

Abstract
Recombination in Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) is a well-documented phenomenon. A high recombination frequency has been reported in experimental conditions both in vitro and in vivo, and its role in driving viral evolution has been postulated by several authors. However field evidences are rare, mainly obtained from large-scale sampling and typically represented by single sequences rather than by groups of circulating “recombinant progenies”. The present work was aimed to investigate the gray area between experimental studies and large-scale epidemiological investigations. The study was performed on ORF5, ORF7 and concatenated sequences obtained in our laboratory or available in GenBank collected between 2009 and 2012 in northern Italy. Six independent recombinant strains out of 66 concatenated sequences (∼9%) were found, demonstrating a high recombination frequency respect to previous field studies but comparable to in vitro experiments. In silico analysis let speculate that this new strain displayed physicochemical features diverse enough to potentially alter its immunological properties. Taken altogether, the results of our study support previous experimental evidences that depict PRRSV to be extremely prone to recombination. The limited temporal and geographical spread of recombinant strains however states in favor of a limited fitness of the recombinant progeny compared to parental strains and the marginal role of this phenomenon in PRRSV evolution.
Keywords: PRRSV, Italy, Recombination, ORF5, ORF7
1. Introduction
Porcine Reproductive and Respiratory Syndrome (PRRS) was first recognized quite contemporaneously in the U.S. and in Europe between the end of 1980s and the early 1990s. Since then PRRS has emerged as the most prevalent disease of swine in the world, causing remarkable economic losses (Neumann et al., 2005, Nieuwenhuis et al., 2012). The agent of the disease, Porcine Reproductive and Respiratory Syndrome Virus (PRRSV), classified in the order Nidovirales, family Arteriviridae, genus Arterivirus, is an enveloped, single-stranded positive-sense RNA virus. The viral genome is approximately 15 kb in length and contains nine open reading frames (ORFs) (Firth et al., 2011, Meulenberg, 2000). Two main genotypes, Type I (European-like) and Type II (North American-like) have been identified sharing 50–70% nucleotides and 50–80% amino acids (Shi et al., 2010a). A great and progressively increasing (Mateu et al., 2006, Pesch et al., 2005) genetic variability has been observed: mean nucleotide diversity within European and American genotypes has been estimated to be about 15% and 12.5%, respectively (Cho and Dee, 2006, Shi et al., 2010a, Shi et al., 2010b). Genetic distance, calculated on ORF5, has reached a maximum of about 30% in Type I and 21% in Type II (Murtaugh et al., 2010). RNA virus evolution is assumed to result primarily from RNA polymerase infidelity. Indeed the PRRSV nucleotide substitution rate has been estimated to vary between 4.7 × 10−2 and 1.55 × 10−3 (Murtaugh et al., 2010, Yoon et al., 2012). Although the role of recombination in evolution of RNA viruses is still debated (Simon-Loriere and Holmes, 2011), several authors assert that recombination is an important mechanism of genetic diversity generation in PRRSV (Liu et al., 2011, Mengeling, 2002, Murtaugh et al., 2010), playing a potential role in conditioning virulence, antigenic escape and diagnostic failure. Several studies have demonstrated recombination in both in vitro (van Vugt et al., 2001, Yuan et al., 1999) and in vivo, in experimental (Liu et al., 2011) and field conditions (Fang et al., 2007, Forsberg et al., 2002, Li et al., 2009, Shi et al., 2010a, Stadejek et al., 2008). In the latter case, results where typically obtained comparing sequences obtained from large-scale (i.e. country level) sampling. The aim of this study was to investigate recombination on a smaller scale in terms of geographic distance and time window (Forsberg et al., 2002).
2. Materials and methods
2.1. Samples
The samples used in this study were drawn from the Istituto Zooprofilattico Sperimentale delle Venezie's historical archive, a regional public veterinary laboratory collecting passive field samples brought by practitioners for diagnostic purposes. All of the 163 samples (serum and lung), coming from 52 pig farms among 12 provinces in northeastern Italy (enclosing a geographic area of about 28,000 km2), found positive at routine RT-PCR to PRRSV between 2010 and 2012 and stored at −80 °C, were analyzed. RNA had been extracted from 200 μl of serum or 200 μl of lung homogenate using the High Pure viral RNA kit and High Pure RNA tissue kit, respectively (Roche Diagnostics, Monza, Italy). Each sample had been routinely tested using a classical two step RT-PCR targeting a genomic fragment within the ORF7 region and allowing the differentiation between the Type I and Type II strains through electrophoresis on acrylamide gels (Persia et al., 2001).
2.2. Sequencing
ORF5 and ORF7 of each sample were amplified using a one-step RT-PCR as described by Oleksiewicz et al. (1998). Briefly, ORF5 sequence was amplified using the primer ORF5F (5′ CAA TGA GGT GGG CIA CAA CC 3′) and ORF5R (5′ TAT GTI ATG CTA AAG GCT AGC AC 3′) while ORF7 was amplified using the primer pair ORF7F (5′ GCC CCT GCC CAI CAC G 3′) and ORF7R (5′ TCG CCC TAA TTG AAT AGG TGA 3′), obtaining an amplicon of 719 bp and 637 bp respectively.
Amplification and band specificity were visualized using a SYBR safe stained 2% agarose gel, after electrophoresis. Amplicons were sequenced with the same primers, in both senses, using the BygDye terminator v.3.1 Cycle Sequencing Kit (Applied Biosystem®, Monza, Italy). Sequences were obtained using ABI PRISM® 3100 Genetic Analyzer (Applied Biosystem®, Monza, Italy). Chromatograms were evaluated by FinchTV (http://www.geospiza.com) and consensus sequences were reconstructed using CromasPro (CromasPro Version 1.5). When both ORFs were available, concatenated sequences were constructed using Mesquite (Maddison and Maddison, 2011).
2.3. Sequence analysis
Sequences obtained plus those (i.e. 11 ORF7 and 64 ORF5) derived from Pesente et al. (2006) were aligned by Guidance (using PRANK as alignment method) (Penn et al., 2010) and score evaluated. For clarification purposes, all ORF7 sequences published by Pesente et al. (2006) were renamed with the accession number assigned to ORF5. JModelTest 2.1.2 (Darriba et al., 2012) was used to select the model of evolution according to Akaike Information Criterion (AIC). Phylogenetic trees based on ORF5 and ORF7 were reconstructed applying the Maximum Likelihood method implemented in PhyML 3.0 (Guindon et al., 2010) assuming the GTR + Γ4 + I nucleotide substitution model. Phylogenetic tree reliability was evaluated using a fast nonparametric version of the aLRT (Shimodaira–Hasegawa [SH]-aLRT), which was developed and implemented in the PhyML 3.0 (Anisimova et al., 2011).
ORF5, ORF7 and concatenated sequence alignments were tested for evidence of recombination using RDP3 (Martin et al., 2010). In order to obtain a conservative estimate, a recombination event was accepted only when detected by two or more methods implemented in the program with a p-value lower than 5 × 10−5. A collection of partitions without recombination was obtained dividing the original alignment at the recombination breakpoint. Phylogenetic trees were reconstructed for each partition using RAXML (Silvestro and Michalak, 2012) and used to calculate per site log likelihoods for each alignment partition. Statistical significance of topological incongruence between segments separated by recombination breakpoints were assessed through SH, KH, ELW and AU tests implemented in CONSEL (Shimodaira and Hasegawa, 2002). A p-value < 0.05 was assumed to indicate statistical significance.
A discrete states phylogeographic reconstruction of PRRSV strains migration pattern was performed using BEAST 1.7.5 (Drummond et al., 2012) as described by Lemey et al. (2009). The 12 provinces where the samples had been collected were considered to be discrete states. An asymmetric substitution model, coupled with the Bayesian Stochastic Search Variable Selection (BSSVS), was implemented. Non-recombinant, concatenated ORF5–ORF7 sequences, for which sampling data was known, were analyzed for this purpose. Bayesian Factor (BF) was calculated in order to define well supported diffusion rates using SPREAD (Bielejec et al., 2011). Rates yielding a BF > 10 were considered adequately supported (Kass and Raftery, 1995). The same software was used to generate the KML file compatible with Google Earth displaying migration history.
2.4. In silico structural analysis
Structural consequences of recombination on GP5 were considered for a recombinant cluster that demonstrated circulation over time in a farm. Nucleotide and amino acid p-distance of recombinant strains from their parents were calculated using MEGA5 (Tamura et al., 2011). Hydrophobicity profile was calculated using ProtScale (Wilkins et al., 1999) assuming the Kyte & Doolittle scale. Secondary structure and transmembrane topology of GP5 were predicted using Psipred (http://bioinf.cs.ucl.ac.uk/psipred/). N-linked glycosylation sites were estimated using NetNGlyc 1.0 Server (Gupta et al., 2004). The possible role of recombination in generating strains with different immunological properties was evaluated through in silico prediction of T- and B-epitopes. Linear B-cell epitopes were predicted using the BepiPred 1.0 Server (Larsen et al., 2006).
For cytotoxic T lymphocytes epitopes, NetCTLpan 1.1 Server (Stranzl et al., 2010) a pan-specific major histocompatibility complex class I epitope predictor, integrating prediction of proteasomal cleavage, antigen transport efficiency and MHC-I binding affinity, was used. All swine MHC-I alleles deposited in the program database were selected to predict 8-,9-,10-,11-mer peptides. MHC-II binders were predicted using NetMHCII 2.2 server (Nielsen and Lund, 2009), searching 15-mer peptides that bound the collection of human MHC-II (loci DR and DQ). Considering that the stronger the binding, the more likely the peptide to become T-cell epitopes (Gustiananda, 2011), highly stringent cut-offs were applied. Peptides were accepted as possible epitopes when their rank score was <1% and IC50nM < 500 (including high and intermediate affinity binder of SLA-I). To limit the presence of false positive results using HLA-II based software, only strong binder (IC50nM < 50) predicted by NetMHCII were accepted, according to Díaz et al. (2009) and Gustiananda (2011).
3. Results
3.1. Recombination analysis
A total of 131 ORF5 and 111 ORF7 were obtained, including those achieved from Pesente et al. (2006). The accession numbers of sequences obtained in our study are provided in Supplementary Data 1. All of the strains were collected from a restricted geographic area of <30,000 km2, with a maximum distance between farms of 270 km. For 66 samples both ORF5 and ORF7 were available allowing the construction of a third database, based on the concatenation (i.e. the joining of two character strings end-to-end) of the respective ORF5 and ORF7 sequences of each strain. All the strains belonged to the Type I subtype I, according to Stadejek's classification (Stadejek et al., 2008, Stadejek et al., 2013). Phylogenetic analysis revealed some incongruences between trees obtained from ORF5 and ORF7 (Fig. 1 ).
Fig. 1.
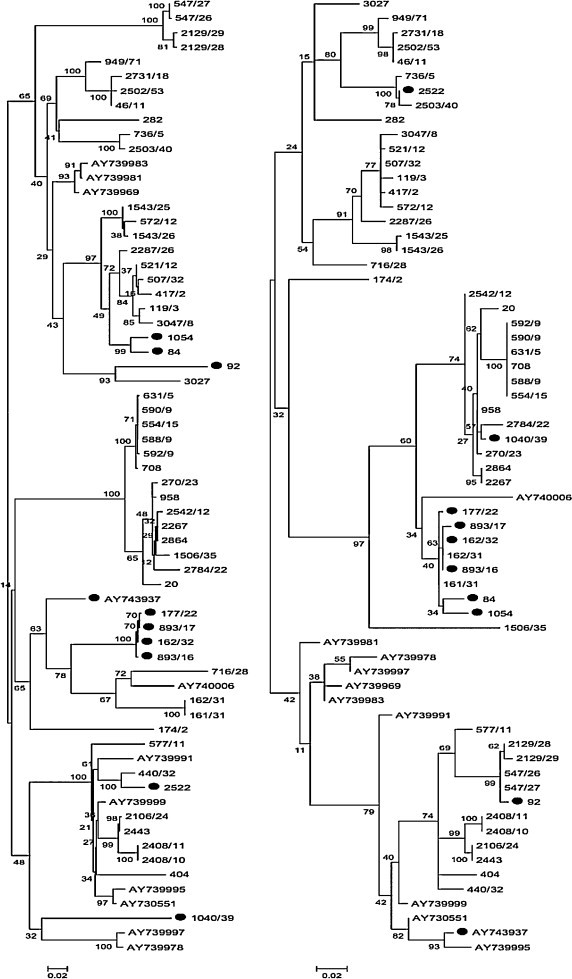
Phylogenetic trees of ORF5 (on the left) and ORF7 (on the right) reconstructed using RAxML. Only strains for which both ORF5 and ORF7 were available were included in this picture. Recombinants are marked with black circles to emphasize the different topology between the two ORFs.
The recombination scan on concatenated sequences coupled with topology comparison revealed 6 statistically significant recombination events (Table 1 and Supplementary Data 2). Four recombinants (i.e. strains 2522, 1040/39, 84 and 92) displayed a single breakpoint between the end of ORF5 and the beginning of ORF7. Unfortunately, a more precise localization could not be performed because the segment spanning these ORFs was not sequenced. Besides, only for strain 2522 both parental viruses were clearly identifiable. Only donors of the ORF7 segment were clearly detected for both 1040/39 and 92 while no closely related sequences were identified in ORF5. However, the analysis of the wider ORF5 database displays a certain relatedness of 1040/39 with strains 390/45. Another recombination event between ORF5 and ORF7 was detected (strain AY743937), although only one parent could be evidently identified (AY739981). Analysis refinement using the ORF5 dataset confirmed the presence of recombination breakpoints in position 210 and 606 and identifies AY739981 and AY74007 as major and minor parents. The ORF7 tree reconstruction revealed a close relationship between AY743937 and AY739995. The last recombinant strain was identified to be the result of two recombination events. Strains 1054 and 84 were classified as recombinants between 2287/26 (ORF5) and 162/31(ORF7). At the same time strain 84 and the closely related 1054 were predicted as donors in a recombination event within ORF5 (segment 123–474): strains 893/17, 177/22, 162/32 and 893/16 were detected as recombinants between 84 (minor parent) and 162/31 (major parent). Remarkably, both parental and recombinants strains were collected from the same farm at different time periods (see Supplementary Data 1). Particularly, recombinant strains were found twice about 3 months apart while their co-circulation with 162/31. On the contrary, the minor parent (strain 84), was recorded only two years later. Predicted recombination events involved both relatively neighbor (<10 km) and distant (more than 100 km) farms. Remarkably, the same strain (i.e. 162/31) was implied in two different recombination events (i.e. events 2 and 4) (Table 1) that took place in the same geographic area. The parental strains involved in these events were sampled from the same farm (i.e. 162/31 and 84) or in nearby farms (i.e. origin farms of 162/31 and 2287/26 were about 10 km apart), supporting the short-range spread of PRRSV strains. At the same time, discrete state phylogeographic analysis evidenced the presence of 12 well-supported migration rates among provinces of northeastern Italy and an extensive PRRSV circulation over time (Supplementary Data 3).
Table 1.
Strains detected as recombinants with RDP3. When identified parental viruses are also reported. Recombination breakpoints are defined by nucleotide position assuming the beginning of ORF5 as position 1.
| Event | Major parent | Minor parent | Recombinant | Recombination breakpoint |
|---|---|---|---|---|
| 1 | 440/32 | 736/5 | 2522 | 606 |
| 2 | 2287/26 | 162/31 | 1054, 84 | 606 |
| 3 | Unknown | 958 | 1040/39 | 606 |
| 4 | AY39981 | AY74007 | AY743937 | 210–606 |
| 5 | 162/31 | 84 | 893/16, 162/32, 893/17, 177/22 | 123–474 |
| 6 | Unknown | 547/27 | 92 | 606 |
3.2. Structural analysis
GP5 of recombinant strains 893/17, 177/22, 162/32 and 893/16, demonstrating a prolonged circulation, was further analyzed to explore the presence of structural differences from parental strains which may affect viral fitness. Strains 84 and 162/31, assumed as representative of minor and major parents, respectively, were also included in the analysis. The calculation of the p-distance demonstrated a relevant amino acidic difference between the two parents: 18.4% in the internal region to recombination breakpoints (AA 41–158) and about 25.3% in the external. Comparable results were obtained considering nucleotides (Table 2 ). Hydrophobicity profile and secondary structure were also affected and recombinants displayed some different secondary structures from both major and minor parents (Supplementary Data 4). However, the transmembrane regions’ prediction revealed no differences among strains even though recombination spanned part of the first ectodomain, the three transmembrane segments and the first part of the main endodomain. Some differences were observed in glycosylation pattern: the strain 162/31, 893/17, 893/16 and 162/32 were predicted to be glycosylated in position 36, 46 and 53. A similar pattern was displayed by strain 84 with the only exception that N-linked glycosylation was present in position 37 instead of 36. Certain dissimilarity was displayed by strain 177/22 which lost the glycosylation in position 46. Consequences of recombination on GP5 antigenicity were also estimated in silico. For computational easiness and clarification, considering the high percentage of identity purposes (p-distance = 0.005 and 0.004 at amino acid and nucleotide level, respectively), 893/17 was assumed as representative of all recombinant strains. Linear B-cell epitopes, although slightly different in extension, were substantially unchanged in position: a region around the first glycosylation site and two sequences within the C-terminal endodomain were identified as possible epitopes in all strains (Fig. 2 ). The first one, which claimed to be a neutralizing epitope (AA 37–44) (Mateu and Diaz, 2008, Plagemann, 2004), spanned the region where the initial recombination breakpoint was estimated. One amino acidic difference between parental viruses was identifiable at amino acid 37 (G-N), with the recombinants sharing the major parent sequence. AA 41 was different in both parental and recombinant strains with the latter sharing the same residue. The analysis of T-cell epitope evidenced that, comparing major, minor and recombinant strains, approximately the same SLA-I alleles recognized the common region of GP5. Two main differences were estimated between major and minor parents in predicted ligands of SLA-I (region 7–14 and 84–93) (Fig. 2). Accordingly, the recombinant epitopic profile can be described as a combination of parental patterns. A similar picture was obtained with regard to HLA-II. Parents differed in region 61–69, 113–121 and 153–162. Recombinants resulted to be approximately a combination of parental strains. However, epitopes in position AA 41–49 and AA 61–69 were lost while one peculiar epitope (AA 103–111) was predicted (Fig. 3 ).
Table 2.
Nucleotide and amino acid p-distance in the segment internal and external to recombination breakpoints. Distance is calculated as mean distance between recombinants’ group and parental viruses.
| Region | Strain | 84 | 162/31 | Recombinants |
|---|---|---|---|---|
| (a) Nucleotide p-distance | ||||
| AA 41–158 | 84 | 0 | 0.191 | 0.031 |
| 162/31 | 0.191 | 0 | 0.18 | |
| Recombinants | 0.031 | 0.18 | 0 | |
| AA 159–40 | 84 | 0 | 0.194 | 0.194 |
| 162/31 | 0.194 | 0 | 0.004 | |
| Recombinants | 0.194 | 0.004 | 0 | |
| (b) Amino acid p-distance | ||||
| AA 41–158 | 84 | 0 | 0.184 | 0.056 |
| 162/31 | 0.184 | 0 | 0.153 | |
| Recombinants | 0.056 | 0.153 | 0 | |
| AA 159–40 | 84 | 0 | 0.253 | 0.253 |
| 162/31 | 0.253 | 0 | 0 | |
| Recombinants | 0.253 | 0 | 0 | |
Fig. 2.
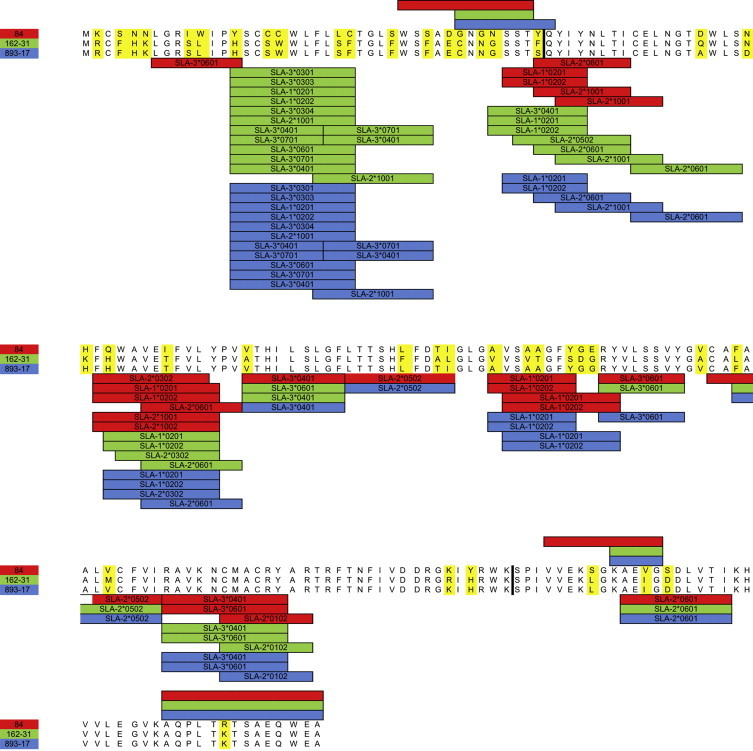
SLA-I alleles are displayed under the respective peptides of GP5 predicted as ligands while linear B-cell epitopes are represented above the corresponding sequence. In both cases a color code is used to associate major (green), minor (red) parents and recombinant strain (blue) to the respective estimated epitopes. Recombination breakpoints are represented as solid black line. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.
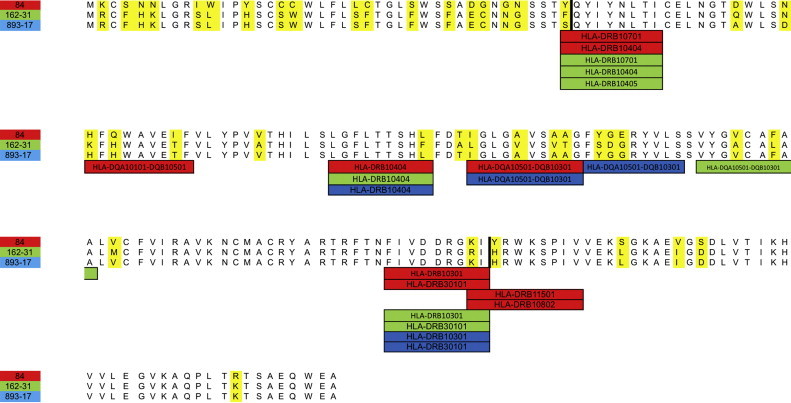
HLA-II alleles are displayed under the respective peptides of GP5 predicted as ligands. A color code is used to associate major (green), minor (red) parents and recombinant strain (blue) to the respective estimated epitopes. Recombination breakpoints are represented as solid black line. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
RNA viruses represent both a fascinating opportunity and a challenge in the study of evolutionary processes (Holmes, 2009). Their populations usually harbor abundant genetic variability, due in large part to the combination of high mutation rate and large population size. Although recombination had been thought to be rare in these viruses, recent studies disavowed this theory, demonstrating not only that it is a frequent phenomenon in some virus families, especially retroviruses and positive-sense single stranded RNA viruses, but also that it can sometimes have a major impact on their emergence, evolution, and epidemiology (Simon-Loriere and Holmes, 2011). A similar picture can be drawn for PRRSV, a highly variable RNA virus, recently emerged as agent of devastating impact on pig production. Although the remarkable genetic distance is mainly attributable to the high substitution rate (Yoon et al., 2012) and to the abrupt increase in population size, consequent to the advent of high density confinement management practice in Europe and North America coupled with increasing pig transportation (Murtaugh et al., 2010), the role of recombination is nowadays increasingly studied. A high recombination frequency was demonstrated both in vitro (van Vugt et al., 2001, Yuan et al., 1999) and in experimentally infected pigs (Liu et al., 2011). Nevertheless, the frequency of naturally occurring recombination was believed to be much lower. In fact, most of the recombination events identified so far in the field were only represented by single sequences rather than by circulating “recombinant progenies”(Fang et al., 2007, Forsberg et al., 2002, Li et al., 2009, Murtaugh et al., 2001, Shi et al., 2010a, Stadejek et al., 2008, Yoshii et al., 2008, Yuan et al., 1999). However the vast majority of field evidences were based on sequences obtained from strains collected at different time points and from distant locations. An underestimate of the recombination rate in the field due to sampling limitations (Shi et al., 2010a) is plausible. Our study attempted to deal with this inadequacy, performing an intensive sampling based on a narrow spatial and temporal scale. Six recombination events were detected in concatenated ORF5–ORF7, affecting ORF5 and the region between ORF5 and ORF7. According to previous studies (Shi et al., 2010a), no recombination events were displayed within ORF7. Six independent recombinant strains out of 66 were found. This proportion of recombinant strains was comparable with those reported in vitro by Yuan and van Vugt for PRRSV (van Vugt et al., 2001, Yuan et al., 1999) and approximate the range reported for some Coronavirus (Lai, 1996). Several hypotheses can be proposed to explain the high frequency of recombination events observed. Firstly, it should be accounted that sequences considered, originating from a small area, are relatively similar (mean p-distance: ORF5 = 0.135; ORF7 = 0.098; concatenated = 0.122). van Vugt et al. (2001) demonstrated that recombination occurs preferentially between genome regions with a high percentage of similarity, which is compatible with the transcription strategy of Arterivirus (Pasternak et al., 2006). Liu et al. (2011) demonstrated that PRRSV is really prone to recombine in experimentally co-infected pigs. The occurrence of transmission of different strains between farms therefore plays a key role in the likelihood of recombination (Forsberg et al., 2002). Analysis of geographic relation between parental strains revealed that viruses originating from both distant and neighboring farms participate to this phenomenon. Accordingly, phylogeographic analysis revealed the presence of 12 well-supported migration rates among provinces of northeastern Italy. These data support the field evidence that multiple strains co-infection is relatively frequent in Italian pig farms. High farm density, intense animal movements and ineffective biosecurity measures are the most probable cause of this situation. A further hypothesis is that, collecting numerous samples on a narrow spatial and temporal scale makes it more likely to find recombinants with low fitness, whose survival and spreading among farms is unlikely, and which otherwise would not have been collected performing a less intensive sampling. The evidence that quite all recombinants were sampled only once and did not demonstrate any persistence or geographical spread supports this hypothesis. A partial exception was represented by strains 893/17, 177/22, 162/32 and 893/16. These recombinants were collected from the same farm approximately 3 months apart demonstrating a certain fitness. In silico analysis revealed a noteworthy amino acid difference from parental strains resulting in different hydrophobicity and secondary structure. Recombinants secondary structure was not a simple combination of the parental ones but presented some peculiar characters, probably as a consequence of interaction between distant amino acidic regions or minor AA substitutions occurred after recombination took place. Both B-cell and T-cell epitopes are constrained by sequence specificity, and mutations within epitopes can result in immune escape. Obviously, mutations within an epitope can directly affect antibody–antigen interactions or epitope-MHC and T-cell receptor interactions. Additionally, mutations outside of the epitope can inhibit antibody binding through conformational changes, or inhibit proper cleavage and processing of T-cell epitopes (Korber et al., 2006). For these reasons, the role of recombination in generating strains with potentially different immunological features was investigated. Two linear B-cell epitopes were predicted within major endodomain and one within the major ectodomain, in substantial agreement with antigenic region identified in vitro by other authors (Vanhee et al., 2011). The first two are unlikely involved in viral neutralization due to their localization (Vanhee et al., 2011). On the contrary, the epitope identified within the GP5 main ectodomain is usually regarded as one of the major neutralization epitopes (Mateu and Diaz, 2008). Interestingly the beginning recombination breakpoint was predicted within this region. All the predicted antigenic regions displayed some amino acidic differences between parental viruses affecting also the sequences of recombinants strains (Fig. 2). It has been demonstrated that different PRRSV isolates differ in susceptibility to neutralization (Martínez-Lobo et al., 2011). Even if a clear correlation with protein sequence was not identified for genotype I, Kim et al. demonstrated that amino acid substitution in specific positions of ectodomain can affect neutralization in genotype II strains (Kim et al., 2013). As a consequence, it is possible to suppose that the recombination events discovered could have influenced viral susceptibility to humoral immunity through changes in epitope sequence, possibly in association with minor conformational changes that might have modified epitope accessibility or immunogenicity (Martínez-Lobo et al., 2011).
Glycosylation pattern showed some differences among parental and recombinant strains. It is well established that glycosylation plays a major role in protecting against humoral response. It has been reported that removal of N-glycosylation site renders the virus more susceptible to neutralization and elicits a significantly greater neutralizing antibodies response (Ansari et al., 2006, Darwich et al., 2010, Vu et al., 2011). However an actual correlation between glycosylation number and neutralization phenotype has not been found yet (Martínez-Lobo et al., 2011). Loss of glycosylation in position 46, predicted in strain 177/22, is usually regarded as deleterious, being it strongly required for both assembly and infectivity in LV (Dokland, 2010). However, although not frequently, field strains without this glycosylation site have been reported (Mateu et al., 2006, Stadejek et al., 2006). Besides, Balka reported a case of reversion to virulence of a PRRSV vaccine strain in which the new virulent strain was characterized by the loss of glycosylation in position 46 associated with glycosylation in position 37 (while vaccine has N-35) (Balka et al., 2008). It is also possible that apparently minor changes in glycosylation profile could significantly affect strain virulence. Also, considering potential T-cell epitopes, as expected, recombinant strains presented a combination of different parental epitopes, besides the one additional MHC-II ligand that was predicted. Despite our conservative settings, in silico prediction of epitopes cannot be considered an accurate tool, even if used with some success also in veterinary medicine (Díaz et al., 2009). However it should be stressed that the main purpose of this analysis was to verify if recombination between parental strains may generate an amino acidic difference relevant enough to affect their original physicochemical and antigenic properties. The results provide strong evidence that through recombination a new strain was generated which displayed characters different enough to potentially alter the development of acquired immunity or decrease the effectiveness of recall response against parental strains. Nevertheless the definition of actual B and T-cell epitopes of these strains, their effect on cross-protection or the effects of recombination on virulence are beyond the scope of this study.
5. Conclusion
This study, conducted in a restricted geographical area (28,000 km2) in a limited time frame, evidenced a frequent occurrence of PRRSV recombination in pig farms, compatible with that reported in vitro and in vivo in experimental conditions. These results provide further confirmation that PRRSV, as other Nidovirales family members, is really prone to undergo recombination events, probably due to their peculiar replicative strategy (Pasternak et al., 2006, Simon-Loriere and Holmes, 2011). On the other hand, the lower frequency of recombinant strains detected on a broader scale in other studies, suggests that recombinants usually have low fitness and rarely gain an evolutionary advantage over their parents. In the present study the prolonged circulation of recombinant viruses, displaying a combination of parental physicochemical and antigenic profiles, was demonstrated in a farm. Their temporal survival and spatial spread remained limited, suggesting a marginal role of recombination in driving PRRSV evolution. Anyhow, recent evidences demonstrated the emergence of virulent Type II strains through recombination (Chen et al., 2013, Shi et al., 2013). Considering the frequency of recombination and its ability to generate strains of unexpected behavior (Li et al., 2009, Liu et al., 2011), further efforts should be deserved to study the consequences of this phenomenon on various aspects as infectivity, virulence, immunogenicity and diagnosability providing more extensive knowledge on the evolutionary driving forces of PRRSV.
Acknowledgements
The work here described was supported by the University of Padua grant, year 2013 (60A08-2170/13).
Footnotes
Supplementary material related to this article can be found, in the online version, at http://dx.doi.org/10.1016/j.virusres.2014.08.005.
Contributor Information
Giovanni Franzo, Email: giovanni.franzo@studenti.unipd.it.
Mattia Cecchinato, Email: mattia.cecchinato@unipd.it.
Marco Martini, Email: marco.martini@unipd.it.
Letizia Ceglie, Email: lceglie@izsvenezie.it.
Alessandra Gigli, Email: agigli@izsvenezie.it.
Michele Drigo, Email: michele.drigo@unipd.it.
Appendix A. Supplementary data
The following are the supplementary data to this article:
References
- Anisimova M., Gil M., Dufayard J.-F., Dessimoz C., Gascuel O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011;60:685–699. doi: 10.1093/sysbio/syr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari I.H., Kwon B., Osorio F.A., Pattnaik A.K. Influence of N-linked glycosylation of porcine reproductive and respiratory syndrome virus GP5 on virus infectivity, antigenicity, and ability to induce neutralizing antibodies. J. Virol. 2006;80:3994. doi: 10.1128/JVI.80.8.3994-4004.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balka G., Hornyák A., Bálint A., Kiss I., Kecskeméti S., Bakonyi T., Rusvai M. Genetic diversity of porcine reproductive and respiratory syndrome virus strains circulating in Hungarian swine herds. Vet. Microbiol. 2008;127:128. doi: 10.1016/j.vetmic.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Bielejec F., Rambaut A., Suchard M.A., Lemey P. SPREAD: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics. 2011;27:2910–2912. doi: 10.1093/bioinformatics/btr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N., Yu X., Wang L., Wu J., Zhou Z., Ni J., Li X., Zhai X., Tian K. Two natural recombinant highly pathogenic porcine reproductive and respiratory syndrome viruses with different pathogenicities. Virus Genes. 2013;46:473–478. doi: 10.1007/s11262-013-0892-4. [DOI] [PubMed] [Google Scholar]
- Cho J.G., Dee S.A. Porcine reproductive and respiratory syndrome virus. Theriogenology. 2006;66:655–662. doi: 10.1016/j.theriogenology.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Darriba D., Taboada G.L., Doallo R., Posada D. JModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwich L., Díaz I., Mateu E. Certainties, doubts and hypotheses in porcine reproductive and respiratory syndrome virus immunobiology. Virus Res. 2010;154:123–132. doi: 10.1016/j.virusres.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Díaz I., Pujols J., Ganges L., Gimeno M., Darwich L., Domingo M., Mateu E. In silico prediction and ex vivo evaluation of potential T-cell epitopes in glycoproteins 4 and 5 and nucleocapsid protein of genotype-I (European) of porcine reproductive and respiratory syndrome virus. Vaccine. 2009;27:5603. doi: 10.1016/j.vaccine.2009.07.029. [DOI] [PubMed] [Google Scholar]
- Dokland T. The structural biology of PRRSV. Virus Res. 2010;154:86. doi: 10.1016/j.virusres.2010.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Suchard M.A., Xie D., Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012;29:1969. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Schneider P., Zhang W.P., Faaberg K.S., Nelson E.A., Rowland R.R.R. Diversity and evolution of a newly emerged North American Type 1 porcine arterivirus: analysis of isolates collected between 1999 and 2004. Arch. Virol. 2007;152:1009–1017. doi: 10.1007/s00705-007-0936-y. [DOI] [PubMed] [Google Scholar]
- Firth A.E., Zevenhoven-Dobbe J.C., Wills N.M., Go Y.Y., Balasuriya U.B., Atkins J.F., Snijder E.J., Posthuma C.C. Discovery of a small arterivirus gene that overlaps the GP5 coding sequence and is important for virus production. J. Gen. Virol. 2011;92:1097–1106. doi: 10.1099/vir.0.029264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg R., Storgaard T., Nielsen H.S., Oleksiewicz M.B., Cordioli P., Sala G., Hein J., Bøtner A. The genetic diversity of European type PRRSV is similar to that of the North American type but is geographically skewed within Europe. Virology. 2002;299:38–47. doi: 10.1006/viro.2002.1450. [DOI] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Gupta R., Jung E., Brunak S. 2004. Prediction of N-glycosylation sites in human proteins.http://www.cbs.dtu.dk/services/NetNGlyc/ Available from: [Google Scholar]
- Gustiananda M. Immunoinformatics analysis of H5N1 proteome for designing an epitope-derived vaccine and predicting the prevalence of pre-existing cellular-mediated immunity toward bird flu virus in Indonesian population. Immunome Res. 2011;7:1. [Google Scholar]
- Holmes E.C. Oxford University Press; USA: 2009. The Evolution and Emergence of RNA Viruses. [Google Scholar]
- Kass E.R., Raftery E.A. Bayes factors. J. Am. Stat. Assoc. 1995;90:773–795. [Google Scholar]
- Kim W.-I., Kim J.-J., Cha S.-H., Wu W.-H., Cooper V., Evans R., Choi E.-J., Yoon K.-J. Significance of genetic variation of PRRSV ORF5 in virus neutralization and molecular determinants corresponding to cross neutralization among PRRS viruses. Vet. Microbiol. 2013;162:10–22. doi: 10.1016/j.vetmic.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Korber B., LaBute M., Yusim K. Immunoinformatics comes of age. PLoS Comput. Biol. 2006;2:0484. doi: 10.1371/journal.pcbi.0020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M.M.C. Recombination in large RNA viruses: coronaviruses. Semin. Virol. 1996;7:381. doi: 10.1006/smvy.1996.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen J.E., Lund O., Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P., Rambaut A., Drummond A.J., Suchard M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009;5:e1000520. doi: 10.1371/journal.pcbi.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Fang L., Xu Z., Liu S., Gao J., Jiang Y., Chen H., Xiao S. Recombination in vaccine and circulating strains of porcine reproductive and respiratory syndrome viruses. Emerg. Infect. Dis. 2009;15:2032–2035. doi: 10.3201/eid1512.090390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Zhou R., Zhang J., Zhou L., Jiang Q., Guo X., Ge X., Yang H. Recombination analyses between two strains of porcine reproductive and respiratory syndrome virus in vivo. Virus Res. 2011;155:473–486. doi: 10.1016/j.virusres.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Maddison W., Maddison P.D.R. 2011. Mesquite: a modular system for evolutionary analysis. [Google Scholar]
- Martin D.P., Lemey P., Lott M., Moulton V., Posada D., Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Lobo F.J., Díez-Fuertes F., Simarro I., Castro J.M., Prieto C. Porcine reproductive and respiratory syndrome virus isolates differ in their susceptibility to neutralization. Vaccine. 2011;29:6928–6940. doi: 10.1016/j.vaccine.2011.07.076. [DOI] [PubMed] [Google Scholar]
- Mateu E., Diaz I. The challenge of PRRS immunology. Vet. J. 2008;177:345–351. doi: 10.1016/j.tvjl.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateu E., Díaz I., Darwich L., Casal J., Martín M., Pujols J. Evolution of ORF5 of Spanish porcine reproductive and respiratory syndrome virus strains from 1991 to 2005. Virus Res. 2006;115:198–206. doi: 10.1016/j.virusres.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengeling W.L. The potential role of genetic recombination in the evolution of new strains of porcine reproductive and respiratory syndrome virus (PRRSV) J. Swine Health Prod. 2002;10:273–275. [Google Scholar]
- Meulenberg J.J. PRRSV, the virus. Vet. Res. 2000;31:11–21. doi: 10.1051/vetres:2000103. [DOI] [PubMed] [Google Scholar]
- Murtaugh M.P., Stadejek T., Abrahante J.E., Lam T.T., Leung F.C. The ever-expanding diversity of porcine reproductive and respiratory syndrome virus. Virus Res. 2010;154:18–30. doi: 10.1016/j.virusres.2010.08.015. [DOI] [PubMed] [Google Scholar]
- Murtaugh M.P., Yuan S., Faaberg K.S. Appearance of novel PRRSV isolates by recombination in the natural environment. Adv. Exp. Med. Biol. 2001;494:31–36. doi: 10.1007/978-1-4615-1325-4_4. [DOI] [PubMed] [Google Scholar]
- Neumann E.J., Kliebenstein J.B., Johnson C.D., Mabry J.W., Bush E.J., Seitzinger A.H., Green A.L., Zimmerman J.J. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. J. Am. Vet. Med. Assoc. 2005;227:385–392. doi: 10.2460/javma.2005.227.385. [DOI] [PubMed] [Google Scholar]
- Nielsen M., Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10:296. doi: 10.1186/1471-2105-10-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis N., Duinhof T.F., van Nes A. Economic analysis of outbreaks of porcine reproductive and respiratory syndrome virus in nine sow herds. Vet. Rec. 2012;170:225. doi: 10.1136/vr.100101. [DOI] [PubMed] [Google Scholar]
- Oleksiewicz M.B., Bøtner A., Madsen K.G., Storgaard T. Sensitive detection and typing of porcine reproductive and respiratory syndrome virus by RT-PCR amplification of whole viral genes. Vet. Microbiol. 1998;64:7–22. doi: 10.1016/S0378-1135(98)00254-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak A.O., Spaan W.J.M., Snijder E.J. Nidovirus transcription: how to make sense…? J. Gen. Virol. 2006;87:1403–1421. doi: 10.1099/vir.0.81611-0. [DOI] [PubMed] [Google Scholar]
- Penn O., Privman E., Ashkenazy H., Landan G., Graur D., Pupko T. GUIDANCE: a web server for assessing alignment confidence scores. Nucleic Acids Res. 2010;38:W23–W28. doi: 10.1093/nar/gkq443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persia D., Pacciarini M., Cordioli P.S. Proceedings of the 10th International Symposium of Veterinary Laboratory Diagnosticians and OIE Seminar on Biotechnologies. 2001. Evaluation of three RT-PCR assays for the detection of porcine and respiratory syndrome virus (PRRSV) in diagnostic samples; pp. 440–441. [Google Scholar]
- Pesch S., Meyer C., Ohlinger V.F. New insights into the genetic diversity of European porcine reproductive and respiratory syndrome virus (PRRSV) Vet. Microbiol. 2005;107:31–48. doi: 10.1016/j.vetmic.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Pesente P., Rebonato V., Sandri G., Giovanardi D., Ruffoni L.S., Torriani S. Phylogenetic analysis of ORF5 and ORF7 sequences of porcine reproductive and respiratory syndrome virus (PRRSV) from PRRS-positive Italian farms: a showcase for PRRSV epidemiology and its consequences on farm management. Vet. Microbiol. 2006;114:214–224. doi: 10.1016/j.vetmic.2005.11.061. [DOI] [PubMed] [Google Scholar]
- Plagemann P. GP5 ectodomain epitope of porcine reproductive and respiratory syndrome virus, strain Lelystad virus. Virus Res. 2004;102:225–230. doi: 10.1016/j.virusres.2004.01.031. [DOI] [PubMed] [Google Scholar]
- Shi M., Holmes E.C., Brar M.S., Leung F.C. Recombination is associated with an outbreak of novel highly pathogenic porcine reproductive and respiratory syndrome viruses in china. J. Virol. 2013;87:10904–10907. doi: 10.1128/JVI.01270-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M., Lam T.T., Hon C.C., Hui R.K., Faaberg K.S., Wennblom T., Murtaugh M.P., Stadejek T., Leung F.C. Molecular epidemiology of PRRSV: a phylogenetic perspective. Virus Res. 2010;154:7–17. doi: 10.1016/j.virusres.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Shi M., Lam T.T., Hon C.C., Murtaugh M.P., Davies P.R., Hui R.K., Li J., Wong L.T., Yip C.-W., Jiang J.-W., Leung F.C. Phylogeny-based evolutionary, demographical, and geographical dissection of North American type 2 porcine reproductive and respiratory syndrome viruses. J. Virol. 2010;84:8700–8711. doi: 10.1128/JVI.02551-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H., Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2002;17:1246. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- Silvestro D., Michalak I. RaxmlGUI: a graphical front-end for RAxML. Organ. Divers. Evol. 2012;12:335. [Google Scholar]
- Simon-Loriere E., Holmes E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011;9:617–626. doi: 10.1038/nrmicro2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadejek T., Oleksiewicz M.B., Potapchuk D., Podgórska K. Porcine reproductive and respiratory syndrome virus strains of exceptional diversity in eastern Europe support the definition of new genetic subtypes. J. Gen. Virol. 2006;87:1835–1841. doi: 10.1099/vir.0.81782-0. [DOI] [PubMed] [Google Scholar]
- Stadejek T., Oleksiewicz M.B., Scherbakov A.V., Timina A.M., Krabbe J.S., Chabros K., Potapchuk D. Definition of subtypes in the European genotype of porcine reproductive and respiratory syndrome virus: nucleocapsid characteristics and geographical distribution in Europe. Arch. Virol. 2008;153:1479–1488. doi: 10.1007/s00705-008-0146-2. [DOI] [PubMed] [Google Scholar]
- Stadejek T., Stankevicius A., Murtaugh M.P., Oleksiewicz M.B. Molecular evolution of PRRSV in Europe: current state of play. Vet. Microbiol. 2013;165:21–28. doi: 10.1016/j.vetmic.2013.02.029. [DOI] [PubMed] [Google Scholar]
- Stranzl T., Larsen M.V., Lundegaard C., Nielsen M. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics. 2010;62:357. doi: 10.1007/s00251-010-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vugt J., Storgaard T., Oleksiewicz M.B., Bøtner A. High frequency RNA recombination in porcine reproductive and respiratory syndrome virus occurs preferentially between parental sequences with high similarity. J. Gen. Virol. 2001;82:2615–2620. doi: 10.1099/0022-1317-82-11-2615. [DOI] [PubMed] [Google Scholar]
- Vanhee M., Van Breedam W., Costers S., Geldhof M., Noppe Y., Nauwynck H. Characterization of antigenic regions in the porcine reproductive and respiratory syndrome virus by the use of peptide-specific serum antibodies. Vaccine. 2011;29:4794–4804. doi: 10.1016/j.vaccine.2011.04.071. [DOI] [PubMed] [Google Scholar]
- Vu H.L.X., Kwon B., Yoon K., Laegreid J., Pattnaik W.W., Osorio A.K.F.A. Immune evasion of porcine reproductive and respiratory syndrome virus through glycan shielding involves both glycoprotein 5 as well as glycoprotein 3. J. Virol. 2011;85:5555–5564. doi: 10.1128/JVI.00189-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins M.R., Gasteiger E., Bairoch A., Sanchez J.C., Williams K.L., Appel R.D., Hochstrasser D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. (Clifton, N.J.) 1999;112:531. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- Yoon S.H., Kim H., Park B., Kim H. Tracing the genetic history of porcine reproductive and respiratory syndrome viruses derived from the complete ORF 5-7 sequences: a Bayesian coalescent approach. Arch. Virol. 2012;157:2143–2151. doi: 10.1007/s00705-012-1408-6. [DOI] [PubMed] [Google Scholar]
- Yoshii M., Okinaga T., Miyazaki A., Kato K., Ikeda H., Tsunemitsu H. Genetic polymorphism of the nsp2 gene in North American type-porcine reproductive and respiratory syndrome virus. Arch. Virol. 2008;153:1323–1334. doi: 10.1007/s00705-008-0098-6. [DOI] [PubMed] [Google Scholar]
- Yuan S.S., Nelsen C.J., Murtaugh M.P., Schmitt B.J., Faaberg K.S. Recombination between North American strains of porcine reproductive and respiratory syndrome virus. Virus Res. 1999;61:87–98. doi: 10.1016/S0168-1702(99)00029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.