

Abstract
We have studied T-cell apoptosis in animal models of human autoimmune disorders of the nervous system and in other tissues devoid of specialized immune-defense mechanisms. Our data suggest that the CNS has high potential for elimination of T-cell-dependent inflammation, whereas this mechanism is less effective in the PNS, and is almost absent in other tissues such as muscle and skin. Interestingly, several conventional and novel immunotherapeutic approaches, such as glucocorticosteroid and high-dose antigen therapy, induce T-cell apoptosis in situ. In vitro experiments suggest different scenarios for the mechanisms by which specific cellular and humoral elements in the nervous system synergize and sensitize T cells for apoptosis in vivo. We also discuss regulatory, proapoptotic mechanisms, such as the Fas–FasL system and galectin-1, that have been utilized in other tissues to mediate immune protection.
Keywords: T cell, experimental autoimmune neuritis, Multiple sclerosis, myositis, Fas
The concept of immune privilege was originally defined as the protection of tissue grafted to certain sites[1] and was later extended to describe the seclusion of particular areas of the body from the systemic immune compartment. However, this concept was abandoned more than a decade ago when it was shown that immune surveillance is indeed operative in such tissues. Even the CNS, previously considered the prototypic privileged site, is constantly patrolled by activated T lymphocytes, which can induce profound damage when they `find' their specific antigen in the context of appropriate restriction molecules (reviewed in [2]). This particular example illustrates the fact that specialized anatomic barriers, such as the blood–brain barrier[3] or the absence of lymphatic drainage, do not necessarily guarantee the absence of immune-mediated damage in these sites4, 5. Furthermore, it has become clear that T-cell trafficking within the CNS can be uncoupled from the development of diseases[6] and that under certain conditions, delayed-type hypersensitivity reactions can give inflammatory lymphocytes access to the CNS ([7]) and even provide therapeutic approaches[8]. Examples of other tissues that utilize specialized immunological defense mechanisms are the retina, the cornea, the anterior chamber of the eye, the testis, and the liver[9]. However, it is likely that in every tissue multiple factors operate that ensure rapid and gentle elimination of inflammation.
In 1972, Kerr and colleagues used morphological criteria to define apoptosis as a specialized form of cell death[10]. Typically, chromatin condensation and cell shrinkage occur in parallel but the integrity of the cell membrane is preserved much longer. Traditionally, apoptotic cells were identified by electron microscopy[11], but new techniques, such as in situ tailing assays, have recently become available12, 13 that detect biochemical events associated with oligonucleosomal DNA fragmentation in apoptotic cells. Although apoptosis is a purely morphological term, it has an array of pathophysiological and functional implications. The importance of apoptosis for the immune system has been reviewed in detail by Cohen and colleagues[14] who have clearly differentiated apoptosis from programmed cell death, which is merely a functional term. Since the plasma membrane itself is not a primary target of apoptosis but rather provides specific signals for phagocytic cells[15], release of proinflammatory cytoplasmic constituents is avoided. It is conceivable that in vulnerable organs, such as the CNS, the parenchyma is extremely susceptible and has low capacity for regeneration. Theoretically, apoptosis would provide an ideal, non-inflammatory mechanism to terminate the autoimmune T-cell attack in vulnerable tissues, assuring a minimum of detrimental bystander damage to the local parenchyma. Do immunologically protected sites take advantage of this mechanism of cell death?
1. Apoptosis of T lymphocytes in inf lamed nervous tissue but not in muscle or skin
Experimental autoimmune encephalomyelitis (EAE) and neuritis (EAN) serve as animal models for the human diseases multiple sclerosis (MS) and Guillain-Barré syndrome16, 17, 18. Both models can be induced by immunization with myelin antigen (active disease) or by intravenous transfer of CD4+ T lymphocytes specific for myelin antigens [adoptive-transfer (AT) model].
Pender and colleagues were the first to draw attention to apoptosis as a possible mechanism of cell destruction in inflammatory brain lesions of Lewis rat EAE ([19]). On morphological grounds, they suggested that most of the apoptotic cells were oligodendrocytes or lymphocytes. Later, T-cell apoptosis was identified by Pender et al. through special pre-embedding techniques[20], and by Schmied and coworkers through combining T-cell immunocytochemistry and molecular labeling techniques[21]. Using the latter approach, we also defined quantitatively the cells that underwent apoptosis in different EAE models in the Lewis rat[21]. Myelin basic protein (MBP) was used as an autoantigen. Of all cells undergoing apoptosis, 64% were identified as T lymphocytes (Fig. 1 C,D), 9% reacted with monoclonal antibodies specific for oligodendrocytes, and the remaining cells could not be stained by immunocytochemistry because they were already in an advanced stage of degeneration (Fig. 1A,B). Apoptosis of other cell types such as macrophages occurred only to a negligible extent21, 22. During the time course of AT-EAE, T-cell apoptosis was rarely observed at early stages on day 4, but reached up to 49% at day 7 when animals had recovered from clinical disease (Fig. 2 ). Similarly, in active EAE T-cell apoptosis was most developed when animals had recovered from disease (Fig. 2). In the meantime T-cell apoptosis has been shown to be similar in encephalomyelitis induced by coronavirus[23]. For obvious reasons, similar systematic studies cannot be performed in human disease. However, from diagnostic brain biopsies and postmortem tissue it is known that apoptotic T cells also occur in MS lesions[24]. Results from studies in patients with acute human disseminated leukoencephalitis are expected soon.
Fig. 1.
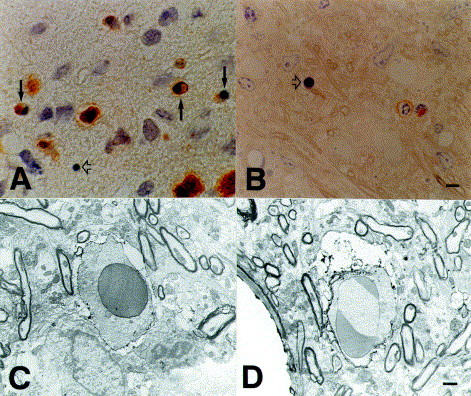
Immunocytochemical characterization of apoptotic cells in Lewis rat autoimmune encephalomyelitis (EAE). (A)
W3/13-positive T cells in the spinal cord. Some of these are in different stages of apoptotic cell death (arrows). In advanced stages of apoptosis, membrane immunoreactivity has been lost (open arrow). (B) 0.5 μm-thick plastic sections after pre-embedding immunocytochemistry for α/β T-cell receptor. Open arrow denotes an apoptotic cell. (C and D) Electron micrographs showing apoptotic T cells with positive staining for α/β T-cell receptor in (C) or common leukocyte antigen (D). Scale bars, 5 μm (A and B), 1 μm (C and D). (C and D) Reproduced from [21] with kind permission of the Am. J. Pathol.
Fig. 2.
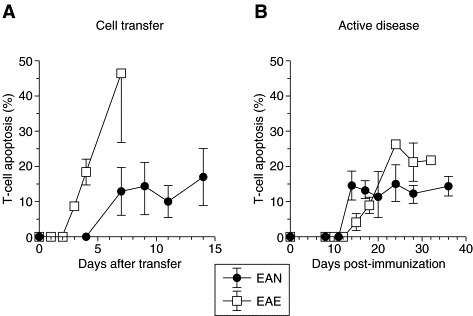
T-cell apoptosis in situ.
Apoptosis during the natural course of experimental autoimmune encephalomyelitis (EAE) or neuritis (EAN) that was induced by cell transfer (A) or by immunization with myelin basic protein (EAE) or peripheral nerve myelin (EAN) (B). Ordinate shows percentage of apoptotic T cells (mean ± SD).
When we followed the neuraxis into the periphery and investigated the PNS, we made some interesting observations: T-cell apoptosis also occurred in AT-EAN and active EAN of the Lewis rat (Fig. 3 A) with a time course similar to EAE, but marked differences in levels[25] (Fig. 2). In EAN, the highest levels of T-cell apoptosis were also found during recovery but never exceeded 10%. With regard to its pathophysiological impor- tance, T-cell apoptosis in EAN still provides a very potent mechanism of cell destruction since the apoptotic process is completed within 4–5 h ([26]) and thus one can assume that up to 50% of the inflammatory infiltrate would be eliminated within 24 h.
Fig. 3.
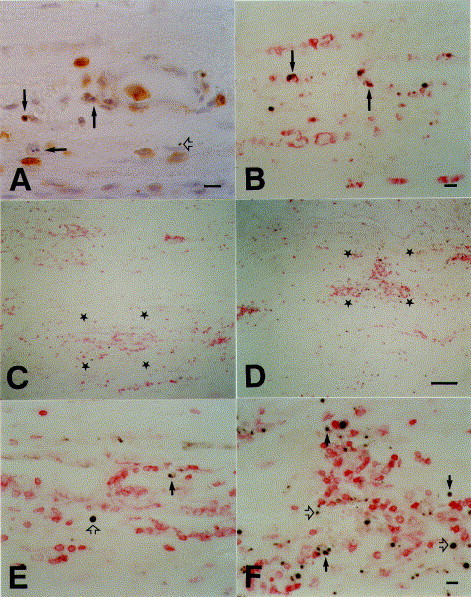
Immunocytochemical characterization of apoptotic cells in Lewis rat experimental autoimmune neuritis (EAN). (A)
B115-1-positive T cells in the sciatic nerve that show nuclear morphology characteristic of apoptosis (arrows). Open arrow denotes advanced stage of apoptosis with loss of membrane immunoreactivity. (B) Double labeling for DNA fragmentation (black) and expression of ED-1, a macrophage marker antigen (red). After antigen therapy macrophages engulf apoptotic DNA. (C and D) Double labeling for DNA fragmentation (black) and expression of B115-1, a T-cell marker antigen (red) in control animals that have received an irrelevant myelin protein (C) or after antigen therapy with i.v. administration of recombinant P2 protein (D). (E and F) Higher magnifications of the regions indicated by stars in C and D respectively. A reduction in inflammatory T cells and increase in apoptotic fragments is visible after antigen therapy. At higher magnification, cells double-stained for T-cell antigen (red) and DNA fragmentation (black) are visible (arrows in E and F). Terminal stages of apoptosis have lost membrane immunoreactivity (open arrows). Scale bars, 10 μm (A, B, E and F), 100 μm (C and D).
The disparity between T-cell apoptosis in the CNS and peripheral tissues became even more pronounced when we studied inflammation in muscle and skin. In the absence of an established, reliable animal model for myositis, we investigated various human inflammatory myopathies, including polymyositis, dermatomyositis, and inclusion body myositis[27]. In these disorders, the inflammatory reaction is diverse and self-sustaining (reviewed in [28]) and putative autoantigens are currently undefined. Autoaggressive T-cells in muscle are CD8+ and not CD4+ as in MS and EAE ([18]). Spontaneous T-cell apoptosis analyzed in diagnostic biopsies taken from untreated patients never exceeded 0.5% ([27]). Thus, inflammation in muscle is not cleared by apoptosis, which is consistent with the non-self-limiting nature of these disorders.
Similar observations were made in skin as follows: in the dermal sensitization site of active EAE, a large amount of cell debris was found in the necrotic area of the abscess. Although the surrounding tissue was heavily inflamed with lymphocytes, no T-cell apoptosis was seen[21]. In draining lymph nodes and spleen, only a very low number of apoptotic cells was observed in EAE and EAN (21, 25). These data underscore the fact that T-cell apoptosis in autoimmune disorders occurs only in immunologically protected sites.
2. Possible mechanisms of T-cell apoptosis in vivo
2.1. Elimination of antigen-specific invading T cells
The inflammatory infiltrate in EAE is composed of primary antigen-specific T cells, which are responsible for the induction of inflammation, and of other non-antigen-specific T cells, which are secondarily recruited into the lesions at later stages when the immunoinflammatory reaction is fully developed[29]. To delineate better the mechanisms of apoptosis in vivo, it is important to know whether antigen-specific cells alone or also additionally recruited bystander T cells are destroyed within the CNS. T cells isolated from established EAE lesions contain only a minor fraction of MBP-specific cells (see discussion in [21]), suggesting that antigen-specific cells are preferentially removed from inflamed CNS tissue. Furthermore, local proliferation of T cells, particularly antigen- specific T cells, occurs only to a very low extent in the lesions[30]. Preferential apoptosis of MBP-reactive cells could account for these observations[31]. To investigate this further, Pender and colleagues induced EAE by a T-cell clone using the Vβ8.2 T-cell receptor, which is the predominant T-cell-receptor element in MBP-induced EAE of the Lewis rat[32]. They then characterized apoptosis and usage of the T-cell receptor in lymphocytes isolated and enriched from spinal cord33, 34. They found that the frequency of Vβ8.2+ T-cells was sevenfold higher in the apoptotic population than in non-apoptotic T cells. Their findings were substantiated by studies with T-cell lines that recognize ovalbumin (OVA), an antigen not present in the Lewis rat, and by experiments in which MBP- and OVA-specific T cells were transferred. These experiments confirmed that the in vitro reactivity was preserved only for non-CNS antigens like OVA. Furthermore, Pender and colleagues could not detect recirculation of Vβ8.2+ T cells in peripheral lymphoid organs. This work suggests via indirect evidence that, in EAE, antigen-specific T cells that utilize the Vβ8.2 T cell receptor are the prime target for destruction by apoptosis. These interesting results might be biased by the altered density of collapsed apoptotic cells[11], which might preclude quantitative recovery during gradient centrifugation. Indeed, the overall frequency of T-cell apoptosis in EAE is much lower than previously reported in histological studies[21]. Furthermore, there is evidence that other T-cell receptors besides Vβ8.2 are involved in reactivity to MBP ([35]). These T cells could also be recruited to the CNS in EAE and would be overlooked in investigations that focus only on Vβ8.2+ T cells.
These studies permit two potential interpretations. First, the nervous system might effectively eliminate T lymphocytes by antigen-specific mechanisms. Second, the CNS tissue might create a hostile environment that drives all T cells into apoptosis as soon as they infiltrate the parenchyma. In ongoing analyses, this question is being addressed by the induction of EAE through the transfer of MBP-specific T cells. These cells contain a stable genomic marker and thus allow unequivocal differentiation between the MBP-reactive and the secondarily recruited T cells in the lesions ( J. Bauer and H. Lassmann, pers. commun.).
2.2. Mechanisms identified in cell-culture experiments
Since T-cell apoptosis appears to be rather organ specific, it is conceivable that local resident cells might render T cells susceptible to apoptosis directly or through soluble factors. Furthermore, local cells might synergize with systemic humoral factors to deliver a proapoptotic stimulus. With regard to the CNS, astrocytes and microglia cells might have a central role. Astrocytes can present autoantigens to autoimmune T-cell lines[36], but astrocytes are only partially competent antigen-presenting cells that are unable to trigger the complete program of T-cell activation[37]. We studied antigen-driven effects of astrocytes on T cells and evaluated the role of steroid hormones[38]. Interestingly, astrocytes exerted a suppressive effect on T-cell activation and this was mediated by cell contact. Glucocorticoids markedly augmented T-cell apoptosis when added to T-cell–astrocyte cultures during late stages of T-cell activation on day 3. However, glucocorticoids had no effect when added at earlier time points or when thymus cells were used as antigen presenters. The effect was specific since it could be inhibited by blockade of the cytosolic steroid receptor through the action of RU486. Other immunomodulatory compounds such as lipocortins, TGFβ, or inhibition of nitric-oxide synthase did not modulate T-cell apoptosis in that system, indicating that these factors are probably not involved. Recently, similar activation of the apoptotic pathway of T cells was noted when freshly isolated microglia that lacked the surface expression of costimulatory molecules were used as antigen-presenting cells[39].
These results argue for a scenario in which nonprofessional antigen-presenting cells prime T cells for an apoptotic stimulus, which might then be delivered by hormonal influences of the microenvironment or systemic changes. Interestingly, steroid hormones might be produced locally[40] or released systemically during the course of EAE ([41]) and blockade of the steroid response augments the severity of EAE and reduces the extent of T-cell apoptosis in the infiltrates[42]. This might also substantiate the therapeutic use of glucocorticosteroids, which are given during acute relapses of autoimmune disorders of the nervous system (see below). The concept of imbalanced signaling of nonprofessional antigen presentation by local glial cells, however, is challenged by data obtained in the model of EAE in radiation bone-marrow chimeras[43]. In this model – owing to mismatches in the major histocompatibility complex – antigen presentation can only take place on hematogeneous inflammatory cells and macrophages that reside in the meninges and the perivascular space. In spite of the absence of antigen presentation on local microglia, astrocytes, and ependymal cells, the autoimmune encephalomyelitis that occurs following the transfer of T-cell lines is qualitatively and quantitatively identical to that in animals with the full repertoire of antigen-presenting cells[44]. Similarly, in this model we found no difference in the incidence of T-cell apoptosis compared with that in fully competent animals. Thus, imbalanced signaling by local nonprofessional antigen-presenting cells is undoubtedly one mechanism that drives T cells into the apoptotic program. This mechanism cannot, however, be sufficient alone for destruction of T cells in inflammatory brain lesions.
2.3. Strategies involved in other tissue sites that use specialized immune-defense mechanisms
The original concept of Medawar[1] that passive mechanisms extend the survival of grafted tissue has been abandoned. It has been clearly demonstrated that immune privilege results from active rather than passive processes (reviewed in [9]). These could arise from local tissue barriers, immunosuppressive microenvironments, or proteins that possess immunomodulatory activity that could eliminate invading T cells by induction of apoptosis.
With regard to immunomodulatory proteins that might mediate T-cell apoptosis at other sites, Fas (CD95, APO-1) and its ligand have gained enormous importance in the control of homeostasis in multicellular organisms. Fas was originally described as a cell-surface protein that belongs to the family of tumor-necrosis-factor receptors that mediates cytolytic cell death45, 46. Crosslinking of the Fas antigen by its trimeric ligand initiates a signaling cascade that leads to apoptotic cell death (reviewed in [47]). The Fas system is also involved in the pathogenesis of liver diseases, where crosslinking of Fas receptors on hepatocytes leads to severe apoptotic liver damage that can be inhibited by therapeutic agents such as linomide[48]. Interactions that use the Fas–FasL system have now been demonstrated in corneal epithelium and in the retina[49] and invading Fas-positive tumor cells can be eliminated by apoptosis in situ. Apart from an active role of Fas–FasL in maintaining immune privilege in grafted testis[50] and prevention of re- jection of islet allografts[51], FasL-bearing cells have recently been demonstrated in brain from MS pa-tients and in other inflammatory CNS diseases52, 53. Dowling et al. [52] showed co-localization of expression of FasL and apoptotic nuclei. Thus, it is conceiv- able that the Fas–FasL system might be involved in apoptosis.
Special reference should also be made to galectin-1 and its role in warranting immune protection in the liver. Galectin-1 belongs to the family of β-galactoside-binding proteins and has been shown to eliminate activated T cells by apoptosis[54]. Obviously, this was mediated by interactions that used N-glycans that were expressed on the CD45 molecule. Galectin-1 is a major lectin on hepatic sinusoidal endothelial cells (reviewed in [55]). Therefore patrolling activated lymphocytes that might accumulate in this organ (reviewed in [55]) could be eliminated after interacting with galectin on sinusoidal cells. It is unknown whether similar galectins are expressed in the nervous system.
To date, detailed studies on T-lymphocyte apoptosis during recovery from inflammation are lacking for autoimmune diseases that involve other specialized and immunologically protected sites such as experimental autoimmune orchitis.
3. Therapeutic induction of T-cell apoptosis in autoimmune diseases: studies in EAN
Therapeutic regimens in patients with immune-mediated disorders of the nervous system aim at reducing inflammation and accelerating recovery. Since high-dose glucocorticosteroids are the mainstay as therapeutically active compounds in this group of disorders and have been used for the treatment of optic neuritis and acute relapses of MS[56], we investigated whether intravenous steroid pulse-therapy could induce T-cell apoptosis in situ [57]. This was studied in the model of AT-EAN that was induced in Lewis rats by transfer of activated, P2-specific T lymphocytes. To delineate whether the effect of steroid hormones is stage specific, two pulses of glucocorticosteroids (10 mg kg–1 body weight) were administered either after the first appearance of symptoms or at the maximum of the disease. Because of the rapid elimination of apoptotic cells[26], rats were sacrificed 6 h later. At both time points there was a massive reduction of T-cell inflammation and an increase of T-cell apoptosis in the inflamed sciatic nerve that was four-to fivefold higher than the rate of apoptosis that occurred spontaneously in control animals. This underscores the fact that the elimination of inflammatory T cells in situ is indeed one mechanism of action in high-dose glucocorticosteroid therapy and is not stage specific. Similar results were recently reported from Penders' group, which studied the effect of glucocorticosteroid hormones in EAE ([58]).
Another approach that has been developed for treatment of experimental autoimmune disorders is antigen-specific therapy. It is based on the observation that T-cell receptor re-engagement at an appropriate stage of the cell cycle eventually leads to T-cell apoptosis[59]. Critchfield et al. treated EAE in mice by intravenous administration of MBP ([60]). The results of in vitro experiments led them to propose that T cells are eliminated by apoptosis. We used an approach similar to that described above for glucocorticosteroid hormones to demonstrate that intravenous treatment with recombinant P2 protein can prevent clinical and histological signs of AT-EAN and active EAN of the Lewis rat, and leads to a profound increase in T-cell apoptosis in the sciatic nerve[61] (Fig. 3C–F). Apoptotic fragments were readily engulfed by macrophages (Fig. 3B). With regard to induction of apoptosis in AT-EAN, antigen therapy was at least as effective as treatment with steroid hormones. Interestingly, the naive P2 protein showed a much higher efficacy than the neuritogenic peptide that spans amino acids 53–78 that is recognized by these neuritogenic T-cell lines in vitro, which supports the concept that antigen presentation must occur in situ [2] and might be executed by activated Schwann cells in the PNS ([62]). Since confrontation with antigen in vitro prompted an increased secretion of typical Th1 cytokines by our Th1 cell lines[61], this argues that cytokine switching is not responsible for the induction of apoptosis. Antigen therapy might prove useful for highly specific treatment of autoimmune disorders of the nervous system, although recent reports have warned that under certain circumstances it might, in fact, exacerbate autoimmune disease[63].
4. Conclusions and perspectives
Disposal of autoaggressive T cells in the nervous system is a very effective mechanism for termination of inflammation of the CNS. Our data strongly suggest that destruction of T cells through apoptosis in inflammatory lesions is a phenomenon that occurs in sites that possess specific immune-defense mecha- nisms and not a general feature for clearance of autoimmune lesions. It might also explain some poorly understood features of CNS autoimmunity. First, the absence of T-cell proliferation in lesions of the nervous system25, 30 might be caused by progression of apoptosis. Furthermore, T-cell apoptosis might eliminate the first wave of MBP-reactive T cells, which are responsible for clinical disease (Fig. 4 ), and thus prevent the expansion of dominant autoaggressive clones, and might form the basis for intramolecular epitope spreading[64] and exposure of cryptic epitopes during later stages of disease. T-cell apoptosis is a sophisticated, naturally occurring mechanism that confers protection of vulnerable sites from tissue damage. At present, we do not know exactly how this is achieved in vivo, but we have now begun to uncover the mechanisms in order to take advantage of its therapeutic applications.
Fig. 4.
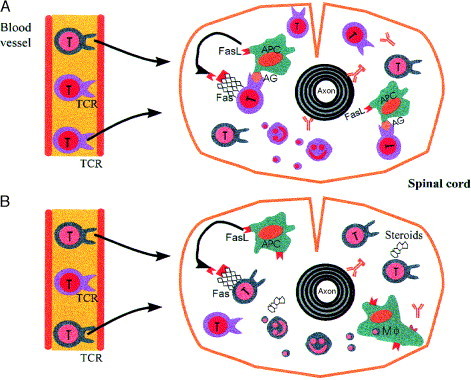
Timecourse of T-cell apoptosis in EAE. (A)
At first, most of the invading T cells are antigen (AG)-specific (violet) and are destroyed after interaction with local antigen-presenting cells (APC). (B) During later stages of the inflammation, `bystander T cells' (green) dominate. Non-antigen-specific mechanisms of elimination, such as the effects and influences of steroid hormones, then gain an increasing importance. Note that apoptotic fragments are phagocytosed by macrophages (Mφ) while they are still intact. Abbreviations: FasL, Fas ligand; TCR, T-cell receptor.
Acknowledgements
The authors wish to thank all those colleagues who participated in different parts of their projects. We are indebted to Dr K.V. Toyka, Würzburg, and Dr H. Wekerle, Martinsried, for many stimulating discussions. Thanks are extended to Dr Cheryl Stucky, Würzburg for help with the language editing of the manuscript.
The work of the authors was supported by grants from the Bundesministerium für Forschung, Deutsche Forschungsgemeinschaft and by The Austrian Science Foundation Project P 10608 Med.
References
- 1.Medawar P. Br. J. Exp. Pathol. 1948;29:58–69. [PMC free article] [PubMed] [Google Scholar]
- 2.Wekerle H. Trends Neurosci. 1986;9:271–277. [Google Scholar]
- 3.Fabry Z., Raine C.S., Hart M.N. Immunol. Today. 1994;15:218–224. doi: 10.1016/0167-5699(94)90247-X. [DOI] [PubMed] [Google Scholar]
- 4.Shrikant P., Benveniste E.N. J. Immunol. 1996;157:1819–1822. [PubMed] [Google Scholar]
- 5.Perry V.H. Curr. Opin. Neurobiol. 1995;5:636–641. doi: 10.1016/0959-4388(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 6.Körner H. Proc. Natl. Acad. Sci. U. S. A. 1995;92:11066–11070. doi: 10.1073/pnas.92.24.11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersson P.B., Perry V.H., Gordon S. Neuroscience. 1995;48:169–186. doi: 10.1016/0306-4522(92)90347-5. [DOI] [PubMed] [Google Scholar]
- 8.Sampson J.H. Proc. Natl. Acad. Sci. U. S. A. 1996;93:10399–10404. doi: 10.1073/pnas.93.19.10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Streilein J.W. Science. 1995;270:1158–1159. doi: 10.1126/science.270.5239.1158. [DOI] [PubMed] [Google Scholar]
- 10.Kerr J.F.R., Wyllie A.H., Currie A.R. Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wyllie A.H., Kerr J.F.R., Currie A.R. Int. Rev. Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 12.Gavrieli Y., Sherman Y., Ben Sasson S.A. J. Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gold R. Lab. Invest. 1994;71:219–225. [PubMed] [Google Scholar]
- 14.Cohen J.J. Annu. Rev. Immunol. 1992;10:267–293. doi: 10.1146/annurev.iy.10.040192.001411. [DOI] [PubMed] [Google Scholar]
- 15.Savill J. Immunol. Today. 1993;14:131–136. doi: 10.1016/0167-5699(93)90215-7. [DOI] [PubMed] [Google Scholar]
- 16.Hartung, H.P., Stoll, G. and Toyka, K.V. (1993) in Peripheral neuropathy (3rd edn) (Dyck, P.J. et al., eds), p, 418, W.B. Saunders
- 17.Lassmann H. Rev. Neurol. (Paris) 1991;147:763–781. [PubMed] [Google Scholar]
- 18.Martin R., McFarland H.F., McFarlin D.E. Annu. Rev. Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 19.Pender M.P. J. Neurol. Sci. 1991;104:81–87. doi: 10.1016/0022-510x(91)90219-w. [DOI] [PubMed] [Google Scholar]
- 20.Pender M.P. J. Autoimmun. 1992;5:401–410. doi: 10.1016/0896-8411(92)90001-7. [DOI] [PubMed] [Google Scholar]
- 21.Schmied M. Am. J. Pathol. 1993;143:446–452. [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen K.B., McCombe P.A., Pender M.P. J. Autoimmun. 1994;7:145–152. doi: 10.1006/jaut.1994.1011. [DOI] [PubMed] [Google Scholar]
- 23.Barac-Latas V., Wege H., Lassmann H. Reg. Immunol. 1995;6:355–357. [Google Scholar]
- 24.Ozawa K. Brain. 1994;117:1311–1322. doi: 10.1093/brain/117.6.1311. [DOI] [PubMed] [Google Scholar]
- 25.Zettl U.K. Acta Neuropathol. 1996;91:360–367. doi: 10.1007/s004010050437. [DOI] [PubMed] [Google Scholar]
- 26.Bursch W. Carcinogenesis. 1990;11:847–853. doi: 10.1093/carcin/11.5.847. [DOI] [PubMed] [Google Scholar]
- 27.Schneider C. J. Neuropathol. Exp. Neurol. 1996;55:1205–1209. doi: 10.1097/00005072-199612000-00003. [DOI] [PubMed] [Google Scholar]
- 28.Dalakas M.C. New Engl. J. Med. 1991;325:1487–1498. doi: 10.1056/NEJM199111213252107. [DOI] [PubMed] [Google Scholar]
- 29.Cross A.H. Lab. Invest. 1990;63:162–170. [PubMed] [Google Scholar]
- 30.Ohmori K. Lab. Invest. 1992;66:54–62. [PubMed] [Google Scholar]
- 31.Bauer J., Wekerle H., Lassmann H. Curr. Opin. Immunol. 1995;7:839–843. doi: 10.1016/0952-7915(95)80057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chluba J. Eur. J. Immunol. 1989;19:279–284. doi: 10.1002/eji.1830190210. [DOI] [PubMed] [Google Scholar]
- 33.Tabi Z., McCombe P.A., Pender M.P. Eur. J. Immunol. 1994;24:2609–2617. doi: 10.1002/eji.1830241107. [DOI] [PubMed] [Google Scholar]
- 34.Tabi Z., McCombe P.A., Pender M.P. Int. Immunol. 1995;7:967–973. doi: 10.1093/intimm/7.6.967. [DOI] [PubMed] [Google Scholar]
- 35.Gold R. Proc. Natl. Acad. Sci. U. S. A. 1995;92:5850–5854. doi: 10.1073/pnas.92.13.5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fontana A., Fierz W., Wekerle H. Nature. 1984;307:273–276. doi: 10.1038/307273a0. [DOI] [PubMed] [Google Scholar]
- 37.Weber F. Brain. 1994;117:59–69. doi: 10.1093/brain/117.1.59. [DOI] [PubMed] [Google Scholar]
- 38.Gold R. Brain. 1996;119:651–659. doi: 10.1093/brain/119.2.651. [DOI] [PubMed] [Google Scholar]
- 39.Ford A.L. J. Exp. Med. 1996;184:1737–1745. doi: 10.1084/jem.184.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yi Hu Z. Proc. Natl. Acad. Sci. U. S. A. 1987;84:8215–8219. doi: 10.1073/pnas.84.23.8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mason D. Immunol. Today. 1991;12:57–60. doi: 10.1016/0167-5699(91)90158-P. [DOI] [PubMed] [Google Scholar]
- 42.Smith T. J. Autoimmun. 1996;9:167–174. doi: 10.1006/jaut.1996.0020. [DOI] [PubMed] [Google Scholar]
- 43.Hickey W.F., Kimura H. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- 44.Lassmann H. Glia. 1993;7:19–24. doi: 10.1002/glia.440070106. [DOI] [PubMed] [Google Scholar]
- 45.Trauth B.C. Science. 1989;245:301–305. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- 46.Yonehara S., Ishii A., Yonehara M. J. Exp. Med. 1989;169:1747. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagata S., Golstein P. Science. 1995;267:1449–1455. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 48.Redondo C. J. Clin. Invest. 1996;98:1245–1252. doi: 10.1172/JCI118908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griffith T.S. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 50.Bellgrau D. Nature. 1995;377:630–632. doi: 10.1038/377630a0. [DOI] [PubMed] [Google Scholar]
- 51.Lau H.T. Science. 1996;273:109–112. doi: 10.1126/science.273.5271.109. [DOI] [PubMed] [Google Scholar]
- 52.Dowling P. J. Exp. Med. 1996;184:1513–1518. doi: 10.1084/jem.184.4.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.D'Souza S.D. J. Exp. Med. 1996;184:2361–2370. doi: 10.1084/jem.184.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perillo N.L. Nature. 1995;378:736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- 55.Crispe I.N., Mehal W.Z. Immunol. Today. 1996;17:522–525. doi: 10.1016/s0167-5699(96)80906-6. [DOI] [PubMed] [Google Scholar]
- 56.Beck R.W. New Engl. J. Med. 1993;329:1764–1769. doi: 10.1056/NEJM199312093292403. [DOI] [PubMed] [Google Scholar]
- 57.Zettl U.K. J. Neuropathol. Exp. Neurol. 1995;54:540–547. doi: 10.1097/00005072-199507000-00008. [DOI] [PubMed] [Google Scholar]
- 58.McCombe P.A. J. Neuroimmunol. 1996;70:93–101. doi: 10.1016/s0165-5728(96)00043-4. [DOI] [PubMed] [Google Scholar]
- 59.Critchfield J.M., Zuniga Pflucker J.C., Lenardo M.J. Cell. Immunol. 1995;160:71–78. doi: 10.1016/0008-8749(95)80011-7. [DOI] [PubMed] [Google Scholar]
- 60.Critchfield J.M. Science. 1994;263:1139–1143. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 61.Weishaupt A. Proc. Natl. Acad. Sci. U. S. A. 1997;94:1338–1342. doi: 10.1073/pnas.94.4.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wekerle H. Eur. J. Immunol. 1986;16:1551–1557. doi: 10.1002/eji.1830161214. [DOI] [PubMed] [Google Scholar]
- 63.Genain C.P. Science. 1996;274:2054–2056. doi: 10.1126/science.274.5295.2054. [DOI] [PubMed] [Google Scholar]
- 64.Lehmann P.V. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]