

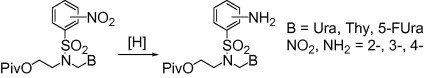
Graphical abstract
Keywords: Azanucleosides, Nucleoside analogs, Antiviral agents, Sulfonamides
Abstract
The acyclic azanucleosides with 2-, 3-, or 4-aminobenzenesulfonyl function at the nitrogen atom of the sugar mimic were prepared by coupling of 2-, 3-, or 4-nitro-N-(2-pivaloyloxyethyl)-N-(pivaloyloxymethyl)benzenesulfonamide with the silylated pyrimidine nucleobases followed by the reduction of the nitro group with sodium dithionite in aqueous solution or the palladium-catalysed transfer hydrogenation. The azanucleosides were evaluated for, but found to be devoid of, activity against several RNA- and DNA-viruses in vitro.
1. Introduction
Nucleoside analogs constitute a very important class of therapeutics used in the treatment of various diseases, mostly viral infections and cancer.1 However, their clinical usefulness can be sometimes limited by toxic side effects, poor oral bioavailability or the emergence of drug resistance.2 Therefore, much attention is still focused on the synthesis and biological activities of novel nucleoside analogs, including acyclonucleosides.3 The introduction of the sulfonamido group [R-SO2–N(H/R1)–] into a sugar4, 5, 6 or a nucleobase7 moiety of the parent nucleoside is one of the reported modifications. Among the sulfonamido nucleosides, however, only a few derivatives with the sulfanilamido group (4-NH2–C6H4–SO2–NH–) have been reported so far. This pharmacophore, found in many biologically active compounds (such as an inhibitor of HIV-1 protease amprenavir),8 is present in 5′-deoxy-5′-sulfanilamido-furanosyl nucleosides A (Fig. 1 ).4b To the best of our knowledge, their biological properties have not been examined.
Figure 1.
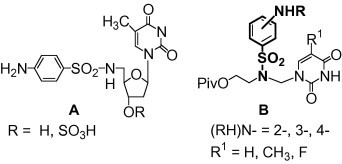
Nucleoside analogs containing 2-, 3-, or 4-aminobenzenesulfonyl function.
Therefore, continuing our research program on azanucleosides,9 we decided to synthesize the pyrimidine aza-derivatives B with 2-, 3-, or 4-aminobenzenesulfonyl function at the nitrogen atom of the sugar mimic (Fig. 1). These compounds can be considered as derivatives of sulfanilamide (4-NH2–C6H4–SO2–NH2). We prepared azanucleosides B in the form of the corresponding pivaloyl esters and/or amides (R = acyl) in order to improve their lipophilicity determining the physiological activity of biologically active compounds.10
2. Results and discussion
In view of the commercial availability of the nitro- or the acetamido-substituted benzenesulfonyl chlorides 2, we decided to prepare the nitro and the acetamido precursors 5, 6, and 7 (Scheme 1 , R = NO2 or NHAc), and then to transform them into the corresponding amino derivatives by reduction or hydrolysis, respectively.
Scheme 1.
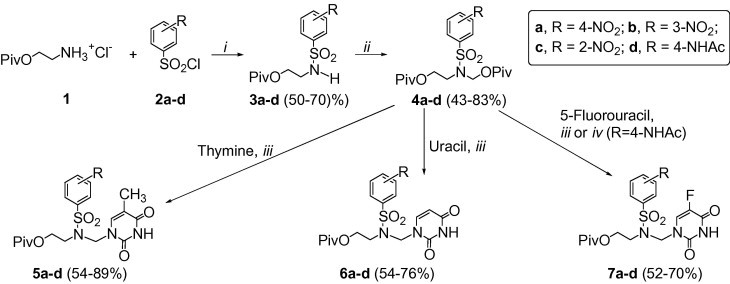
Reagents and conditions: (i) pyridine, dichloromethane, room temperature, overnight; (ii) PivOCH2Cl, K2CO3, DMF, room temperature, 5 days; (iii) a—nucleobase, BSA, acetonitrile, room temperature, 1 h; b—TMSOTf, acetonitrile, room temperature, 2 days. (iv) a—5-fluorouracil, BSA, acetonitrile, room temperature, 1 h; b—SnCl4, acetonitrile, dichloromethane, room temperature, 2 days.
The key substrates for the synthesis of 5, 6, and 7, that is, the corresponding N-(pivaloyloxymethyl)sulfonamides 4, were obtained from the reaction of 2-pivaloyloxyethylamine hydrochloride 1 with benzenesulfonyl chlorides 2 followed by the alkylation of 3 with chloromethyl pivalate (Scheme 1). N-(Pivaloyloxymethyl)sulfonamides 4 were transformed into 5, 6, or 7 by the one-pot base silylation/nucleoside coupling methodology (Scheme 1).11 Derivatives 4 reacted with the silylated thymine, uracil or 5-fluorouracil in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) to give the corresponding azanucleosides 5, 6, and 7 in 54–89% yields. The only exception was the coupling of the acetamido derivative 4d with 5-fluorouracil; instead of the desired 7d, 4-acetamido-N-(2-pivaloyloxyethyl)-N-(acetamidomethyl)benzenesulfonamide12 (not shown) was formed as the only product under these conditions. This compound resulted from the reaction of 4d with N-monosilylated or free acetamide, which are formed during silylation of the nucleobase with N,O-bis(trimethylsilyl)acetamide (BSA).13 Replacement of TMSOTf with tin(IV) chloride let us to prepare 7d in 52% yield.
The 1H–13C HMBC correlations observed for 5a, and the 1H–1H ROESY correlations for 5b or 5d confirmed the N-1 substitution pattern of azanucleosides 5–7 (Fig. 2 ).
Figure 2.
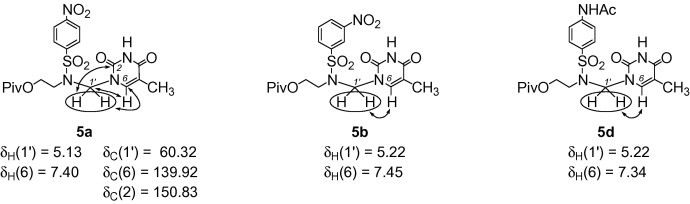
1H–13C HMBC correlations observed for 5a, and 1H–1H ROESY correlations for 5b and 5d.
Nucleoside analogs with the nitro group in a sugar moiety14 (mostly derivatives of uracil(a), (b), (c), (d), (e)) have been reduced to the corresponding amino derivatives by the heterogeneous catalytic hydrogenation (in the presence of Raney-Nickel,(a), (b), (c) palladium on charcoal,(f), (g) or platinum(IV) oxide14h), or with the following reducing agents: sodium dithionite,4b tin in acetic acid,14d or sodium borohydride (in the presence of nickel(II) chloride14e or palladium on charcoal14g).15 The number of reports on the reduction of the 5-fluorouracil nitro nucleosides is limited. They have been reduced by heterogeneous catalytic hydrogenation in the presence of palladium on charcoal, or with sodium borohydride/palladium on charcoal mixture; unfortunately yields of the reaction products have not been given.14g
Considering the presence of the 4-nitrobenzenesulfonamido function in a molecule, the reduction conditions of 5′-deoxy-5′-(4-nitrobenzenesulfonamido)thymidine (i.e., sodium dithionite in alkaline medium) has attracted our considerable attention.4b To the best of our knowledge, this reducing agent has not been employed for the reduction of nucleoside nitro analogs being derivatives of uracil or 5-fluorouracil so far. Therefore, we decided to examine this method for the transformation of the nitro derivatives 5, 6, and 7 into the amino azanucleosides B. Initially, the reduction of the thymine derivatives 5a–c was examined (Scheme 2 ). Taking into account the NO2-isomerism of 5a–c, the reaction was found to be not general. The reduction of 5a (4-NO2) or 5b (3-NO2) with sodium dithionite in alkaline solution at 90 °C was accompanied by hydrolysis of the ester group to give the hydroxy derivatives 8a (59%) or 8b (52%), respectively (Scheme 2, conditions (i). Reduction of the 2-NO2 isomer 5c under the same conditions afforded a complex mixture, from which the amino derivative 8c was isolated in 5% yield. Modification of the literature procedure by the use of aqueous sodium dithionite at 90 °C resulted in the formation of all isomers 9a–c as pivaloyl esters in yields exceeding 50% (Scheme 2, conditions (ii). The 2-NH2 isomer 9c was treated with ammonium hydroxide at 70 °C to obtain 8c in 79% yield (Scheme 2, conditions (iii)).
Scheme 2.
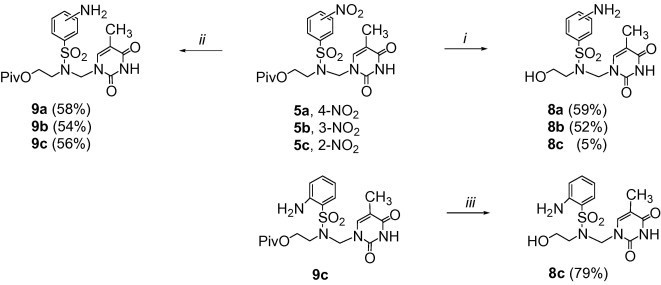
Reagents and conditions: (i) Na2S2O4, 4% NaOH aq, 90 °C, 1 h; (ii) Na2S2O4 aq, 90 °C, 1 h; (iii) NH3 aq, MeOH, 70 °C, 1 day.
Taking into account a kind of nucleobase present in the nitro compounds tested, the dithionite reduction was not general as well. In contrast to the thymine derivatives 5a–c, treating of the nitro derivatives of uracil or 5-fluorouracil, that is, 6a or 7a, respectively, with sodium dithionite in alkaline medium or in aqueous solution did not afford the corresponding amino azanucleosides at all.
Although the heterogeneous catalytic hydrogenation is one of the most often used methods for the reduction of nucleoside nitro analogs, the palladium-catalysed hydrogenation (H2, 10% Pd/C, ambient pressure) of 6a–c or 7a–c did not give the expected results; multi-component mixtures (by TLC) were produced. Therefore, we applied the palladium-catalysed transfer hydrogenation16 to achieve the amino analogs 10 and 11 (Scheme 3, Scheme 4 ). Treating of the uracil derivatives 6a–c with cyclohexene/10% Pd/C mixture in ethanol at 60 °C gave the corresponding amino derivatives 10a–c in 58–89% yields (Scheme 3).
Scheme 3.
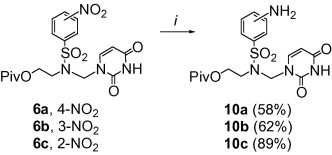
Reagents and conditions: (i) cyclohexene, 10% Pd/C, EtOH, 60 °C, 1 day.
Scheme 4.
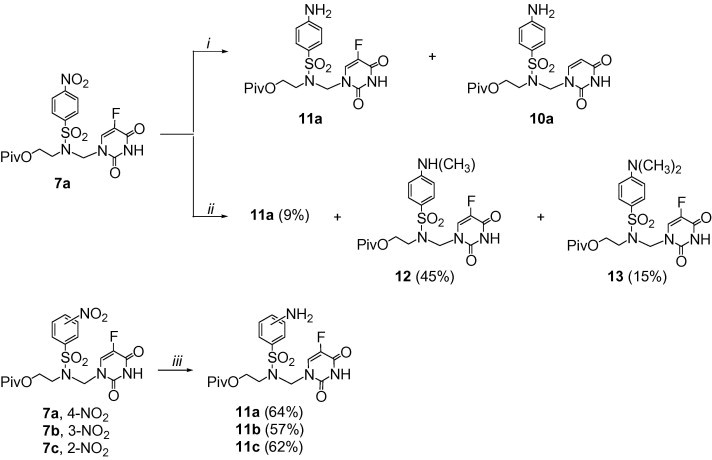
Reagents and conditions: cyclohexene, 10% Pd/C, 60 °C, 1 day: (i) EtOH; 11a:10a = 3:2 (1H NMR); (ii) MeOH; (iii) 1,4-dioxane.
The reduction of the 5-fluorouracil derivatives 7 by the palladium-catalysed transfer hydrogenation was more complex and its outcome depended on the solvent used (Scheme 4). The reduction of 7a in ethanol provided an inseparable mixture of the expected amino derivative 11a and the uracil derivative 10a in a 3:2 ratio17 (Scheme 4, conditions (i)); 10a was a product of hydrogenolysis of the carbon–fluorine bond.18 The formation of 10a was not observed when the reaction was carried out in methanol at 60 °C (Scheme 4, conditions (ii)), but 11a was the minor product (yield 9%); the N-methyl derivative 12 (45%) and the N,N-dimethyl one 13 (15%) were also obtained. We assume that 12 and 13 were formed from the reductive methylation of 11a or 12 with formaldehyde, respectively, which was produced from the catalytic dehydrogenation of methanol.16a All difficulties with the reduction of 7 were solved when 1,4-dioxane was used as the reaction medium (Scheme 4, conditions (iii)); all isomers 11a–c were prepared in the yields of 57–64%.
An alternative approach toward azanucleosides B consisted in ammonolysis or an alkaline hydrolysis of the 4-acetamido derivatives (Scheme 5 ). Ammonolysis of 5d or 6d with ammonium hydroxide at 70 °C resulted in the selective cleavage of the pivaloyl ester; azanucleosides 14a or 14b were obtained in 94% or 85% yield, respectively. Both the O- and N-protecting groups were removed when the thymine derivative 5d was treated with aqueous sodium hydroxide at 90 °C to provide 8a in 39% yield. The latter conversion was much less effective than the previously performed reduction of the nitro derivative 5a with sodium dithionite under alkaline conditions (Scheme 2).
Scheme 5.
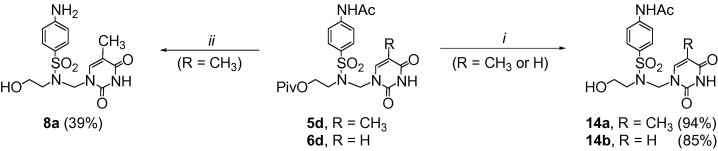
Reagents and conditions: (i) NH3 aq, MeOH, 70 °C, 1 day; (ii) NaOH aq (4%), 90 °C, 2 h.
3. Antiviral evaluation
All azanucleosides with nitro, amino, or acetamido group were evaluated for activity against several RNA- and DNA-viruses, using the following cell-based assays: (a) Vero cells infected with parainfluenza-3 virus, reovirus-1, Sindbis virus, Coxsackie B4 virus, or Punta Toro virus; (b) Crandell-Rees Feline Kidney (CRFK) cells infected with feline corona virus or feline herpes virus; (c) human embryonic lung (HEL) fibroblasts infected with herpes simplex virus-1 (KOS), herpes simplex virus-2 (G), acyclovir-resistant herpes simplex virus-1 (TK− KOS ACVr), vaccinia virus, or vesicular stomatitis virus; (d) HeLa cells infected with vesicular stomatitis virus, coxsackie B4 virus or respiratory syncytial virus; and (e) Madin Darby canine kidney (MDCK) cells infected with influenza virus, subtype A/H1N1, A/H3N2 or B.
The cytotoxicity of the test compounds toward the uninfected host cells was expressed as the minimal compound concentration (MCC) that caused a microscopically detectable alteration of normal cell morphology, or 50% cytotoxic concentration (CC50), causing a 50% decrease in cell viability, as determined by a colorimetric formazan-based MTS assay.19, 20 None of the tested compounds displayed cytotoxicity on HEL cells or HeLa cells at concentrations up to 100 μM. Among the tested compounds, 5d and 6d (the O-pivaloylated 4-NHAc derivatives of thymine or uracil, respectively) showed cytotoxicity against Vero cells at the concentration of 200 μM; with the MCC value of 40 μM, 14a (analog of 5d with the hydroxy group instead of the O-pivaloyloxy one at the sugar mimic) was found to be much more cytotoxic than 5d, while the cytotoxicity of 14b (the hydroxy analog of 6d) was not detected at concentrations up to 100 μM. The remaining compounds showed no cytotoxicity for Vero cells at concentrations up to 100 μM. The nitro azanucleosides, and among them mainly derivatives of 5-fluorouracil, proved cytotoxic for MDCK cells and CRFK cells. The following cytotoxic concentration values were determined for the nitro derivatives of the 5-fluorouracil series: (i) 7a (the 4-NO2 isomer): MCC = 20 μM (MDCK cells), CC50 = 69 μM (MDCK cells), CC50 = 44 μM (CRFK cells); (ii) 7b (the 3-NO2 isomer): MCC ⩾ 100 μM (MDCK cells), CC50 > 100 μM (MDCK cells), CC50 = 95 μM (CRFK cells); and (iii) 7c (the 2-NO2 isomer): MCC = 4 μM (MDCK cells), CC50 > 100 μM (MDCK cells), CC50 = 14 μM (CRFK cells).21 The thymine 4-NO2 derivative 5a (CC50 = 53 μM) and the uracil 4-NO2 derivative 6a (CC50 = 49 μM) displayed cytotoxicity on CRFK cells. The remaining compounds tested did not show cytotoxicity toward MDCK or CRFK cells at concentrations up to 100 μM.
The antiviral activity was expressed as the 50% effective concentration, or concentration producing 50% inhibition of virus-induced cytopathic effect, as determined by visual scoring of the CPE, or by measuring the cell viability with the colorimetric formazan-based MTS assay. Antiviral effect of the tested compounds was examined at the following concentration values: assays (a), (i) 40 μM for 5d and 6d, (ii) 8 μM for 14a, or (iii) 100 μM for other compounds; assays (b), (i) 20 μM for 5a, 6a, 7a, and 7b, (ii) 4 μM for 7c, or (iii) 100 μM for other compounds; and assays (c)–(e), 100 μM for all tested compounds. In evaluations (a), both 5c (the thymine 2-NO2 derivative) and 7b (the 5-fluorouracil 3-NO2 derivative) displayed EC50 = 100 μM against Coxsackie B4 virus and Punta Toro virus; activity of the compounds in other viruses was not detected at concentration of 100 μM. No antiviral effects were detected for any of the remaining compounds against any of the viruses evaluated.
4. Conclusion
Acyclic azanucleosides with 2-, 3-, or 4-aminobenzene-sulfonyl function at the nitrogen atom of the sugar mimic were obtained via reduction of the corresponding nitro precursors; depending on the nucleobase present in the molecule of the nitro derivative, sodium dithionite or cyclohexene–10% Pd/C mixture was employed as the reducing agent. The studies showed that nitro analogs sensitive to reduction conditions, such as 5-fluorouracil derivatives, could be transformed into the corresponding amines by the palladium-catalysed transfer hydrogenation with good yields. Generally, our findings on the reduction of the nucleoside nitro analogs by the heterogeneous transfer hydrogenation may be of help in synthesizing of many amino nucleosides from nitro precursors.
5. Experimental
5.1. Materials and methods
Pre-coated Merck silica gel 60 F-254 plates were used for both thin-layer chromatography (TLC, 0.2 mm), and the preparative thin-layer chromatography (2 mm); the spots were detected under UV light (254 nm). Column chromatography was performed using silica gel (200–400 mesh, Merck). High Resolution Mass Spectra (Electrospray Ionisation, ESI) were performed on a Mariner® spectrometer in positive ionization mode. The IR spectra were recorded on a Perkin-Elmer System 2000 spectrometer in KBr disc; resolution was 2 cm−1; absorption maxima (ν max) are given in cm−1 and quoted as ‘s’ strong, ‘m’ medium, ‘br’ broad. In the case of highly overlapped IR bands deconvolution with Grams Research software was preformed. The 1H NMR spectra were measured on a Varian Gemini-200BB at 200 MHz or on a Varian Mercury-400BB spectrometer at 400 MHz. The 13C NMR spectra were recorded on a Varian Gemini-200BB spectrometer at 50 MHz. 1H and 13C chemical shifts (δ) are reported in parts per million (ppm) relative to the solvent signals: CDCl3, δ H (residual CHCl3) 7.26 ppm, δ C 77.16 ppm; or DMSO-d 6, δ H (residual DMSO-d 5) 2.50 ppm, δ C 39.52 ppm; signals are quoted as ‘s’ (singlet), ‘d’ (doublet), ‘t’ (triplet), ‘m’ (multiplet), and ‘br s’ (broad singlet). Coupling constants J are reported in Hertz. The 1H–13C HMBC (Heteronuclear Multiple Bond Correlation) spectrum of 5a was measured on a Varian Mercury-400BB spectrometer in DMSO-d 6. The 1H–1H ROESY (Rotating frame Overhause Effect Spectroscopy) spectra of 5b or 5d were measured on a Varian Mercury-400BB spectrometer in CDCl3 or DMSO-d 6, respectively. Anhydrous MgSO4 was employed as a drying agent. Solvents were distilled off under reduced pressure on a rotating evaporator.
The methodology used for measuring the antiviral activity has been described previously.22
5.2. 2-Pivaloyloxyethylamine hydrochloride (1)
A mixture of ethanolamine hydrochloride (10.30 g, 105 mmol) and pivaloyl chloride (105 mmol, 13.0 mL) was heated on an oil bath at 90 °C until the production of hydrogen chloride ceased (ca. 4 h); after this time the mixture solidified. The residual hydrogen chloride was removed under reduced pressure. The residue was kept at a vacuum desiccator over KOH overnight to give the crude 1 (18.26 g) as a pale yellow, amorphous solid. An analytical sample was obtained by crystallization from dry acetone. δ H (200 MHz; DMSO-d 6) 1.17 (s, 9H), 3.06 (m, 2H), 4.17 (t, 3 J H–H 5.6, 2H), 8.29 (br s, 3H, ). δ H (50 MHz; DMSO-d 6) 26.83, 37.65, 40.61, 60.79, 177.36. ν max (KBr) 2916–3112br s (NH3 +), 1724s (C O), 1575m , 1500 . LRMS m/z calcd for C7H16NO2 (M+H)+ 146.1, found 146.1.
5.3. General method for the synthesis of N-(2-pivaloyloxyethyl)benzenesulfonamides (3)
A mixture of the crude 1, dry pyridine and dry dichloromethane, in the ratio of 10 mmol/30 mmol/18 mL, respectively, was cooled in an ice-water bath and the corresponding benzenesulfonyl chloride 2 (11 mmol) was added in one portion. The mixture was kept overnight at room temperature and then washed with water, diluted hydrochloric acid (5%), brine, and dried. The solvent was distilled off. The residue was purified by column chromatography or crystallization to afford 3; the solvents are given in parentheses below.
5.3.1. 4-Nitro-N-(2-pivaloyloxyethyl)benzenesulfonamide (3a)
According to the general procedure, 3a was obtained from 1 (5.0 g, 27.5 mmol) and 4-nitrobenzenesulfonyl chloride 2a (6.7 g, 30.3 mmol). Crystallization (hexane/ethyl acetate, 2:1, v/v) gave 3a (6.17 g, 68%) as a white solid; mp 134–135 °C. δ H (400 MHz, CDCl3) 1.17 (s, 9H), 3.29–3.33 (m, 2H), 4.12–4.15 (m, 2H), 5.03 (t, 3 J H–H 6.0, 1H, NH), 8.04–8.09 (m, 2H), 8.36–8.40 (m, 2H). δ C (50 MHz, CDCl3) 27.11, 38.80, 42.74, 62.63, 124.56, 128.33, 145.93, 150.18, 178.62. ν max (KBr) 3248m (NH), 1702s (C O), 1533s (NO2), 1350s (NO2), 1341s (SO2), 1174s (SO2). HRMS m/z calcd for C13H18N2O6NaS (M+Na)+ 353.0778, found 353.0767.
5.3.2. 3-Nitro-N-(2-pivaloyloxyethyl)benzenesulfonamide (3b)
According to the general procedure, 3b was obtained from 1 (6.0 g, 33 mmol) and 3-nitrobenzenesulfonyl chloride 2b (8.04 g, 36.3 mmol). Crystallization (hexane/ethyl acetate, 2.5:1, v/v) gave 3b (6.87 g, 63%) as a white solid; mp 91–92 °C. δ H (400 MHz, CDCl3) 1.17 (s, 9H), 3.30–3.34 (m, 2H), 4.13–4.15 (m, 2H), 5.02 (t, 3 J H–H 6.0, 1H, NH), 7.74–7.79 (m, 1H), 8.19–8.22 (m, 1H), 8.44–8.46 (m, 1H), 8.70–8.73 (m, 1H). δ C (50 MHz, CDCl3) 27.15, 38.86, 42.60, 62.61, 122.28, 127.37, 130.81, 132.61, 142.43, 148.47, 178.72. ν max (KBr) 3260m (NH), 1705s (C O), 1527s (NO2), 1350s (NO2), 1345s (SO2), 1165s (SO2). HRMS m/z calcd for C13H18N2O6NaS (M+Na)+ 353.0778, found 353.0763.
5.3.3. 2-Nitro-N-(2-pivaloyloxyethyl)benzenesulfonamide (3c)
According to the general procedure, 3c was obtained from 1 (6.0 g, 33 mmol) and 2-nitrobenzenesulfonyl chloride 2c (8.04 g, 36.3 mmol). Chromatographic purification (chloroform/acetone, 98:2, v/v) gave 3c (7.58 g, 70%) as a colorless oil. δ H (200 MHz, CDCl3) 1.14 (s, 9H), 3.33–3.42 (m, 2H), 4.09–4.15 (m, 2H), 5.77 (t, 3 J H–H 5.8, 1H, NH), 7.70–7.77 (m, 2H), 7.84–7.87 (m, 1H), 8.10–8.15 (m, 1H). δ C (50 MHz, CDCl3) 27.16, 38.86, 42.94, 62.49, 125.60, 131.00, 133.14, 133.73, 133.95, 148.15, 178.31. ν max (KBr) 3242m (NH), 1704s (C O), 1536s (NO2), 1351s (NO2), 1344s (SO2), 1171s (SO2). HRMS m/z calcd for C13H18N2O6NaS (M+Na)+, 353.0778, found 353.0794.
5.3.4. 4-Acetamido-N-(2-pivaloyloxyethyl)benzenesulfonamide (3d)
According to the general procedure, 3d was obtained from 1 (5.0 g, 27.5 mmol) and 4-acetamidobenzenesulfonyl chloride 2d (7.07 g, 30.3 mmol). Chromatographic purification (chloroform/acetone, 95:5, v/v) gave 3d (4.7 g, 50%) as a white solid; mp 90–91 °C. δ H (400 MHz, CDCl3) 1.17 (s, 9H), 2.22 (s, 3H), 3.20–3.24 (m, 2H), 4.08–4.10 (m, 2H), 4.78 (t, 3 J H–H 6.0, 1H, NH), 7.48 (br s, 1H, NH), 7.61–7.65 (m, 2H), 7.63–7.77 (m, 2H). δ C (50 MHz, CDCl3) 24.80, 27.25, 38.92, 42.46, 62.84, 119.69, 128.30, 134.49, 142.30, 169.24, 178.72. ν max (KBr) 3307m (NH), 3269m (NH), 1725s (C O), 1676s (C O), 1594s, 1533br s, 1327s (SO2), 1158s (SO2). HRMS m/z calcd for C15H22N2O5NaS (M+Na)+ 365.1142, found 365.1159.
5.4. General method for the synthesis of N-(2-pivaloyloxyethyl)-N-(pivaloyloxymethyl)benzenesulfonamides (4)
Chloromethyl pivalate (15 mol) was added to a stirred mixture of N-(2-pivaloyloxyethyl)benznenesulfonamide 3 (5 mol) and anhydrous potassium carbonate (25 mmol) in dry DMF (10 mL) at room temperature. The mixture was stirred for 5 days at room temperature and then was poured into ice-cold water (50 mL). The organic phase was extracted with dichloromethane (3× 50 mL). The extracts were combined and washed with water, and dried. The solvent was distilled off. The residue was purified by column chromatography or crystallization to give 4; the solvents are given in parentheses below.
5.4.1. 4-Nitro-N-(2-pivaloyloxyethyl)-N-(pivaloyloxymethyl)benzenesulfonamide (4a)
According to the general procedure, 4a was obtained from 3a (5.0 g, 15 mmol). Crystallization (hexane/ethyl acetate, 3:1, v/v) afforded 4a (5.15 g, 76%) as a white solid; mp 105–107 °C. δ H (CDCl3, 400 MHz) 0.99 (s, 9H), 1.22 (s, 9H), 3.49 (t, 3 J H–H 5.6, 2H), 4.29 (t, 3 J H–H 5.6, 2H), 5.58 (s, 2H), 8.06–8.11 (m, 2H), 8.34–8.39 (m, 2H). δ C (CDCl3, 50 MHz) 26.77, 27.15, 38.74, 45.59, 61.94, 71.76, 124.33, 128.89, 145.68, 150.23, 177.62, 178.20. ν max (KBr) 1735s (C O), 1725s (C O), 1533s (NO2), 1356s (NO2), 1352s (SO2), 1152s (SO2). HRMS m/z calcd for C19H28N2O8NaS (M+Na)+ 467.1459, found 467.1458.
5.4.2. 3-Nitro-N-(2-pivaloyloxyethyl)-N-(pivaloyloxymethyl)benzenesulfonamide (4b)
According to the general procedure, 4b was obtained from 3b (3.1 g, 9.2 mmol). Crystallization (hexane/ethyl acetate, 13:1, v/v) afforded 4b (2.42 g, 59%) as a white solid; mp 84–85 °C. δ H (200 MHz, CDCl3) 0.96 (s, 9H), 1.20 (s, 9H), 3.48 (t, 3 J H–H 5.4, 2H), 4.27 (t, 3 J H–H 5.4, 2H), 5.57 (s, 2H), 7.71–7.79 (m, 1H), 8.20–8.24 (m, 1H), 8.43–8.47 (m, 1H), 8.69–8.71 (m, 1H). δ C (50 MHz, CDCl3) 26.84, 27.29, 38.81, 38.89, 45.69, 62.11, 71.93, 122.86, 127.58, 130.68, 133.21, 142.34, 148.53, 177.83, 178.34. ν max (KBr) 1739s (C O), 1728s (C O), 1537s (NO2), 1359s (NO2), 1353s (SO2), 1145s (SO2). HRMS m/z calcd for C19H28N2O8NaS (M+Na)+ 467.1459, found 467.1454.
5.4.3. 2-Nitro-N-(2-pivaloyloxyethyl)-N-(pivaloyloxymethyl)benzenesulfonamide (4c)
According to the general procedure, 4c was obtained from 3c (3.1 g, 9.2 mmol). Chromatographic purification (dichloromethane) gave 4c (1.77 g, 43%) as a white solid; mp 45–47 °C. δ H (200 MHz, CDCl3) 0.99 (s, 9H), 1.19 (s, 9H), 3.71 (t, 3 J H–H 5.4, 2H), 4.24 (t, 3 J H–H 5.4, 2H), 5.52 (s, 2H), 7.63–7.78 (m, 3H), 8.06–8.15 (m, 1H). δ C (50 MHz, CDCl3) 26.86, 27.26, 38.78, 38.85, 46.73, 62.22, 71.89, 124.43, 131.51, 132.04, 133.48, 134.24, 148.19, 177.81, 178.34. ν max (KBr) 1733s (C O), 1721s (C O), 1540s (NO2), 1372s (NO2), 1349s (SO2), 1150s (SO2). HRMS m/z calcd for C19H28N2O8NaS (M+Na)+, 467.1459, found 467.1458.
5.4.4. 4-Acetamido-N-(2-pivaloyloxyethyl)-N-(pivaloyloxymethyl)benzenesulfonamide (4d)
According to the general procedure, 4d was obtained from 3d (2.0 g, 5.8 mmol). Chromatographic purification (chloroform/acetone, 98:2, v/v) gave 4d (2.2 g, 83%) as a white solid; mp 73–76 °C. δ H (200 MHz, CDCl3) 1.00 (s, 9H), 1.19 (s, 9H), 2.19 (s, 3H), 3.41 (t, 3 J H–H 5.6, 2H), 4.24 (t, 3 J H–H 5.6, 2H), 5.52 (s, 2H), 7.64–7.68 (m, 2H), 7.75–7.80 (m, 2H), 7.95 (br s, 1H, NH) δ C (50 MHz, CDCl3) 24.87, 26.98, 27.31, 38.87, 45.38, 62.53, 72.41, 119.39, 129.00, 134.51, 142.50, 168.77, 178.42, 178.47. ν max (KBr) 3336m (NH), 1738s (C O), 1709s (C O), 1679s (C O), 1593s, 1537br s, 1351s (SO2), 1158s (SO2). HRMS m/z calcd for C21H32N2O7NaS (M+Na)+ 479.1822, found 479.1828.
5.5. General procedure for the coupling of 4 with thymine, uracil, or 5-fluorouracil
A mixture of a nucleobase (thymine, uracil, or 5-fluorouracil) (2.0 mmol) and N,O-bis-(trimethylsilyl)acetamide (BSA, 815 mg, 4.0 mmol, 1.0 mL) in dry acetonitrile (10 mL) was stirred at room temperature for 1 h under an argon atmosphere. Then, a solution of 4 (1.0 mmol) in dry acetonitrile (1 mL) and subsequently a Lewis acid [TMSOTf (370 mg, 1.7 mmol, 0.3 mL), or 1 M solution of tin(IV) chloride in dichloromethane (3 mL)] were added. The reaction mixture was kept at room temperature for 2 days. Ethyl acetate (50 mL) and a saturated aqueous solution of sodium bicarbonate (1 mL) were added. The mixture was stirred for 1 h and then filtered through a Celite pad. The organic phase was separated, washed with brine, and dried. The volatiles was distilled off. The residue was purified by column chromatography or crystallization to give the corresponding azanucleoside 5, 6, or 7; the solvents are given in parentheses below.
5.5.1. 1-[N-(4-Nitrobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (5a)
According to the general procedure, 5a was obtained from 4a (445 mg, 1 mmol) and thymine in the presence of TMSOTf. Chromatographic purification (chloroform/acetone, 9:1, v/v) gave 5a (315 mg, 67%) as a white solid; mp 172–173 °C. δ H (200 MHz, DMSO-d 6) 1.11 [s, 9H, –C(O)C(CH 3)3], 1.72 [s, 3H, –CH C(CH 3)–], 3.74–3.79 (m, 2H, PivO–CH2–CH 2–), 4.13–4.18 (m, 2H, PivO–CH 2–CH2–), 5.13 (s, 2H, –N–CH 2–N–), 7.40 [br s, 1H, –CH C(CH3)–], 8.06–8.10 (m, 2H), 8.35–8.39 (m, 2H), 11.26 [br s, 1H, NH]. δ C (50 MHz, DMSO-d 6,) 11.83 [–CH C(CH3)–], 26.69 [–C(O)C(CH3)3], 37.50 [–C(O)C(CH3)3], 48.09 (PivO–CH2–CH2–), 60.32 (–N–CH2–N–), 61.61 (PivO–CH2–CH2–), 108.91 [–CH C(CH3)–], 124.49 (Ar), 128.22 (Ar), 139.92 [–CH C(CH3)–], 144.93 (Ar), 149.68 (Ar), 150.83 [–N–C(O)–NH–], 163.62 [–NH–C(O)–C(CH3) ], 177.03 [–C(O)C(CH3)3]. ν max (KBr) 1722s (C O), 1712s (C O), 1683s (C O), 1533s (NO2), 1354s (NO2). HRMS m/z calcd for C19H24N4O8NaS (M+Na)+ 491.1207, found 491.1206.
5.5.2. 1-[N-(3-Nitrobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (5b)
According to the general procedure, 5b was obtained from 4b (445 mg, 1 mmol) and thymine in the presence of TMSOTf. Crystallization (methanol/diethyl ether, 5:1, v/v) gave 5b (370 mg, 79%) as a white solid; mp 160–162 °C. The filtrate was concentrated to dryness, and the residue was purified by column chromatography (chloroform/acetone, 95:5, v/v) to give the additional amount of 5b (49 mg, 10%) as a white solid. δ H (DMSO-d 6 200 MHz) 1.12 [s, 9H, –C(O)C(CH 3)3], 1.71 [d, 4 J H–H 1.0, 3H, –CH C(CH 3)], 3.76–3.82 (m, 2H, PivO–CH 2–CH2– or PivO–CH2–CH 2–), 4.14–4.19 (m, 2H, PivO–CH2–CH 2– or PivO–CH 2–CH2–), 5.22 (s, 2H, –N–CH 2–N–), 7.45 [q, 4 J H–H 1.0, 1H, –CH C(CH3)–], 7.85–7.93 (m, 1H, Ar-H), 8.25–8.29 (m, 1H, Ar-H), 8.43–8.53 (m, 2H, Ar-H), 11.23 (br s, 1H, NH). δ C (DMSO-d 6, 50 MHz) 11.87, 26.82, 38.17, 48.32, 60.74, 61.95, 109.13, 121.36, 127.78, 131.62, 132.74, 140.23, 141.32, 147.74, 151.00, 163.74, 177.24. ν max (KBr) 1718s (C O), 1710s (C O), 1681s (C O), 1531s (NO2), 1351s (NO2). HRMS m/z calcd for C19H24N4O8NaS (M+Na)+ 491.1207, found 491.1196.
5.5.3. 1-[N-(2-Nitrobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (5c)
According to the general procedure, 5c was obtained from 4c (445 mg, 1 mmol) and thymine in the presence of TMSOTf. Chromatographic purification (chloroform/acetone, 95:5, v/v) gave 5c (248 mg, 54%) as a white solid; mp 129–130 °C. δ H (200 MHz, DMSO-d 6) 1.11 (s, 9H), 1.63 (br s, 3H), 3.83–8.87 (m, 2H), 4.13–4.18 (m, 2H), 5.30 (s, 2H), 7.17 (br s, 1H), 7.76–8.03 (m, 4H), 11.30 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 11.93, 26.81, 38.15, 48.06, 59.70, 61.44, 109.11, 124.66, 129.07, 132.34, 132.80, 134.88, 139.54, 147.44, 151.26, 163.72, 177.23. ν max (KBr) 1738s (C O), 1712s (C O), 1674s (C O), 1540s (NO2), 1366s (NO2). HRMS m/z calcd C19H24N4O8NaS (M+Na)+ 491.1207, found 491.1226.
5.5.4. 1-[N-(4-Acetamidobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (5d)
According to the general procedure, 5d was obtained from 4d (228 mg, 0.5 mmol) and thymine in the presence of TMSOTf. Chromatographic purification (chloroform/acetone, 8:2, v/v) gave 5d (152 mg, 63%) as a white solid; mp 115–117 °C. δ H (200 MHz, CDCl3) 1.18 [s, 9H, –C(O)C(CH 3)3), 1.89 [d, 4 J H–H 1.2, 3H, –CH C(CH 3)–], 2.20 [s, 3H, –NH–C(O)CH 3], 3.69 (t, 3 J H–H 5.4, 2H, PivO–CH 2–CH2– or PivO–CH2–CH 2–), 4.18 (t, 3 J H–H 5.4, 2H, PivO–CH2–CH 2– or PivO–CH 2–CH2–), 5.22 (s, 2H, –N–CH 2–N–), 7.34 [q, 4 J H–H 1.2, 1H, –CH C(CH3)–], 7.63–7.71 (m, 4H, Ar-H), 7.92 [br s, 1H, –NH–C(O)CH3], 9.07 [br s, 1H, –C(O)–NH–C(O)–] δ C (50 MHz, CDCl3) 12.42, 24.82, 27.31, 38.88, 48.10, 60.94, 62.47, 111.37, 119.56, 128.14, 134.33, 139.83, 142.86, 151.37, 164.08, 168.99, 178.40. ν max (KBr) 3320m (NH), 1742s (C O), 1735s (C O), 1716s (C O), 1685s (C O), 1664m, 1592m, 1533m. HRMS m/z calcd for C21H28N4O7NaS (M+Na)+ 503.1571, found 503. 1595.
5.5.5. 1-[N-(4-Nitrobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-1H,3H-pyrimidin-2,4-dione (6a)
According to the general procedure, 6a was obtained from 4a (445 mg, 1 mmol) and uracil in the presence of TMSOTf. Chromatographic purification (chloroform/acetone, 9:1, v/v) gave 6a (343 mg, 76%) as a white solid; mp 213–215 °C. δ H (200 MHz, DMSO-d 6) 1.11 (s, 9H), 3.75 (t, 3 J H–H 5.2, 2H), 4.14 (t, 3 J H–H 5.2, 2H), 5.25 (s, 2H), 5.61 (d, 3 J H–H 8.0, 1H), 7.62 (d, 3 J H–H 8.0, 1H), 8.07–8.12 (m, 2H), 8.34–8.40 (m, 2H), 11.26 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.82, 38.13, 48.20, 60.85, 61.76, 101.59, 124.67, 128.32, 144.65, 144.93, 149.91, 151.04, 163.24, 177.20 ν max (KBr) 1731s (C O), 1716s (C O), 1707s (C O), 1685s, 1533s (NO2), 1349s (NO2). HRMS m/z calcd for C18H22N4O8NaS (M+Na)+ 477.1051, found 477.1068.
5.5.6. 1-[N-(3-Nitrobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-1H,3H-pyrimidin-2,4-dione (6b)
According to the general procedure, 6b was obtained from 4b (1.25 g, 2.8 mmol) and uracil in the presence of TMSOTf. Chromatographic purification (chloroform/acetone, 95:5, v/v) gave 6b (971 mg, 76%) as a white solid; mp 142–143 °C. δ H (200 MHz, DMSO-d 6) 1.11 (s, 9H), 3.77–3.79 (m, 2H), 4.12–4.17 (m, 2H), 5.27 (s, 2H), 5.60 (d, 3 J H–H 8.0, 1H), 7.65 (d, 3 J H–H 8.0, 1H), 7.86–7.94 (m, 1H), 8.25–8.30 (m, 1H), 8.45–8.55 (m, 2H), 11.25 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.82, 38.13, 48.24, 60.98, 61.91, 101.54, 121.46, 127.85, 131.62, 132.70, 141.11, 144.73, 147.86, 151.04, 163.17, 177.19. ν max (KBr) 1729s (C O), 1715s (C O), 1703s (C O), 1683s, 1534s (NO2), 1355s (NO2,). HRMS m/z calcd for C18H22N4O8NaS (M+Na)+ 477.1051, found 477.1065.
5.5.7. 1-[N-(2-Nitrobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-1H,3H-pyrimidin-2,4-dione (6c)
According to the general procedure, 6c was obtained from 4c (445 mg, 1 mmol) and uracil in the presence of TMSOTf. Chromatographic purification (chloroform/acetone, 95:5, v/v) gave 6c (245 mg, 54%) as a white solid; mp 145–146 °C. δ H (200 MHz, DMSO-d 6) 1.09 (s, 9H), 3.78–3.83 (m, 2H), 4.09–4.15 (m, 2H), 5.35 (s, 2H), 5.53 (d, 3 J H–H 8.0, 1H), 7.44 (d, 3 J H–H 8.0, 1H), 7.79–8.05 (m, 4H), 11.31 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.80, 38.12, 47.71, 60.00, 61.39, 101.78, 124.85, 129.27, 131.96, 132.86, 135.01, 143.98, 147.46, 151.32, 163.18, 177.20. ν max (KBr) 1737s (C O), 1704s (C O), 1686s (C O), 1656s, 1541s (NO2), 1367s (NO2). HRMS m/z calcd for C18H22N4O8NaS (M+Na)+ 477.1051, found 477.1070.
5.5.8. 1-[N-4-(Acetamidobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-1H,3H-pyrimidin-2,4-dione (6d)
According to the general procedure, 6d was obtained from 4d (228 mg, 0.5 mmol) and uracil in the presence of TMSOTf. Chromatographic purification (chloroform/acetone, 8:2, v/v) gave 6d (148 mg, 66%) as a white solid; mp > 121 °C (dec). δ H (200 MHz, CDCl3) 1.19 (s, 9H), 2.21 (s, 3H), 3.68 (t, 3 J H–H 5.6, 2H), 4.18 (t, 3 J H–H 5.6, 2H), 5.25 (s, 2H), 5.72 (d, 3 J H–H 8.2, 1H), 7.60 (d, 3 J H–H 8.2, 1H), 7.63–7.70 (m, 5H), 8.97 (br s, 1H, NH) δ C (50 MHz, CDCl3) 24.88, 27.34, 38.87, 48.05, 61.23, 62.47, 102.85, 119.63, 128.20, 134.25, 142.80, 144.17, 151.12, 163.12, 168.85, 178.37. ν max (KBr) 3318m (NH), 1748s (C O), 1731s (C O), 1712s (C O), 1681s (C O), 1632m, 1592m, 1533m. HRMS m/z calcd for C20H26N4O7NaS (M+Na)+ 489.1414, found 489.1438.
5.5.9. 1-[N-(4-Nitrobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-fluoro-1H,3H-pyrimidin-2,4-dione (7a)
According to the general procedure, 7a was obtained from 4a (445 mg, 1 mmol) and 5-fluorouracil in the presence of TMSOTf. Crystallization (methanol) gave 7a (275 mg, 58%) as a white solid; mp 154–155 °C. δ H (200 MHz, DMSO-d 6) 1.10 (s, 9H), 3.73–3.78 (m, 2H), 4.12–4.17 (m, 2H), 5.22 (s, 2H), 7.94 (d, 3 J H–F 6.6, 1H), 8.10–8.15 (m, 2H), 8.38–8.42 (m, 2H), 11.84 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.82, 38.16, 48.34, 61.10, 61.56, 124.73, 128.44, 128.81 (d, 2 J C–F 34.9), 139.47 (d, 1 J C–F 229.9), 144.85, 149.74, 149.98, 157.15 (d, 2 J C–F 25.8), 177.20. ν max (KBr) 1718s (C O), 1705s (C O), 1666s (C O), 1537s (NO2), 1349s (NO2). HRMS m/z calcd for C18H21N4O8FNaS (M+Na)+ 495.0956, found 495.0937.
5.5.10. 1-[N-(3-Nitrobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-fluoro-1H,3H-pyrimidin-2,4-dione (7b)
According to the general procedure, 7b was obtained from 4b (445 mg, 1 mmol) and 5-fluorouracil in the presence of TMSOTf. Chromatographic purification (chloroform/acetone, 85:15, v/v) gave 7b (322 mg, 68%) as a white solid; mp 132–133 °C. δ H (200 MHz, CDCl3) 1.13 (s, 9H), 3.68–3.74 (m, 2H), 4.16–4.21 (m, 2H), 5.30 (s, 2H), 7.74–7.83 (m, 2H), 8.11–8.16 (m, 1H), 8.41–8.47 (m, 1H), 8.52–8.54 (m, 1H), 9.93 (br s, 1H, NH). δ C (50 MHz, CDCl3) 27.17, 38.79, 48.10, 61.34, 61.67, 121.94, 127.86 (d, 2 J C–F 33.4), 128.02, 131.25, 132.54, 140.67 (d, 1 J C–F 238.6), 141.60, 148.57, 150.21, 157.19 (d, 2 J C–F 26.6), 178.28. ν max (KBr) 1724s (C O), 1703s (C O), 1676s (C O), 1534s (NO2), 1353s (NO2). HRMS m/z calcd for C18H21N4O8FNaS (M+Na)+, 495.0956, found 495.0974.
5.5.11. 1-[N-(2-Nitrobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-fluoro-1H,3H-pyrimidin-2,4-dione (7c)
According to the general procedure, 7c was obtained from 4c (445 mg, 1 mmol) and 5-fluorouracil in the presence of TMSOTf. Chromatographic purification (chloroform/acetone, 95:5, v/v) gave 7c (329 mg, 70%) as a white solid; mp 162–163 °C. δ H (200 MHz, DMSO-d 6) 1.10 (s, 9H), 3.80–3.86 (m, 2H), 4.12–4.17 (m, 2H), 5.32 (s, 2H), 7.69–7.72 (m, 1H), 7.78–7.95 (m, 2H), 8.01–8.05 (m, 2H), 11.84 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.79, 38.15, 47.96, 60.25, 61.26, 124.79, 128.20 (d, 2 J C–F 33.8), 129.32, 131.96, 132.86, 135.03, 139.38 (d, 1 J C–F 230.3), 147.45, 149.95, 157.10 (d, 2 J C–F 25.8), 177.21. ν max (KBr) 1724s (C O), 1694s (C O), 1666s (C O), 1542s (NO2), 1368s (NO2). HRMS m/z calcd for C18H21N4O8FNaS (M+Na)+ 495.0956, found 495.0970.
5.5.12. 1-[N-(4-Acetamidobenzenesulfonyl]-N-(2-pivaloyloxyethyl)aminomethyl]-5-fluoro-1H,3H-pyrimidin-2,4-dione (7d)
According to the general procedure, 7d was obtained from 4d (228 mg, 0.5 mmol) and 5-fluorouracil in the presence of tin(IV) chloride. Chromatographic purification (chloroform/methanol, 98:2, v/v) gave 7d (127 mg, 52%) as a white solid; mp 168 °C (subl.). δ H (200 MHz, DMSO-d 6) 1.09 (s, 9H), 2.09 (s, 3H), 3.60–3.65 (m, 2H), 4.07–4.12 (m, 2H), 5.16 (s, 2H), 7.71–7.77 (m, 4H), 7.86 (d, 3 J H–F 6.4, 1H), 10.38 (br s, 1H, NH), 11.89 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 24.18, 26.78, 47.78, 61.03, 61.70, 118.61, 127.97, 128.74 (d, 2 J C–F 34.55), 132.60, 139.35 (d, 1 J C–F 230.60), 143.66, 149.82, 157.19 (d, 2 J C–F 25.45), 169.10, 177.14. ν max (KBr) 3383m (NH), 1742s (C O), 1719s (C O), 1705s (C O), 1678s (C O), 1591m, 1533m. HRMS m/z calcd for C20H25N4O7FNaS (M+Na)+ 507.1320, found 507.1342.
5.6. General method for the reduction of the thymine derivatives 5a–c with sodium dithionite in alkaline medium
A mixture of 5a–c (0.4 mmol), sodium dithionite (350 mg, 2 mmol) and 4% aqueous solution of sodium hydroxide (20 mL) was heated at 90 °C for 1 h. Then, the mixture was cooled to room temperature, neutralized with an aqueous hydrochloric acid (5%), and extracted with ethyl acetate (5× 10 mL). The extracts were combined, washed with brine (5 mL) and dried. The solvent was distilled off. The residue was purified by column chromatography (chloroform/methanol/NH3 aq, 95:5:0.1, v/v/v) to give 8a–c.
5.6.1. 1-[N-(4-Aminobenzenesulfonyl)-N-(2-hydroxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (8a)
Method A. According to the general procedure, 8a was obtained from 5a (100 mg, 0.2 mmol). Chromatographic purification afforded 8a (42 mg, 59%) as a white solid; mp 179–180 °C. δ H (DMSO-d 6, 200 MHz) 1.75 (s, 3H), 3.19–3.28 (m, 2H), 3.39–3.45 (m, 2H), 4.76 (t, 3 J H–H 5.2, 1H, OH), 5.07 (s, 2H), 6.09 (br s, 2H, NH 2), 6.57–6.62 (m, 2H), 7.36–7.44 (m, 2H), 7.36 (br s, 1H), 11.29 (br s, 1H, NH). δ C (DMSO-d 6, 50 MHz) 11.89, 50.31, 59.53, 60.27, 108. 45, 112.56, 123.66, 128.58, 139.92, 150.89, 153.02, 163.81. ν max (KBr) 3435m (NH), 3374m (OH), 3335m (NH), 1719 (C O), 1687 (C O), 1630m, 1361s (SO2), 1151s (SO2). HRMS m/z calcd for C14H18N4O5NaS (M+Na)+ 377.0890, found 377.0898.
Method B. (Scheme 5). A mixture of 5d (47 mg, 0.1 mmol) and an aqueous sodium hydroxide (4%, 3 mL) was heated at 90 °C for 2 h. Then, the mixture was cooled to room temperature, neutralized with aqueous hydrochloric acid (5%), and extracted with ethyl acetate (5× 5 mL). The extracts were combined, washed with brine (5 mL) and dried. The solvent was distilled off. The residue was purified by column chromatography (chloroform/methanol/NH3 aq, 95:5:0.1, v/v/v) to afford 8a (14 mg, 39%).
5.6.2. 1-[N-(3-Aminobenzenesulfonyl)-N-(2-hydroxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (8b)
According to the general procedure, 8b was obtained from 5b (143 mg, 0.3 mmol). Chromatographic purification afforded 8b (74 mg, 52%) as a white solid; mp 168–169 °C. δ H (200 MHz, DMSO-d 6) 1.74 (s, 3H), 3.28–3.47 (m, 4H), 4.82 (t, 3 J H–H 5.0, 1H, OH), 5.12 (s, 2H), 5.61 (br s, 2H, NH 2), 6.77–6.87 (m, 2H), 6.98 (s, 1H), 7.15–7.23 (m, 1H), 7.33 (m, 1H), 11.30 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6), 12.09, 50.76, 59.77, 60.45, 108.89, 110.98, 113.01, 117.89, 129.84, 139.95, 139.97, 149.66, 151.15, 164.02. ν max (KBr) 3471m (NH), 3405m (OH), 3362m (NH), 1716s (C O), 1685s (C O), 1640m, 1361s (SO2), 1151s (SO2). HRMS m/z calcd for C14H18N4O5NaS (M+Na)+ 377.0890, found 377.0889.
5.6.3. 1-[N-(2-Aminobenzenesulfonyl)-N-(2-hydroxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (8c)
Method A. According to the general procedure, 8c was obtained from 5c (187 mg, 0.4 mmol). Chromatographic purification afforded 8c (7 mg, 5%) as a white solid; mp 138–139 °C. δ H (200 MHz, DMSO-d 6) 1.71 (s, 3H), 3.14–3.57 (m, 4H), 4.80 (br s, 1H, OH), 5.21 (s, 2H), 6.02 (br s, 2H, NH 2), 6.58–6.66 (m, 1H), 6.82–6.86 (m, 1H), 7.26–7.31 (m, 2H), 7.46–7.50 (m, 1H), 11.00 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 12.05, 49.94, 59.32, 59.97, 108.73, 115.54, 117.45, 118.74, 129.26, 134.33, 139.92, 146.89, 151.20, 164.00. ν max (KBr) 3471m (NH), 3406m (OH), 3351m (NH), 1714s (C O), 1386s (C O), 1625m, 1361s (SO2), 1143s (SO2). HRMS m/z calcd for C14H18N4O5NaS (M+Na)+ 377.0890, found 377.0903.
Method B. A mixture of 9c (107 mg, 0.24 mmol), concentrated ammonium hydroxide (5 mL), and methanol (5 mL) was heated in a sealed tube at 70 °C for 1 day. The volatiles were evaporated to dryness under reduced pressure. The residue was purified by column chromatography (chloroform/methanol, 95:5, v/v) to afford 8c (68 mg, 79%).
5.7. General method for the reduction of the thymine derivatives 5a–c with sodium dithionite under neutral conditions
A mixture of 5a–c (0.3 mmol), sodium dithionite (260 mg, 1.5 mmol) and water (15 mL) was heated at 90 °C for 1 h. The mixture was cooled to room temperature and extracted with ethyl acetate (5× 10 mL). The extracts were combined, washed with brine (5 mL) and dried. The solvent was distilled off. The residue was purified by column chromatography (chloroform/methanol, 95:5, v/v) to yield 9a–c.
5.7.1. 1-[N-(4-Aminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (9a)
According to the general procedure, 9a was obtained from 5a (140 mg, 0.3 mmol). Chromatographic purification afforded 9a (70 mg, 58%) as a white solid; mp 186–190 °C (dec). δ H (200 MHz, CDCl3) 1.17 (s, 9H), 1.91 (d, 4 J H–H 1.0, 3H), 3.62 (d, 3 J H–H 5.4, 2H), 4.15 (d, 4 J H–H 5.4, 2H), 4.26 (br s, 2H, NH 2), 5.21 (s, 2H), 6.62–6.66 (m, 2H), 7.47–7.51 (m, 2H), 7.37 (d, 4 J H–H 1.0, 1H), 9.88 (br s, 1H, NH). δ C (50 MHz, CDCl3) 12.43, 22.27, 38.82, 47.59, 60.65, 62.41, 111.34, 114.21, 127.45, 129.11, 139.77, 151.31, 151.40, 164.08, 178.30. ν max (KBr) 3483m (NH), 3381m (NH), 1730s (C O), 1717s (C O), 1663s (C O), 1630m, 1594m, 1314s (SO2), 1142s (SO2). HRMS m/z calcd for C19H26N4O6NaS (M+Na)+ 461.1465, found 461.1488.
5.7.2. 1-[N-(3-Aminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (9b)
According to the general procedure, 9b was obtained from 5b (141 mg, 0.3 mmol). Chromatographic purification afforded 9b (71 mg, 54%) as a white solid; mp > 163 °C (dec). δ H (200 MHz, DMSO-d 6) 1.11 (s, 9H), 1.72 (s, 3H), 3.56–3.61 (m, 2H), 4.08–4.13 (m, 2H), 5.16 (s, 2H), 5.62 (br s, 2H, NH 2), 6.77–6.87 (m, 2H), 6.98 (br s, 1H), 7.14–7.26 (m, 2H), 11.34 (s, 1H, NH). δ C (50 MHz, DMSO-d 6) 12.03, 26.81, 38.13, 47.29, 59.94, 61.70, 109.00, 110.87, 112.88, 117.96, 129.88, 139.82, 139.97, 149.72, 151.24, 163.88, 177.20. ν max (KBr) 3404m (NH), 3353m (NH), 1729s (C O), 1713s (C O), 1686s (C O), 1628m, 1599m, 1339s (SO2), 1157s (SO2). HRMS m/z calcd for C19H26N4O6NaS (M+Na)+ 461.1465, found 461.1471.
5.7.3. 1-[N-(2-Aminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (9c)
According to the general procedure, 9c was obtained from 5c (187 mg, 0.4 mmol). Chromatographic purification afforded 9c (97 mg, 56%) as a white solid; mp 137–138 °C. δ H (200 MHz, DMSO-d 6) 1.11 (s, 9H), 1.68 (d, 4 J H–H 1.0, 3H), 3.61–3.66 (m, 2H), 4.01–4.07 (m, 2H), 5.24 (s, 2H), 6.02 (br s, 2H, NH 2), 6.59–6.66 (m, 1H), 6.82–6.86 (m, 1H), 7.20 (q, 4 J H–H 1.0, 1H), 7.26–7.30 (m, 1H), 7.46–7.50 (m, 1H), 11.26 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 12.01, 26.83, 38.12, 46.69, 59.63, 61.75, 108.82, 115.66, 117.54, 118.80, 129.15, 134.44, 139.78, 146.86, 151.33, 163.90, 177.22. ν max (KBr) 3475m (NH), 3369m (NH), 1733s (C O), 1719s (C O), 1691s (C O), 1621m, 1599m, 1324s (SO2), 1144s (SO2). HRMS m/z calcd for C19H26N4O6NaS (M+Na)+ 461.1465, found 461.1471.
5.8. General method for the palladium-catalysed transfer hydrogenation of derivatives 6a–c or 7a–c
A mixture of 6a–c or 7a–c (200 mg), cyclohexene (8 mL), palladium on charcoal (10% Pd/C, 100 mg), and a solvent (ethanol, methanol, or 1,4-dioxane; 15 mL) was heated in a sealed tube at 60 °C for 1 day under an argon atmosphere. The catalyst was filtered off, and the filtrate was concentrated to dryness. The residue was purified by column or preparative thin-layer chromatography to afford the corresponding products (see Scheme 3, Scheme 4, Scheme 5); the eluting solvents are given in parentheses below.
5.8.1. 1-[N-(4-Aminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-1H,3H-pyrimidin-2,4-dione (10a)
According to the general procedure, 10a was obtained from 6a (100 mg, 0.22 mmol) in ethanol. Column chromatography (chloroform/acetone, 95:5, v/v) provided 10a (54 mg, 58%) as a white solid; mp 147–149 °C. δ H (200 MHz, DMSO-d 6) 1.18 (s, 9H), 3.59–3.65 (m, 2H), 4.13–4.18 (m, 2H), 4.24 (br s, 2H, NH 2), 5.24 (s, 2H), 5.73 (m, 3 J H–H 8.0, 1H), 6.62–6.68 (m, 2H), 7.46–7.54 (m, 2H), 7.64 (d, 3 J H–H 8.0, 1H), 8.74 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 27.31, 38.83, 47.66, 61.02, 62.49, 102.81, 114.30, 127.29, 129.16, 144.17, 151.22, 151.34 163.24, 178.27. ν max (KBr) 3483m (NH), 3389m (NH), 1723s (C O), 1710s (C O), 1687s (C O), 1629m, 1598m, 1324s (SO2), 1146s (SO2). HRMS m/z calcd for C18H24N4O6NaS (M+Na)+ 447.1309, found 447.1330.
5.8.2. 1-[N-(3-Aminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-1H,3H-pyrimidin-2,4-dione (10b)
According to the general procedure, 10b was obtained from 6b (200 mg, 0.44 mmol) in ethanol. Column chromatography (chloroform/acetone, 98:2, v/v) provided 10b (117 mg, 62%) as a white solid; mp 147–148 °C. δ H (200 MHz, DMSO-d 6) 1.11 (s, 9H), 3.55–3.60 (m, 2H), 4.06–4.11 (m, 2H), 5.18 (s, 2H), 5.6–5.64 (m, 3H), 6.79–6.87 (m, 2H), 6.92–6.98 (m, 1H), 7.16–7.23 (m, 1H), 7.56 (d, 3 J H–H 7.8, 1H), 11.30 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.84, 38.14, 47.53, 60.63, 61.86, 101.47, 110.96, 112.85, 118.00, 129.93, 139.69, 144.45, 149.70, 151.26, 163.36, 177.19. ν max (KBr) 3373m (NH), 3305m (NH), 1740s (C O), 1696s (C O), 1679s (C O), 1633m, 1599m, 1356s (SO2), 1155s (SO2). HRMS m/z calcd for C18H24N4O6NaS (M+Na)+ 447.1309, found 447.1330.
5.8.3. 1-[N-(2-Aminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-1H,3H-pyrimidin-2,4-dione (10c)
According to the general procedure, 10c was obtained from 6c (200 mg, 0.44 mmol) in ethanol. Column chromatography (chloroform/acetone, 95:5, v/v) provided 10c (168 mg, 89%) as a white solid; mp 146–147 °C. δ H (200 MHz, DMSO-d 6) 1.10 (s, 9H), 3.57–3.63 (m, 2H), 3.97–4.02 (m, 2H), 5.30 (s, 2H), 5.58 (d, 3 J H–H 8.0, 1H), 6.03 (s, 2H, NH 2), 6.59–6.66 (m, 1H), 6.8–6.87 (m, 1H), 7.27–7.35 (m, 1H), 7.46–7.50 (m, 1H), 7.56 (d, 3 J H–H 8.0, 1H), 11.32 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.84, 38.10, 46.68, 60.31, 61.84, 101.39, 115.70, 117.65, 118.55, 129.13, 134.50, 144.43, 146.93, 151.39, 163.39, 177.18. ν max (KBr) 3471m (NH), 3373m (NH), 1717s (C O), 1702s (C O), 1695s (C O), 1670m, 1618m, 1340s (SO2), 1155s (SO2). HRMS m/z calcd for C18H24N4O6NaS (M+Na)+ 447.1309, found 447.1287.
5.8.4. 1-[N-(4-Aminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-fluoro-1H,3H-pyrimidin-2,4-dione (11a)
According to the general procedure, 11a was obtained from 7a (50 mg, 0.11 mmol) in 1,4-dioxane. Preparative thin-layer chromatography (chloroform/acetone, 85:15, v/v) provided 11a (30 mg, 64%) as a white solid; mp 153–155 °C. δ H (200 MHz, DMSO-d 6) 1.10 (s, 9H), 3.50–3.57 (m, 2H), 4.03–4.10 (m, 2H), 5.10 (s, 2H), 6.14 (s, 2H, NH 2), 6.58–6.62 (m, 2H), 7.42–7.46 (m, 2H), 7.82 (d, 3 J H–F 6.8, 1H), 11.67 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.84, 38.15, 47.53, 61.04, 61.92, 112.79, 123.45, 128.26, (d, 2 J C–F 32.7), 128.84, 138.83 (d, 1 J C–F 228.8), 149.86, 153.50, 157.24 (d, 2 J C–F 25.8), 177.21. ν max (KBr) 3478m (NH), 3364m (NH), 1731s (C O), 1716s (C O), 1691s (C O), 1672m, 1619m, 1599m, 1330s (SO2), 1145s (SO2). HRMS m/z calcd for C18H23N4O6FNaS (M+Na)+ 465.1215, found 465.1235.
5.8.5. 1-[N-(3-Aminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-fluoro-1H,3H-pyrimidin-2,4-dione (11b)
According to the general procedure, 11b was obtained from 7b (50 mg, 0.11 mmol) in 1,4-dioxane. Preparative thin-layer chromatography (chloroform/acetone, 85:15, v/v) provided 11b (27 mg, 57%) as a white solid; mp 151–152 °C. δ H (200 MHz, DMSO-d 6) 1.11 (s, 9H), 3.58–3.62 (m, 2H), 4.07–4.12 (m, 2H), 5.16 (s, 2H), 5.63 (br s, 2H, NH 2), 6.78–6.89 (m, 2H), 6.91–7.06 (m, 1H), 7.16–7.23 (m, 1H), 7.77 (d, 3 J H–F 6.6, 1H), 11.89 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.86, 38.19, 47.79, 60.86, 61.68, 110.85, 112.89, 118.10, 128.53 (d, 2 J C–F 33.8), 129.98, 139.40 (d, 1 J C–F 229.5), 139.74, 149.81, 149.89, 157.45 (d, 2 J C–F 25.8), 177.24. ν max (KBr) 3365m (NH), 3315m (NH), 1738s (C O), 1710s (C O), 1691s (C O), 1669m, 1599m, 1355s (SO2), 1165s (SO2). HRMS m/z calcd for C18H23N4O6FNaS (M+Na)+ 465.1215, found 465.1224.
5.8.6. 1-[N-(2-Aminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-fluoro-1H,3H-pyrimidin-2,4-dione (11c)
According to the general procedure, 11c was obtained from 7c (50 mg, 0.11 mmol) in 1,4-dioxane. Preparative thin-layer chromatography (chloroform/acetone, 85:15, v/v) provided 11c (29 mg, 62%) as a white solid; mp > 170 °C (dec). δ H (400 MHz, DMSO-d 6) 1.09 (s, 9H), 3.64–3.66 (m, 2H), 3.99–4.01 (m, 2H), 5.24 (s, 2H), 6.03 (br s, 2H, NH 2), 6.60–6.64 (m, 1H), 6.83 (d, 3 J H–H 8.4, 1H), 7.27–7.31 (m, 1H), 7.48 (d, 3 J H–H 8.0, 1H), 7.74 (d, 3 J H–F 6.8, 1H), 11.89 (br s, 1H, NH). δ C (100 MHz, DMSO-d 6) 26.85, 38.14, 47.10, 60.60, 61.08, 115.75, 117.62, 118.69, 128.63, (d, 2 J C–F 33.4), 129.20, 134.58, 139.23 (d, 1 J C–F 229.8), 146.92, 150.01, 157.27 (d, 2 J C–F 25.8), 177.24. ν max (KBr) 3488m (NH), 3367m (NH), 1735s (C O), 1720s (C O), 1699s (C O), 1664m, 1629m, 1599m, 1324s (SO2), 1141s (SO2). HRMS m/z calcd for C18H23N4O6FNaS (M+Na)+ 465.1215, found 465.1222.
5.8.7. 1-[N-(4-Methylaminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-fluoro-1H,3H-pyrimidin-2,4-dione (12)
According to the general procedure, 12 was obtained from 7a (100 mg, 0.21 mmol in methanol. Column chromatography (chloroform/acetone, 95:5, v/v) provided 12 (43 mg, 45%) as a white solid; mp 157–158 °C. δ H (200 MHz, DMSO-d 6) 1.10 (s, 9H), 2.72 (d, 3 J H–H 5.0, 3H), 3.50–3.56 (m, 2H), 4.04–4.10 (m, 2H), 5.10 (s, 2H), 6.56–6.61 (m, 2H), 7.48–7.52 (m, 2H), 6.71 (q, 3 J H–H 5.0, 1H, NH), 7.82 (d, 3 J H–F 6.8, 1H), 11.48 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.81, 29.10, 38.11, 47.53, 60.98, 61.91, 110.80, 123.35, 128.64, (d, 2 J C–F 33.8), 128.69, 139.32 (d, 1 J C–F 229.1), 149.88, 153.53, 157.27 (d, 2 J C–F 25.4), 177.17. ν max (KBr) 3430m (NH), 1738s (C O), 1722s (C O), 1684s (C O), 1664m, 1599m, 1319s (SO2), 1150s (SO2). HRMS m/z calcd for C19H25N4O6FNaS (M+Na)+ 479.1371, found 479.1367.
5.8.8. 1-[N-(4-Dimethylaminobenzenesulfonyl)-N-(2-pivaloyloxyethyl)aminomethyl]-5-fluoro-1H,3H-pyrimidin-2,4-dione (13)
According to the general procedure, 13 was obtained from 7a (100 mg, 0.21 mmol in methanol. Column chromatography (chloroform/acetone, 95:5, v/v) provided 13 (15 mg, 15%) as a white solid; mp 164–165 °C. δ H (200 MHz, DMSO-d 6) 1.09 (s, 9H), 2.99 (s, 6H), 3.53–3.59 (m, 2H), 4.05–4.10 (m, 2H), 5.11 (s, 2H), 6.72–6.77 (m, 2H), 7.54–7.59 (m, 2H), 7.82 (d, 3 J H–F 6.8, 1H), 11.83 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 26.84, 38.18, 39.64, 47.68, 60.99, 61.95, 111.10, 123.75, 128.65 (d, 2 J C–F 33.6), 128.46, 139.33 (d, 1 J C–F 229.5), 149.87, 153.02, 157.30 (d, 2 J C–F 27.2), 177.22. ν max (KBr) 1735s (C O), 1715s (C O), 1694s (C O), 1671m, 1598m, 1319s (SO2), 1172s (SO2). HRMS m/z calcd for C20H27N4O6FNaS (M+Na)+ 493.1528, found 493.1552.
5.9. General procedure for the ammonolysis of 5d or 6d
A mixture of 5d or 6d, concentrated ammonium hydroxide, and methanol in the ratio of 0.5 mmol/10 mL/10 mL, respectively, was heated in a sealed tube at 70 °C for 1 day. The volatiles were evaporated to dryness under reduced pressure. The residue was purified by column chromatography (chloroform/methanol, 95:5, v/v) to give 14a or 14b, respectively.
5.9.1. 1-[N-(4-Acetamidobenzenesulfonyl)-N-(2-hydroxyethyl)aminomethyl]-5-methyl-1H,3H-pyrimidin-2,4-dione (14a)
According to the general procedure, 14a was obtained from 5d (136 mg, 0.28 mmol). Chromatographic purification afforded 14a (105 mg, 94%) as a white solid; mp > 219 °C (dec). δ H (200 MHz, DMSO-d 6) 1.73 (br s, 3H), 2.10 (s, 3H), 3.17–3.72 (m, 4H), 4.82 (br s, 1H, OH), 5.13 (s, 2H), 7.41 (br s, 1H), 7.69–7.85 (m, 5H), 10.80 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 12.06, 24.18, 50.72, 59.70, 60.53, 108.80, 118.63, 123.94, 132.89, 140.26, 143.68, 151.15, 164.06, 169.30. ν max (KBr) 3406s (OH), 3312m (NH), 1715s (C O), 1698s (C O), 1664s (C O), 1594m, 1537m, 1336s (SO2), 1155s (SO2). HRMS m/z calcd for C16H20N4O6NaS (M+Na)+ 419.0996, found 419.0998.
5.9.2. 1-[N-(4-Acetamidobenzenesulfonyl)-N-(2-hydroxyethyl)aminomethyl]-1H,3H-pyrimidin-2,4-dione (14b)
According to the general procedure, 14b was obtained from 6d (81 mg, 0.18 mmol). Chromatographic purification afforded 14b (69 mg, 85%) as a white solid; mp > 212 °C (dec). δ H (200 MHz, DMSO-d 6) 2.10 (s, 3H), 3.21–3.59 (m, 4H), 4.83 (br s, 1H, OH), 5.16 (s, 2H), 5.62 (d, 3 J H–H 8.0, 1H), 7.65 (d, 3 J H–H 8.0, 1H), 7.70–7.75 (m, 2H), 7.81–7.85 (m, 2H), 8.44 (br s, 1H, NH), 10.75 (br s, 1H, NH). δ C (50 MHz, DMSO-d 6) 24.05, 50.61, 59.53, 60.75, 101.21, 118.45, 127.75, 132.52, 143.46, 144.66, 150.94, 163.30, 169.04. ν max (KBr) 3394s (OH), 3327m (NH), 1718s (C O), 1698s (C O), 1671s (C O), 1591s, 1522s, 1154s (SO2), 1342s (SO2). HRMS m/z calcd for C15H18N4O6NaS (M+Na)+ 405.0839, found 405.0859.
Acknowledgments
The synthetic part of this work was financially supported by Warsaw University of Technology. We thank Dr. Wojciech Sas, Warsaw University of Technology, for his support, and fruitful discussions. We thank Leentje Persoons, Frieda De Meyer and Vicky Broeckx for excellent technical help, and the International Consortium for Antivirals (ICAV) and Fonds voor Wetenschappelijk Onderzoek Vlaanderen for financial support (Project No. G.0188.07).
References and notes
- 1.(a) De Clercq E. J. Clin. Virol. 2004;30:115. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]; (b) Galmarini C.M., Mackey J.R., Dumontet C. Lancet Oncol. 2002;3:415. doi: 10.1016/s1470-2045(02)00788-x. [DOI] [PubMed] [Google Scholar]; (c) Opio C.K., Lee W.M., Kirkpatrick P. Nat. Rev. Drug Disc. 2005;4:535. doi: 10.1038/nrd1780. [DOI] [PubMed] [Google Scholar]
- 2.(a) Johnson A.A., Ray A.S., Hanes J., Suo Z., Colacino J.M., Anderson K.S., Johnson K.A. J. Biol. Chem. 2001;276:40847. doi: 10.1074/jbc.M106743200. [DOI] [PubMed] [Google Scholar]; (b) De Clercq E. J. Antimicrob. Chemother. 2003;51:1079. doi: 10.1093/jac/dkg205. [DOI] [PubMed] [Google Scholar]; (c) Jeha S., Gandhi V., Chan K.W., McDonald L., Ramirez I., Madden R., Rytting M., Brandt M., Keating M., Plunkett W., Kantarjian H. Blood. 2004;103:784. doi: 10.1182/blood-2003-06-2122. [DOI] [PubMed] [Google Scholar]
- 3.Gumina G., Olgen S., Chu C.K. In: Antiviral Nucleosides: Chiral Synthesis and Chemotherapy. Chu C.K., editor. Elsevier B.V.; 2003. pp. 161–169. [Google Scholar]
- 4.(a) Markham A.F., Newton C.R., Porter R.A., Sim I.S. Antiviral Res. 1982;2:319. doi: 10.1016/0166-3542(82)90001-8. For 5′-deoxy-5′-sulfonamidofuranosyl nucleosides, see: [DOI] [PubMed] [Google Scholar]; (b) Cosstick R., Jones A.S., Walker R.T. Tetrahedron. 1984;40:427. [Google Scholar]; (c) Elliott R.D., Brockman R.W., Montgomery J.A. J. Med. Chem. 1986;29:1052. doi: 10.1021/jm00156a025. [DOI] [PubMed] [Google Scholar]; (d) Sim I.S., Picton C., Cosstick R., Jones A.S., Walker R.T., Chamiec A.J. Nucleosides Nucleotides. 1988;7:129. [Google Scholar]; (e) Martin J.A., Duncan I.B., Hall M.J., Wong-Kai-In P., Lambert R.W., Thomas G.J. Nucleosides Nucleotides. 1989;8:753. [Google Scholar]; (f) Homma H., Watanabe Y., Abiru T., Murayama T., Nomura Y., Matsuda A. J. Med. Chem. 1992;35:2881. doi: 10.1021/jm00093a022. [DOI] [PubMed] [Google Scholar]; (g) Criton M., Dewyntert G., Aouf N., Montero J.-L., Imbach J.-L. Nucleosides Nucleotides. 1995;14:1795. [Google Scholar]; (h) Sharma M., Li Y.X., Ledvina M., Bobek M. Nucleosides Nucleotides. 1995;14:1831. [Google Scholar]; (i) Urjasz W., Celewicz L., Golankiewicz K. Nucleosides Nucleotides. 1996;15:1189. [Google Scholar]; (j) Martin J.A., Thomas G.J., Merret J.H., Lambert R.W., Bushnel D.J., Dundson S.J., Freeman A.C., Hopkins R.A., Johns I.R., Keech E., Simmonite H., Wong-Kai-In P., Holland M. Antiviral Chem. Chemother. 1998;1:1. [PubMed] [Google Scholar]; (k) Scherman M.S., Winans K.A., Stern R.J., Jones V., Bertozzi C.R., McNeil M.R. Antimicrob. Agents Chemother. 2003;47:378. doi: 10.1128/AAC.47.1.378-382.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Vanada J., Bennet E., Wilson D., Bishoff H., Barry C., Aldrich C. Org. Lett. 2006;8:4707. doi: 10.1021/ol0617289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Van Calenbergh S., Van Den Eeckhout E., Herdewijn P., De Bruyn A., Verlinde C., Hol W., Callens M., Van Aerschot A., Rozenski J. Helv. Chim. Acta. 1994;77:631. For 3′-deoxy-3′-sulfonamidofuranosyl nucleosides, see: [Google Scholar]; (b) Yamana K., Ohashi Y., Nunota K., Nakano H. Tetrahedron. 1997;53:4265. [Google Scholar]
- 6.(a) Nair V., Walsh R.H. J. Org. Chem. 1974;39:3045. doi: 10.1021/jo00934a024. For N-(p-toluenesulfonyl)pyrrolidines, see: [DOI] [PubMed] [Google Scholar]; (b) Ng K.E., Orgel L.E. J. Med. Chem. 1989;32:1754. doi: 10.1021/jm00128a015. [DOI] [PubMed] [Google Scholar]; (c) Pickering L., Malhi B.S., Coe P.L., Walker R.T. Nucleosides Nucleotides. 1994;13:1493. [Google Scholar]; (d) Westwood N.B., Walker R.T. Tetrahedron. 1998;54:13391. [Google Scholar]; (e) Negwer, M.; Scharmow, H.-G. In Organic-Chemical Drugs and their Synonyms; Wiley-VCH Verlag GmbH, 2001, p 1454.
- 7.Krizmanić I., Višnjevac A., Luić M., Glavaš-Obrovac L., Žinić M., Žinić B. Tetrahedron. 2003;59:4947. and references cited therein. [Google Scholar]
- 8.(a) Scozzafava A., Owa T., Mastrolorenzo A., Supuran C.T. Curr. Med. Chem. 2003;10:925. doi: 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]; (b) Supuran C.T., Casini A., Scozzafava A. Med. Res. Rev. 2003;23:535. doi: 10.1002/med.10047. [DOI] [PubMed] [Google Scholar]; (c) Supuran C.T., Innocenti A., Mastrolorenzo A., Scozzafava A. Mini-Rev. Med. Chem. 2004;4:189. doi: 10.2174/1389557043487402. [DOI] [PubMed] [Google Scholar]
- 9.(a) Koszytkowska-Stawińska M., Sas W. Tetrahedron Lett. 2004;45:5437. [Google Scholar]; (b) Koszytkowska-Stawińska M., Sas W., De Clercq E. Tetrahedron. 2006;62:10325. [Google Scholar]; (c) Koszytkowska-Stawińska M., Kaleta K., Sas W., De Clercq E. Nucleosides Nucleotides Nucl. 2007;26:51. doi: 10.1080/15257770601052281. [DOI] [PubMed] [Google Scholar]; (d) Koszytkowska-Stawińska M., Kołaczkowska E., Adamkiewicz E., De Clercq E. Tetrahedron. 2007;63:10587. [Google Scholar]
- 10.(a) Pitman I.H. Med. Res. Rev. 1981;1:189. doi: 10.1002/med.2610010204. [DOI] [PubMed] [Google Scholar]; (b) Gao H., Mitra A. Synthesis. 2000:329. [Google Scholar]; (c) Anastasi C., Quelever G., Burlet S., Garino C., Souard F., Kraus J.-L. Curr. Med. Chem. 2003;10:1825. doi: 10.2174/0929867033457034. [DOI] [PubMed] [Google Scholar]; (d) Calogeropoulou T., Detsi A., Lekkas E., Koufaki M. Curr. Top. Med. Chem. 2003;3:1467. doi: 10.2174/1568026033451763. [DOI] [PubMed] [Google Scholar]; (e) De Clercq E., Field H.J. Br. J. Pharmacol. 2006;147:1. doi: 10.1038/sj.bjp.0706446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vorbrüggen, H.; Ruh-Pohlenz, C. In Handbook of Nucleoside Synthesis; John Wiley: New York, 2001; pp 29-33.
- 12.4-Acetamido-N-(2-pivaloyloxyethyl)-N-(acetamidomethyl)benzenesulfonamide: (36% yield); δH (200 MHz, CDCl3) 1.17 (s, 9H), 1.95 (s, 3H), 2.20 (s, 3H), 3.52 (t, 3JH–H 5.6, 2H), 4.19 (t, 3JH–H 5.6, 2H), 4.72 (d, 3JH–H 6.2, 2H), 6.55 (t, 3JH–H 6.2, 1H, NH), 7.62–7.68 (m, 2H), 7.70–7.74 (m, 2H), 8.12 (br s, 1H, NH). δC (50 MHz, CDCl3) 23.26, 24.77, 38.86, 46.65, 53.26, 62.20, 119.67, 128.30, 134.40, 142.64, 169.13, 170.98, 178.66. HRMS m/z calcd for C18H27N3O6NaS (M+Na)+ 436.1513, found 436.1528.
- 13.(a) Cristalli G., Volpini R., Vittorio S., Camaioni E., Rafaiani G., Potenza S., Vita A. Nucleosides Nucleotides. 1996;15:1567. Such competition of N-monosilylated or free acetamide with persilylated nucleobases for sugar cation upon the nucleosides synthesis has been reported. [Google Scholar]; (b) Ochoa C., Provensio R., Jimeno M.L., Balzarini J., De Clercq E. Nucleosides Nucleotides. 1998;17:901. doi: 10.1080/07328319808003462. [DOI] [PubMed] [Google Scholar]
- 14.(a) Watanabe K.A., Beránek J., Friedman H.A., Fox J.J. J. Org. Chem. 1965;30:2735. doi: 10.1021/jo01019a055. [DOI] [PubMed] [Google Scholar]; (b) Friedman H.A., Watanabe K.A., Fox J.J. J. Org. Chem. 1967;32:3775. doi: 10.1021/jo01287a010. [DOI] [PubMed] [Google Scholar]; (c) Matsuda A., Watanabe K.A. Nucleosides Nucleotides. 1996;15:205. [Google Scholar]; (d) Ohta N., Minamoto K., Yamamoto T., Koide N., Naoya S., Sakodo R. Nucleosides Nucleotides. 1996;15:833. [Google Scholar]; (e) Rozens E., Katkevica D., Bizdena E., Stroemberg R. J. Am. Chem. Soc. 2003;125:12125. doi: 10.1021/ja0360900. [DOI] [PubMed] [Google Scholar]; (f) Glinski R.P., Sporn M.B. Biochemistry. 1972;11:405. doi: 10.1021/bi00753a017. [DOI] [PubMed] [Google Scholar]; (g) Liu B., Hu L. Bioorg. Med. Chem. 2003;11:3889. doi: 10.1016/s0968-0896(03)00426-7. [DOI] [PubMed] [Google Scholar]; (h) Baer H.H., Bayer M. Can. J. Chem. 1971;49:568. [Google Scholar]
- 15.(a) Bigge C.F., Kalaritis P., Deck J.R., Mertes M.P. J. Am. Chem. Soc. 1980;102:2033. For the reduction of nucleoside analogues with the nitro group in a nucleobase moiety, see: [Google Scholar]; (b) Dziwiszek K., Schinazi R.F., Chou T.-C., Su T.-L., Dzik J. Nucleosides Nucleotides. 1994;13:77. [Google Scholar]; (c) De Riccardis F., Bonala R.R., Johnson F. J. Am. Chem. Soc. 1999;121:10453. [Google Scholar]; (d) Chakraborti D., Colis L., Schneider R., Bau A. Org. Lett. 2003;5:2861. doi: 10.1021/ol034904b. [DOI] [PubMed] [Google Scholar]; (e) Takamura-Enya T., Enomoto S., Wakabayashi K. J. Org. Chem. 2006;71:5599. doi: 10.1021/jo0605243. [DOI] [PubMed] [Google Scholar]
- 16.(a) Johnstone R.A.W., Wilby A.H. Chem. Rev. 1985;85:129. For reviews, see: [Google Scholar]; (b) Rylander, P.N. In Hydrogenation Methods; Academic Press, 1985; pp 16-17, 104–107, and 163.
- 17.The 11a:10a ratio was estimated from the 1H NMR spectrum of the mixture by the comparison of the intensities of the signals corresponding to the H-6 proton of 10a [7.64 (d, 3JH–H 8.0 Hz)] and to the H-6 one of 11a [7.82 (d, 3JH–F 6.8 Hz)].
- 18.(a) Duschinsky R., Pleven E. J. Am. Chem. Soc. 1957;79:4559. [Google Scholar]; (b) Colla L., Herdewijn P., De Clercq E., Balzarini J., Vanderhaeghe H. Eur. J. Med. Chem. Chim. Ther. 1985;20:295. [Google Scholar]
- 19.The reference antivirals displayed the following MCC values: (a) Vero cells: brivudin, >250 μM; [(S)-DHPA], >250 μM; ribavirin, >250 μM; (b) HEL cells: brivudin, >250 μM; ribavirin, >250 μM; acyclovir, >250 μM; ganciclovir, >100 μM; (c) HeLa cells: brivudin, >250 μM; (S)-9-(2,3-dihydroxy-propyl)adenine [(S)-DHPA], >250 μM; ribavirin, >250 μM; (d) MDCK cells: oseltamivir carboxylate, >100 μM; ribavirin, 100 μM; amantadin, >100 μM; rimantadin, >100 μM.
- 20.The reference antivirals displayed the following CC50 values: (a) MDCK cells: oseltamivir carboxylate, >100 μM; ribavirin, >100 μM; amantadin, >100 μM; rimantadin, >100 μM; (b) CRFK cells: hippeastrum hybrid agglutinin (HHA), >100 μg/mL; urtica dioica agglutinin (UDA), >100 μg/mL; ganciclovir, >100 μM.
- 21.The estimated values indicate that in the case of both the host cell cultures, cytotoxicity of the 5-fluorouracil nitro derivatives decreases in the following order: 2-NO2 isomer > 4-NO2 isomer > 3-NO2 isomer. However, based on our current knowledge, it is difficult to discuss on the relationship between NO2-isomerism and cytotoxicity of the compounds.
- 22.(a) De Clercq E., Descamps J., Verhelst G., Walker R.T., Jones A.S., Torrence P.F., Shugar D. J. Infect. Dis. 1980;141:563. doi: 10.1093/infdis/141.5.563. [DOI] [PubMed] [Google Scholar]; (b) De Clercq E., Cools M., Balzarini J., Marquez V.E., Borcherding D.R., Borchardt R.T., Drach J.C., Kitaoka S., Konno T. Antimicrob. Agents Chemother. 1989;33:1291. doi: 10.1128/aac.33.8.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]