previously reported [17], we found that the reduction of MEF2 activity by PHB2 was reversed by active CaMK IV, but Xp-PHB2-mut-mediated repression could not be effectively relieved by active CaMK IV (Fig. 4B). This finding suggests that the phosphorylation of PHB2 by active CaMK IV de-represses the transcriptional inhibitory effect of PHB2 on MEF2 activity, which could be one of the mechanisms underlying CaMK-mediated activation of MEF2.
Fig. 4.
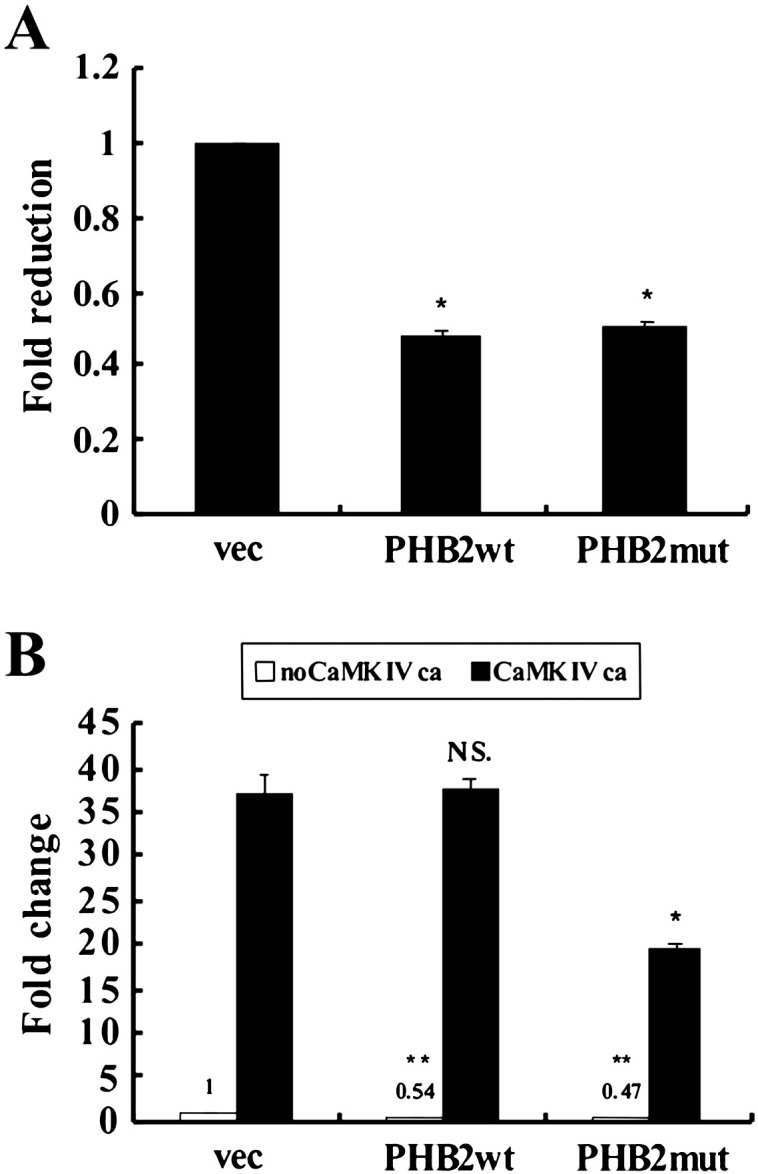
CaMKIV derepresses the inhibitory effect of PHB2 on MEF2 through phosphorylation. C2C12 cells were co-transfected with 3xMEF2-luc together with an empty vector, wild type or mutant PHB2 (A) and with CaMKIVca in various combinations as indicated (B). After 36 h of growth in GM and 24 h in DM, cells were harvested and the luciferase activity determined. The data denote fold changes in the luciferase activity of samples transfected with PHB2 or CaMKIVca alone or combination versus that transfected with an empty vector. The experiments were performed in triplicate and independently three times with similar results. The results from a representative experiment are presented. *p < 0.001, compared to the sample transfected with an empty vector; N.S, not significant compared to the sample transfected with CaMKIVca alone; ** p < 0.001, compared to the sample transfected with CaMKIVca alone or PHB-wt and CaMKIVca combination. Abbreviations: wt, wild type; mut, mutant; ca, constitutively active; vec, an empty vector.
4. Discussion
This study reports the physical interaction of PHB2 and CaMK IV as evidenced by yeast two-hybrid assay and cellular co-immunoprecipitations. The data are consistent with a model of which the CaMK IV phosphorylates serine 91, a residue positioned in the PHB domain of PHB2, thereby modulating PHB2 interaction with MEF2, possibly via interaction of HDAC co-repressor complex. Our findings provide a new avenue in exploring the molecular mechanism by which PHB2 regulates gene repression.
PHB2 is 299 amino acids in length. Its amino terminus containing hydrophobic stretches anchors PHB2 to the membrane [20], whereas the C-terminal part of PHB2 seems always responsible for binding other partners. Our data show that the long C-terminus region of PHB2 (amino acid 120 to 299) interacts with CaMK IV. This region of PHB2 has been also demonstrated to contain several domains which bind to several other proteins including PHB1 [21], HDAC1 [13], Ring finger 2 (RNF2) [7], estrogen receptor (ER) [8] and Akt [6]. As compared with Akt binding ability to PHB2, a C-terminal domain of PHB2 (amino acids 232–299), which is not necessary for Akt binding, is indispensable but not sufficient for specific interaction with CaMK IV. PHB2 has been shown to have multiple binding partners in multiple cellular compartments. For example, mitochondrial PHB1 [21] and spningosine-1-phosphate (S1P) [22] interact and cooperate with PHB2 to maintain normal mitochondrial functions; PHB2 also bond to several transcription factors, such as ER [8], MyoD and MEF2 [6], and inhibited their transcriptional activities. It has been shown that cytoplasmic brefeldin A-inhibited guanine nucleotide-exchange protein 3 (Big 3) interacts with PHB2 and traps PHB2 in the cytoplasm to inhibit its nuclear translocation, which rescues ER alpha transcriptional activity from PHB2 inhibition [23]. Our present and previous data showed PHB2 interacts with CaMK IV and Akt, suggesting its potential function in regulating cellular signal transduction events.
It is interesting to note that, in addition to the cellular proteins, PHB2 also interacts with exogenous proteins, especially viral proteins. The C-terminal cytoplasmic domain of the HIV-1 glycoprotein binds PHB1/PHB2 complex for its replicative spread in non-permissive cells [24]. The interaction between severe acute respiratory syndrome coronavirus (SARS-CoV) nonstructural protein 2 and PHB1/PHB2 complex is involved in the disruption of intracellular host signaling during SARS-CoV infections [25]. These findings suggest that PHB2 could interact with versatile proteins, through which the biologic effects of PHB2 are mediated. Moreover, the various cellular localization of PHB2 and its potential to interact with versatile partners, together with its ubiquitous expression and high evolutionary conservation, imply the versatile and important biological functions of PHB2, but of which have yet to be completely elucidated.
Several studies have indicated that the serine and tyrosine residues in PHB2 are phosphorylated [14], but phosphorylation of these sites in vitro and in vivo and upstream kinases has not been validated. We for first time mapped the exact serine phosphorylation site of PHB2 to serine91 and demonstrated that CaMK IV is one of its upstream serine/threonine kinases. Serine91 is present in the conserved PHB domain (amino acids 39–201) and is highly evolutionarily conserved in human, mouse, rat, xenopus, zebrafish, drosophila and S. Pombe. However S. cerevisiae contains an alanine at this position and plants lost this phosphorylation motif completely, suggesting a gain of function at this point of divergence. It is interesting to note that this CaMKIV consensus motif is not conserved in PHB1, while threonine258 in PHB1, which is phosphorylated by Akt, is not conserved in PHB2 [26]. So far, there is no evidence to show that Akt could phosphorylate PHB2. Our previous data showed that Akt could interact with PHB2 but did not phosphorylate it [6]. These findings are striking, given the high degree of identity between the relevant sites in PHB1 and PHB2, and suggest that PHBs are differentially regulated by distinct mechanisms.
MEF2 transcription factors play central roles in the activation of the genetic programs that control cell differentiation, proliferation, morphogenesis, survival and apoptosis of a wide range of cell types [27]. CaMK IV is a potent activator of MEF2 activity. One of the mechanisms by which CaMK IV activates MEF2 is the CaMK IV phosphorylation of the transcriptional repressor HDAC5, resulting in the de-repression of the MEF2-dependent gene transcription [28], [29]. HDAC5 represses MEF2-dependent gene transcription by direct binding to MEF2 [30]. Phosphorylation of HDAC5 by CaMK IV leads to disruption of the interaction between MEF2 and HDAC5, which results in the translocation of HDA5 from nucleus to cytoplasm and releases the repression [28], [29], [31]. Although strong evidence suggest a role for CaMK in the transmission of signals that converge on HDACs, the existence of distinct signaling mechanisms that control HDACs remains possible. Indeed, our results provide another potential mechanism underlying the activation of MEF2 by CaMKIV: CaMKIV phosphorylates another MEF2 repressor PHB2 to relieve its inhibition of MEF2. Our previous study showed that PHB2 interacts with MEF2 and represses MEF2 activity by further recruiting HDAC1. To explore whether similar mechanism is subjected to HDAC5 after PHB2 is phosphorylated by CaMK IV, we performed the subcellular localization and co-immunoprecipitation assays. However, we neither could detect obvious translocation of PHB2 induced by CaMK IVca using GFP fluorescence assay (unpublished data) nor demonstrate CaMK IVca decreases the interaction between PHB2 and MEF2 or PHB2 and HDAC1 using co-immunoprecipitation assay (unpublished data). Thus, how CaMK IV signaling rescues MEF2 from repression by PHB2 is currently under further investigation.
Despite the role in skeletal muscle and myocardial cells, the regulatory effects of CaMKIV on MEF2 play important roles in endothelial cells [31] and T cells [32] as well. It is conceivable that PHB2 may be also involved in the regulation of signal transduction events in these cells, since PHB2 is widely expressed in the endothelial and T cells. As such, further elucidation of the signal pathways that regulate this transcription factor may lead to specific pharmacological agents that target MEF2-resposive genes by means of PHB2.
In summary, we have identified that CaMK IV, as a binding partner of PHB2, phosphorylates PHB2 at Ser 91, and leads to release PHB2-mediated inhibition of MEF2-dependent gene transcription. Our studies reveal a nuclear pathway by which CaMK IV modulates the functions of PHB2 as a transcriptional co-repressor and provides new insight into the understanding how CaMK IV contributes to the regulation of gene transcription.
Acknowledgments
We thank Dr. Zhenguo Wu (HKUST) for reagents, protocols and comments. We thank Xiaoming Gong for helpful suggestions and critical reading of the manuscript. This work was supported by two grants from National Nature Science Foundation of China (No. 30600098 and No. 30671091) and two foundations from Science and Technology Department of Jilin Province (No. 200705134 and No.20070729-4).
References
- 1.Nijtmans L.G., Artal S.M., Grivell L.A., Coates P.J. Cell. Mol. Life Sci. 2002;59:143–155. doi: 10.1007/s00018-002-8411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nijtmans L.G., de Jong L., Artal Sanz M., Coates P.J., Berden J.A., Back J.W., Muijsers A.O., van der Spek H., Grivell L.A. EMBO J. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osman C., Merkwirth C., Langer T. J. Cell Sci. 2009;122:3823–3830. doi: 10.1242/jcs.037655. [DOI] [PubMed] [Google Scholar]
- 4.Artal-Sanz M., Tavernarakis N. Trends Endocrinol. Metab. 2009;20:394–401. doi: 10.1016/j.tem.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Terashima M., Kim K.M., Adachi T., Nielsen P.J., Reth M., Kohler G., Lamers M.C. EMBO J. 1994;13:3782–3792. doi: 10.1002/j.1460-2075.1994.tb06689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun L., Liu L., Yang X.J., Wu Z. J. Cell Sci. 2004;117:3021–3029. doi: 10.1242/jcs.01142. [DOI] [PubMed] [Google Scholar]
- 7.Lee S.J., Choi D., Rhim H., Choo H.J., Ko Y.G., Kim C.G., Kang S. Mol. Cell. Biochem. 2008;319:69–77. doi: 10.1007/s11010-008-9878-2. [DOI] [PubMed] [Google Scholar]
- 8.Delage-Mourroux R., Martini P.G., Choi I., Kraichely D.M., Hoeksema J., Katzenellenbogen B.S. J. Biol. Chem. 2000;275:35848–35856. doi: 10.1074/jbc.M001327200. [DOI] [PubMed] [Google Scholar]
- 9.Wang S., Zhang B., Faller D.V. EMBO J. 2004;23:2293–2303. doi: 10.1038/sj.emboj.7600231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takata H., Matsunaga S., Morimoto A., Ma N., Kurihara D., Ono-Maniwa R., Nakagawa M., Azuma T., Uchiyama S., Fukui K. Curr. Biol. 2007;17:1356–1361. doi: 10.1016/j.cub.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 11.Molkentin J.D., Olson E.N. Curr. Opin. Genet. Dev. 1996;6:445–453. doi: 10.1016/s0959-437x(96)80066-9. [DOI] [PubMed] [Google Scholar]
- 12.Lu J., McKinsey T.A., Zhang C.L., Olson E.N. Mol. Cell. 2000;6:233–244. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 13.Kurtev V., Margueron R., Kroboth K., Ogris E., Cavailles V., Seiser C. J. Biol. Chem. 2004;279:24834–24843. doi: 10.1074/jbc.M312300200. [DOI] [PubMed] [Google Scholar]
- 14.Ross J.A., Nagy Z.S., Kirken R.A. J. Biol. Chem. 2008;283:4699–4713. doi: 10.1074/jbc.M708232200. [DOI] [PubMed] [Google Scholar]
- 15.Racioppi L., Means A.R. Trends Immunol. 2008;29:600–607. doi: 10.1016/j.it.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 16.McKinsey T.A., Zhang C.L., Olson E.N. Trends Biochem. Sci. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 17.Xu Q., Yu L., Liu L., Cheung C.F., Li X., Yee S.P., Yang X.J., Wu Z. Mol. Biol. Cell. 2002;13:1940–1952. doi: 10.1091/mbc.02-02-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Passier R., Zeng H., Frey N., Naya F.J., Nicol R.L., McKinsey T.A., Overbeek P., Richardson J.A., Grant S.R., Olson E.N. J. Clin. Invest. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enslen H., Tokumitsu H., Stork P.J., Davis R.J., Soderling T.R. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10803–10808. doi: 10.1073/pnas.93.20.10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merkwirth C., Langer T. Biochim. Biophys. Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 21.Tatsuta T., Model K., Langer T. Mol. Biol. Cell. 2005;16:248–259. doi: 10.1091/mbc.E04-09-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strub G.M., Paillard M., Liang J., Gomez L., Allegood J.C., Hait N.C., Maceyka M., Price M.M., Chen Q., Simpson D.C., Kordula T., Milstien S., Lesnefsky E.J., Spiegel S. FASEB J. 2011;25:600–612. doi: 10.1096/fj.10-167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J.W., Akiyama M., Park J.H., Lin M.L., Shimo A., Ueki T., Daigo Y., Tsunoda T., Nishidate T., Nakamura Y., Katagiri T. Cancer Sci. 2009;100:1468–1478. doi: 10.1111/j.1349-7006.2009.01209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emerson V., Holtkotte D., Pfeiffer T., Wang I.H., Schnolzer M., Kempf T., Bosch V. J. Virol. 2010;84:1355–1365. doi: 10.1128/JVI.01641-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornillez-Ty C.T., Liao L., Yates J.R., 111, Kuhn P., Buchmeier M.J. J. Virol. 2009;83:10314–10318. doi: 10.1128/JVI.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han E.K., McGonigal T., Butler C., Giranda V.L., Luo Y. Anticancer Res. 2008;28:957–963. [PubMed] [Google Scholar]
- 27.Potthoff M.J., Olson E.N. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 28.McKinsey T.A., Zhang C.L., Lu J., Olson E.N. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKinsey T.A., Zhang C.L., Olson E.N. Proc. Natl. Acad. Sci. U.S.A. 2000;97:14400–14405. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemercier C., Verdel A., Galloo B., Curtet S., Brocard M.P., Khochbin S. J. Biol. Chem. 2000;275:15594–15599. doi: 10.1074/jbc.M908437199. [DOI] [PubMed] [Google Scholar]
- 31.Liu G., Han J., Profirovic J., Strekalova E., Voyno-Yasenetskaya T.A. Angiogenesis. 2009;12:1–15. doi: 10.1007/s10456-008-9123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blaeser F., Ho N., Prywes R., Chatila T.A. J. Biol. Chem. 2000;275:197–209. doi: 10.1074/jbc.275.1.197. [DOI] [PubMed] [Google Scholar]