

Graphical abstract
Novel iso-d-2′,3′-dideoxythianucleoside derivatives 1–4 were designed and synthesized from d-glucose as a bioisostere of lamivudine. Among compounds tested, cytosine analogue 3 showed a potent anti vesicular stomatitis virus (VSV) activity. This result implies that iso-2′,3′-dideoxy sugar templates might play a role of a sugar surrogate of nucleosides for the development of anti-RNA virus agent.
Keywords: Iso-d-2′,3′-dideoxythianucleosides; Lamivudine; Anti-VSV activity; Sulfur participation; Bioisostere; Mitsunobu reaction
Abstract
Novel iso-d-2′,3′-dideoxythianucleoside derivatives 1–4 were designed and asymmetrically synthesized as a bioisostere of lamivudine to search for new anti-HIV agents. The information about using sulfur participation occurred on DAST fluorination and Mitsunobu reaction will be of great help in synthesizing sulfur-containing compounds. Final compounds 1–4 were evaluated against HIV-1 and 2, HSV-1 and 2, EMCV, Cox. B3, VSV, FluA (Taiwan), FluA (Johan.), FCV, and FIP. Only cytosine analogue 3 showed a potent anti-VSV activity (EC50 = 9.43 μg/mL). This result implies that iso-2′,3′-dideoxy sugar templates might play a role of a sugar surrogate of nucleosides for the development of anti-RNA virus agent.
1. Introduction
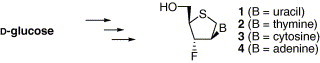
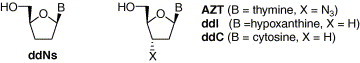
Since zidovudine (AZT)1 has been known as a potent anti-human immunodeficiency virus (anti-HIV) agent, a number of nucleoside analogues have been designed, synthesized, and their antiviral activities were evaluated against a variety of viruses including HIV for the development of antiviral agents. Particularly, 2′,3′-dideoxynucleosides (ddNs) have exhibited potent anti-HIV activity. Among them, didanosine (ddI),2 zalcitabine (ddC),3 and stavudine (d4T)4 have been launched as drugs for the treatment of AIDS patients (Fig. 1 ). However, these 2′,3′-ddNs have several drawbacks such as easy cleavage of their glycosidic bond under acidic conditions similar to a gastric environment and catabolism by adenosine deaminase (ADA), adenosine-5′-monophosphate (AMP) deaminase, and purine nucleoside phosphorylase (PNP).
Figure 1.
The general structure of 2′3′-dideoxynucleosides and representative HIV drugs.
Also, lamivudine (3TC)5 has been clinically used for the treatment of AIDS and HBV (hepatitis B virus) infection (Fig. 2 ). Structural characteristics of lamivudine are that there is no hydroxyl substituent at both its 2′- and 3′-positions like 2′,3′-ddN analogues and are two heteroatoms on the sugar moiety. In addition, lamivudine belongs to l-nucleoside unlike other anti-AIDS drugs. Isonucleosides6 in which their base is moved into C2′-position from anomeric position have been synthesized and evaluated for anti-HIV activity. Among them, iso-ddA and iso-ddG were found to be active against HIV-1 without cytotoxicity and this class of nucleosides were known to possess the intrinsic metabolic advantages such as resistance to ADA and enhanced stability of glycosidic bond under acidic and enzymatic conditions compared to natural nucleosides and 2′,3′-ddNs including lamivudine.(a), 7
Figure 2.
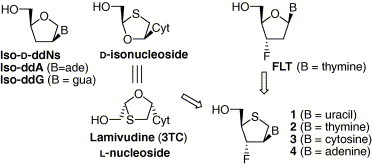
The rationale for the design of the desired nucleosides 1–4.
On the other hand, a fluorine atom has been used as a good bioisostere because many fluorinated nucleosides such as 2′-F-ddA,8 2′-F-ara-ddC,9 and FLT10 were found to exhibit significant antiviral activities. Furthermore, it is worthy to note that l-nucleoside analogues such as lamivudine can be considered as iso-d-nucleoside analogue as shown in Figure 2.
Therefore, as a part of our continuous efforts searching for novel antiviral agents, it was interesting to design and synthesize iso-d-2′,3′-dideoxythianucleoside derivatives with a 3′-fluoro substituent and to evaluate them against various viruses because CH–F group might act as a bioisostere of sugar ring oxygen of lamivudine and/or as a hydrogen bonding acceptor at the active site of their target enzyme, and because the d-sugar skeleton of iso-d-2′,3′-dideoxythianucleosides resembles l-1,3-oxathiolane ring of lamivudine. Here, we wish to report the asymmetric synthesis and biological activities of novel fluorinated iso-d-2′,3′-dideoxythianucleosides 1–4.
2. Results and discussion
2.1. Chemistry
A strategy for the synthesis of glycone of iso-d-2′,3′-dideoxy-3′-fluorothianucleosides is outlined in Scheme 1 . It was envisioned that 1,4-anhydro-4-thioarabitol 6 derived from d-glucose via 1,2;5,6-di-O-isopropylidene-d-glucofuranose (5) could be an appropriate intermediate to synthesize d-4-fluoro-3-thiophenol 12, which could be condensed with nucleobases to afford lamivudine analogues 1–4.
Scheme 1.
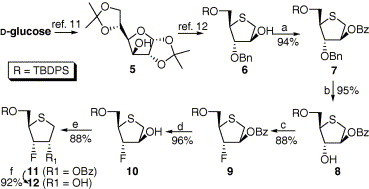
Reagents and conditions: (a) BzCl, DMAP, pyridine, 88 °C, 3 h; (b) BCl3, CH2Cl2, −78 °C, 40 min; (c) DAST, CH2Cl2, −10 °C, 30 min; (d) 1 M NaOMe, CH2Cl2, MeOH, rt, 4 h; (e) BzOH, PPh3, DEAD, THF, 60 °C, 5 h; (f) 1 M NaOMe, MeOH, CH2Cl2, rt, 7 h.
Diacetone glucose 5 was derived from d-glucose by the known one-pot synthesis method reported by Moravcova and his co-workers.11 Compound 5 was converted into 1,4-anhydro-4-thioarabitol 6 according to the known procedure developed by Yoshimura and his co-workers over 10 steps.12 Surprisingly, benzoylation of compound 6 with benzoyl chloride, DMAP, and pyridine at room temperature was very slowly converted to the corresponding benzoate 7. Elevated temperature (88 °C) completed the benzoylation reaction in 3 h in 94% yield. Debenzylation of 7 with BCl3 at −78 °C gave α secondary alcohol 8 whose stereochemistry has the appropriate configuration to introduce α-fluoro substituent when treated with (diethylamino)sulfur trifluoride (DAST). Fluorination of 8 with DAST gave the fluorinated compound 9 in 88% yield with the desired ‘α’ stereochemistry through double inversion mechanism by the nucleophilic participation of the ring sulfur atom.13 Removal of benzoyl group of 9 using cat. NaOMe in MeOH gave the corresponding alcohol 10 in nearly quantitative yield, which was used for coupling with benzoic acid under Mitsunobu conditions.14 During Mitsunobu reaction the nucleophilic participation of sulfur atom occurred only to the extent of 3–5%. The major product 11 which underwent a single inversion was formed in 88% yield with its epimer (3–5%) at 3-position. The structures of major and minor product, 11 and its epimer, were confirmed by comparing 1H NMR data of the corresponding alcohols generated from each epimer after debenzoylation with those of compound 10. Debenzoylation of 11 afforded the glycosyl donor 12, which was ready for the condensation with nucleobases.
Synthesis of pyrimidine nucleoside analogues under Mitsunobu conditions is illustrated in Scheme 2 . Coupling of 12 with N 3-benzoyluracil and N 3-benzoylthymine, PPh3, and DEAD gave the desired β-nucleosides 13 (62%) and 14 (66%), respectively. It is worthy to note that the participation of sulfur atom did not occur at all during coupling of glycosyl donor 12 with nucleobases. Deprotection of N-benzoyl group with 1 M NaOMe produced TBDPS-protected compounds 15 and 16 in 82% and 94% yield, respectively. Finally, treatment of 15 and 16 with TBAF gave the desired final compounds 1 and 2, respectively. Confirmation of N-alkylated nucleosides 1 and 2 was determined by 13C value of anomeric carbon of each nucleoside.15 Compound 1 was converted into lamivudine analogue, cytosine nucleoside 3, via acetylation of the hydroxyl group followed by successive three conventional steps (1,2,4-triazole, POCl3, pyridine; 1,4-dioxane, NH4OH; NH3, MeOH)16 in overall 54% yield.
Scheme 2.
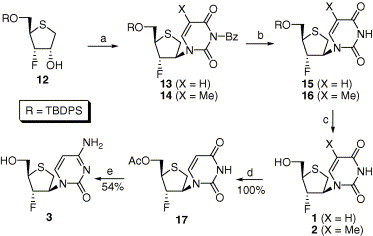
Reagents and conditions: (a) N3-benzoyl uracil or N-benzoyl thymine, PPh3, DEAD, THF, rt, overnight, 62% for 13, 66% for 14; (b) 1 M NaOMe, MeOH, CH2Cl2, rt, 6 h, 82% for 15, overnight, 94% for 16; (c) 1 M TBAF, THF, rt, 2 h, 97% for 1, 98% for 2; (d) Ac2O, pyridine, rt, overnight; (e) (i) 1,2,4-triazole, POCl3, pyridine, rt, overnight; (ii) 1,4-dioxane, 28% NH4OH, rt, overnight; (iii) NH3, MeOH, rt, overnight.
Synthesis of adenine nucleoside analogue 4 is shown in Scheme 3 . Whereas reaction of d-4-fluoro-3-thiophenol 12 with N 6-benzoyladenine at room temperature for 2 d under Mitsunobu conditions (PPh3 and DEAD) did not give a clear spot expected as the desired product on TLC, reaction with 6-chloropurine as a nucleobase instead of N 6-benzoyladenine afforded the protected 6-chloropurine nucleoside 18 in 88% yield. Participation degree of sulfur atom on the sugar ring seems like depending on electron density on the sugar ring, ring strain of the generated episulfonium cation, and properties of nucleophile and leaving group. Mitsunobu reaction of fluorinated compounds (10 and 12) with benzoic acid or nucleobases might have difficulty in forming episulfonium cation because the sugar ring is already electron-poor by electron-withdrawing fluoro group. Therefore, the major product was single inverted one (11, 13, 14, and 18). On the other hand, in case of DAST reaction of compound 8 there is no strong electron-withdrawing group such as a fluorine atom on the sugar ring, implying that participation of the sulfur atom is expected and the major product is double inverted one (9). The remaining things are conversion of 6-chloropurine into adenine base and desilylation of TBDPS group in sequence. Surprisingly, treatment of compound 18 with methanolic ammonia at 80 °C overnight accomplished not only amination at 6-position of 6-chloropurine base but also desilylation of TBDPS group, giving the final product 4 in 68% yield.
Scheme 3.
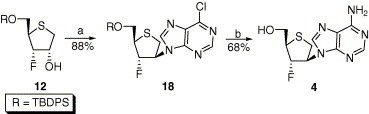
Reagents and conditions: (a) 6-chloropurine, PPh3, DEAD, rt, overnight; (b) NH3, MeOH, 80 °C, overnight.
2.2. Biological activity
Antiviral activities of all iso-d-2′,3′-dideoxy-3′-fluorothianucleoside derivatives 1–4 synthesized as a bioisostere of lamivudine were measured against a variety of viruses such as HIV (human immunodeficiency virus) type 1 and 2, EMCV (encephalomyocarditis virus), Cox. B3 (Coxsackie B virus type 3), VSV (vesicular stomatitis virus), and HSV (herpes simplex virus) type 1 and 2 (Table 1 ). EC50 of compounds 1, 2, and 4 exceeded 100 μg/mL and also did not show any cytotoxicity up to 100 μg/mL at all tested cells such as MT4, HeLa, and Vero cells. On the other hand, cytosine nucleoside derivative 3 exhibited potent anti-VSV activity (EC50 = 9.43 μg/mL), whereas inactive (EC50 > 100 μg/mL) and non-cytotoxic (CS50 and CC50 > 100 μg/mL) against the other viruses. However, selectivity index (SI) of 3 showed low value of 1.56 due to its high cytotoxicity (CC50 = 14.54 μg/mL) in HeLa cell. It is very interesting that cytosine nucleoside analogue 3 designed as a bioisostere of lamivudine showed anti-VSV activity instead of the expected anti-HIV activity. Considering that VSV belongs to RNA virus, it is worthy to note that 3 classified into ddNs exhibited potent anti-VSV activity.
Table 1.
Antiviral activities of the final compounds 1–4 against HIV type 1 and 2, EMCV, Cox. B3, VSV, and HSV type 1 and 2a
| Compound | MT4b CS50c | HIV (EC50) |
HeLaf CC50g | Anti-RNA (EC50) |
Verok CC50 | Anti-HSV (EC50) |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| IIIBd | RODe | EMCVh | Cox.B3i | VSVj | HSV-1l | HSV-2m | ||||
| 1 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 2 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 3 | >100 | >100 | >100 | 14.54 | >14.54 | >14.54 | 9.43 | >100 | >100 | >100 |
| 4 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| Ribavirin | >300 | 23.45 | 64.09 | 10.32 | ||||||
| ddI | >100 | 6.62 | 16.00 | >10 | 0.17 | 0.47 | ||||
The potency is given as an EC50 (50% effective concentration, μg/mL) and evaluated by the degree of inhibition of virus-induced cytopathogenic effects (CPE).
HTLV-1-infected human T-lymphocyte.
50% cytostatic concentration.
Human immunodeficiency virus type 1 strain IIIB.
Human immunodeficiency virus type 1 strain ROD.
Human cervical carcinoma cell.
50% cytotoxic concentration.
Encephalomyocarditis virus.
Coxsackie B virus type 3.
Vesicular stomatitis virus.
African Green monkey kidney cell.
Herpes simplex virus type 1.
Herpes simplex virus type 2.
Antiviral evaluation of compounds 1–4 was also performed against FluA (influenza virus type A) (H1N1) strain Taiwan, FluA (H3N2) strain Johannesburg, FCV (Feline coronavirus) strain WSU, and FIP (Feline infectious peritonitis virus) strain WSU, but all compounds did not show antiviral activity and cytotoxicity up to 100 μg/mL (not shown in Table 1).
3. Conclusion
We have designed and synthesized novel iso-d-2′,3′-dideoxythianucleoside derivatives 1–4 as a bioisostere of lamivudine to search for new anti-HIV agents. Among compounds tested, only cytosine analogue 3 showed potent anti-VSV activity even if it pertains to ddNs classification and VSV is RNA virus. This observation implies that iso-d-2′,3′-dideoxy sugar templates might act as a sugar surrogate of nucleosides for the development of anti-RNA virus agent. The information about participation of sulfur atom during fluorination reaction using DAST and Mitsunobu reaction will be of great help in synthesizing sulfur-containing compounds.
4. Experimental
4.1. General procedure
Melting points are uncorrected. Ultraviolet (UV) spectra were recorded on a JASCO V-530 UV/vis spectrophotometer. Optical rotations were measured on a JASCO DIP-370 digital polarimeter. NMR data were recorded on a Bruker AC200 and Varian Unity AS 500 spectrometer, using CDCl3, or CD3OD and chemical shifts were reported in parts per million (ppm) with reference to the respective residual solvent or deuteriated peaks (δ H 3.30 and δ C 49.0 for CD3OD, δ H 7.26 and δ C 77.0 for CDCl3). Coupling constants are reported in hertz. The abbreviations used are as follows: s (singlet), d (doublet), q (quartet), m (multiplet), dd (doublet of doublets), br s (broad singlet), ddd (doublet of doublets of doublets), or td (triplet of doublets). FAB mass spectra were recorded on Jeol HX 110 spectrometer. Elemental analyses were performed by the General Instrument Laboratory of Ewha Womans University, Korea. All the reactions described below were performed under nitrogen or argon atmosphere and monitored by thin-layer chromatography (TLC). TLC was performed on Merck precoated 60 F254 plates. Column chromatography was performed using silica gel 60 (230–400 mesh, Merck). All anhydrous solvents were distilled over CaH2 or Na/benzophenone prior to use.
4.2. (+)-Benzoic acid (3S,4S,5R)-4-benzyloxy-5-(tert-butyl-diphenyl-silanyloxymethyl)-tetrahydro-thiophen-3-yl ester (7)
To a stirred solution of alcohol 6 (6.070 g, 12.68 mmol) and 4-dimethylaminopyridine (15 mg, 0.12 mmol) in anhydrous pyridine (30 mL) was dropwise added benzoyl chloride (1.62 mL, 13.96 mmol) at 0 °C and the reaction mixture was heated to 88 °C for 3 h. After the volatiles were removed under reduced pressure, the resulting residue was partitioned between ethyl acetate and 0.5 M HCl aqueous solution, and the organic layer was washed with saturated NaHCO3 aqueous solution and brine successively, dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography using hexane and ethyl acetate (15:1) as the eluent to give the corresponding benzoate 7 (6.927 g, 94%) as a colorless oil: +2.1 (c 1.58, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.97–7.27 (m, 20H, 4× Ar), 5.71 (td, 1 H, J = 3.0, 5.0 Hz, 3-H), 4.73 (s, 2H, OCH 2Ph), 4.47 (t, 1H, J = 3.0 Hz, 4-H), 3.89 (dd, 1H, J = 8.5, 10.0 Hz, TBDPSOCHH), 3.77 (dd, 1H, J = 6.0, 10.5 Hz, TBDPSOCHH), 3.69 (ddd, 1H, J = 3.0, 6.5, 9.0 Hz, 5-H), 3.46 (dd, 1H, J = 4.5, 12.0 Hz, SCHH), 3.04 (dd, 1H, J = 3.0, 12.0 Hz, SCHH), 1.60 (s, 9H, t-Bu); 13C NMR (125 MHz, CDCl3) δ 165.9, 138.0, 135.8, 135.7, 133.5, 133.5, 133.5, 130.0, 130.0, 129.9, 129.9, 128.7, 128.7, 128.1, 128.0, 127.9, 127.9, 85.2, 80.0, 72.2, 66.0, 53.3, 34.6, 27.0, 19.5; LRMS(FAB) m/z 605 (M++Na); Anal. Calcd for C35H38O4SSi: C, 72.13; H, 6.57; S, 5.50. Found: C, 72.46; H, 6.71; S, 5.84.
4.3. (−)-Benzoic acid (3S,4S,5R)-5-(tert-butyl-diphenyl-silanyloxymethyl)-4-hydroxy-tetrahydro-thiophen-3-yl ester (8)
To a stirred solution of benzoate 7 (6.927 g, 11.89 mmol) in anhydrous CH2Cl2 (50 mL) was slowly added boron trichloride (28.5 mL, 28.50 mmol, 1 M solution in CH2Cl2) at −78 °C and the reaction mixture was stirred at the same temperature for 40 min. Pyridine (28.5 mL) and methanol (28.5 mL) were successively added and the reaction mixture was kept at room temperature for 1 h. The mixture was partitioned between methylene chloride and 0.5 M HCl aqueous solution, and the organic layer was washed with saturated NaHCO3 aqueous solution and brine successively, dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography using hexane and ethyl acetate (5:1) as the eluent to give the corresponding alcohol 8 (5.570 g, 95%) as a colorless oil: −17.5 (c 0.87, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.03-7.31 (m, 15H, 3× Ar), 5.46 (q, 1H, J = 6.5 Hz, 3-H), 4.47 (t, 1H, J = 6.0 Hz, 4-H), 3.92 (irregular t, 1H, J = 8.5, 10.0 Hz, TBDPSOCHaHb), 3.84 (dd, 1H, J = 6.0, 10.0 Hz, TBDPSOCHaHb), 3.49 (br s, 1H, SCHaHb), 3.35 (br s, 1H, SCHaHb), 2.96 (br s, 1H, 5-H), 2.41 (br s, 1H, OH), 1.07 (s, 9H, t-Bu); 13C NMR (125 MHz, CDCl3) δ 166.2, 135.6, 135.5, 133.3, 132.9, 132.9, 129.9, 129.9, 129.8, 129.5, 128.4, 127.8, 127.8, 80.4, 79.4, 66.8, 50.6, 31.1, 26.8, 19.2; LRMS(ESI) m/z 515 (M+Na)+; Anal. Calcd for C28H32O4SSi: C, 68.26; H, 6.55; S, 6.51. Found: C, 68.02; H, 6.42; S, 6.76.
4.4. (−)-Benzoic acid (3S,4S,5R)-5-(tert-butyl-diphenyl-silanyloxymethyl)-4-fluoro-tetrahydro-thiophen-3-yl ester (9)
To a stirred solution of alcohol 8 (3.050 g, 6.19 mmol) in anhydrous CH2Cl2 (20 mL) was dropwise added (diethylamino)sulfur trifluoride (DAST, 1.23 mL, 9.31 mmol) at −10 °C and the reaction mixture was stirred at the same temperature for 30 min. After saturated NaHCO3 aqueous solution was added, the reaction mixture was stirred for 5 min and partitioned between CH2Cl2 and water. The organic layer was dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography using hexane and ethyl acetate (45:1) as the eluent to give the corresponding fluoro compound 9 (2.688 g, 88%) as a colorless oil: −22.9 (c 0.83, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.98–7.30 (m, 15H, 3× Ar), 5.76–5.70 (m, 1H, 3-H), 5.39 (td, 1H, J = 3.0, 49.0 Hz, 4-H), 3.93–3.89 (m, 1H, CHaHbOTBDPS), 3.81–3.71 (m, 2H, 5-H and CHaHbOTBDPS), 3.42 (ddd, 1H, J = 2.0, 5.5, 12.0 Hz, SCHaCHb), 3.04 (dd, 1H, J = 4.0, 12.0 Hz, SCHaCHb), 1.06 (s, 9H, t-Bu); 13C NMR (125 MHz, CDCl3) δ 165.3, 135.5, 135.4, 133.4, 133.1, 133.0, 129.9, 129.8, 129.2, 128.5, 127.8, 127.7, 96.8 (d, J = 181.8 Hz), 78.4 (d, J = 30.0 Hz), 64.8 (d, J = 6.8 Hz), 51.7 (d, J = 21.4 Hz), 32.9 (d, J = 2.3 Hz), 26.7, 19.2; 19F NMR (470 MHz, CDCl3) δ −185.63; LRMS(ESI) m/z 517 (M+Na)+; Anal. Calcd for C28H31FO3SSi: C, 67.98; H, 6.32; S, 6.48. Found: C, 67.88; H, 6.54; S, 6.41.
4.5. (+)-(3S,4S,5R)-5-(tert-Butyl-diphenyl-silanyloxymethyl)-4-fluoro-tetrahydro-thiophen-3-ol (10)
To a stirred solution of fluoro compound 9 (2.655 g, 5.37 mmol) in methanol (7 mL) and CH2Cl2 (3 mL) was added 1 M NaOMe (0.54 mL, 0.54 mmol) at 0 °C and the reaction mixture was stirred for 4 h at room temperature. The mixture was partitioned between CH2Cl2 and water, dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography using hexane and ethyl acetate (8:1) as the eluent to give the corresponding alcohol 8 (2.005 g, 96%) as a colorless oil: +34.9 (c 0.65, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.74–7.40 (m, 10H, 2× Ar), 5.02 (td, 1H, J = 2.0, 49.5 Hz, 4-H), 4.51–4.48 (m, 1H, 3-H), 3.87–3.84 (m, 1H, CHaHbOTBDPS), 3.72–3.63 (m, 2H, 5-H and CHaHbOTBDPS), 3.23 (br td, 1H, J = 3.5, 12.0 Hz, SCHaCHb), 2.99 (br d, 1H, SCHaCHb), 1.09 (s, 9H, t-Bu); 13C NMR (125 MHz, CDCl3) δ 135.8, 135.6, 132.4, 132.1, 130.0, 130.0, 127.9, 127.9, 100.1 (d, J = 182.3 Hz), 76.3 (d, J = 27.4 Hz), 64.6 (d, J = 9.1 Hz), 53.1 (d, J = 22.9 Hz), 37.8, 26.7, 19.2; 19F NMR (470 MHz, CDCl3) δ −178.32; LRMS(ESI) m/z 413 (M+Na)+; Anal. Calcd for C21H27FO2SSi: C, 64.58; H, 6.97; S, 8.21. Found: C, 64.34; H, 7.16; S, 8.47.
4.6. (+)-Benzoic acid (3R,4S,5R)-5-(tert-butyl-diphenyl-silanyloxymethyl)-4-fluoro-tetrahydro-thiophen-3-yl ester (11)
To a stirred solution of alcohol 10 (1.360 g, 3.48 mmol), triphenyl phosphine (3.099 g, 11.82 mmol), and benzoic acid (1.440 g, 11.79 mmol) in anhydrous THF (20 mL) was dropwise added diethyl azodicarboxylate (1.9 mL, 12.07 mmol, DEAD) at 0 °C over 5 min, and the reaction mixture was heated to 60 °C for 5 h. After cooling, the reaction mixture was evaporated in vacuo. The resulting residue was purified by silica gel column chromatography using hexane and ethyl acetate (8:1 → 6:1) as the eluent to give benzoate 11 (1.516 g, 88%) as a colorless oil: +44.1 (c 1.03, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.15–7.42 (m, 15H, 3× Ar), 5.50–5.42 (m, 1H, 3-H), 5.32 (td, 1H, J = 2.5, 52.5 Hz, 4-H), 3.81–3.70 (m, 3H, CH 2OTBDPS and 5-H), 3.25 (dd, 1H, J = 6.0, 10.0 Hz, SCHaCHb), 3.14 (td, 1H, J = 1.0, 10.0 Hz, SCHaCHb), 1.13 (s, 9H, t-Bu); 13C NMR (50 MHz, CDCl3) δ 165.8, 135.8, 135.7, 133.5, 133.1, 133.0, 130.0, 129.6, 128.6, 128.0, 93.8 (d, J = 187.0 Hz), 75.0 (d, J = 16.5 Hz), 65.1 (d, J = 7.8 Hz), 49.9 (d, J = 20.6 Hz), 29.6 (d, J = 3.7 Hz), 26.7, 19.4; 19F NMR (470 MHz, CDCl3) δ −198.04; LRMS(ESI) m/z 389 (M−Bz)+; Anal. Calcd for C28H31FO3SSi: C, 67.98; H, 6.32; S, 6.48. Found: C, 68.20; H, 6.32; S, 6.59.
4.7. (+)-(3R,4S,5R)-5-(tert-Butyl-diphenyl-silanyloxymethyl)-4-fluoro-tetrahydro-thiophen-3-ol (12)
Compound 11 (1.453 g, 2.94 mmol) was converted to 12 (1.052 g, 92%) as a colorless oil according to the similar procedure used in the preparation of compound 10: +45.4 (c 1.06, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.69–7.40 (m, 10H, 2× Ar), 5.04 (td, 1H, J = 2.5, 51.5 Hz, 4-H), 4.37–4.29 (m, 1H, 3-H), 3.75–3.59 (m, 3H, CH 2OTBDPS and 5-H), 3.02 (dd, 1H, J = 6.0, 10.5 Hz, SCHaCHb), 2.86 (irregular t, 1H, J = 9.0, 10.0 Hz, SCHaCHb), 1.08 (s, 9H, t-Bu); 13C NMR (50 MHz, CDCl3) δ 135.7, 135.7, 133.1, 133.0, 130.0, 127.9, 127.9, 96.3 (d, J = 180.6 Hz), 74.7 (d, J = 18.9 Hz), 65.0 (d, J = 7.4 Hz), 49.4 (d, J = 19.9 Hz), 32.5 (d, J = 4.4 Hz), 26.9, 19.4; 19F NMR (470 MHz, CDCl3) δ −136.53; LRMS(FAB) m/z 413 (M+Na)+; Anal. Calcd for C21H27FO2SSi: C, 64.58; H, 6.97; S, 8.21. Found: C, 64.71; H, 7.26; S, 8.51.
4.8. (+)-3-Benzoyl-1-(3S,4S,5R)-5-(tert-butyl-diphenyl-silanyloxymethyl)-4-fluoro-tetrahydro-thiophen-3-yl]-1H-pyrimidine-2,4-dione (13)
To a stirred solution of alcohol 12 (218 mg, 0.56 mmol), triphenyl phosphine (439 mg, 1.67 mmol), and N 3-benzoyluracil (361 mg, 1.67 mmol) in anhydrous THF (6 mL) was dropwise added DEAD (0.26 mL, 1.65 mmol) at 0 °C over 5 min, and the reaction mixture was stirred at room temperature overnight. After the volatiles were removed in vacuo, the resulting residue was purified by silica gel column chromatography using hexane and ethyl acetate (3:1) as the eluent to give N 3-benzoyluracil nucleoside 13 (203 mg, 62%) as a colorless sticky oil: UV (CHCl3) λ max 254 nm; +3.6 (c 1.06, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.95-7.38 (m, 15H, 3× Ar), 7.32 (d, 1H, J = 8.0 Hz, H-6), 5.84 (d, 1H, J = 8.5 Hz, H-5), 5.34 (td, 1H, J = 7.0, 53.0 Hz, 4-H), 5.03–4.95 (m, 1H, 3-H), 3.91 (dd, 1H, J = 6.0, 11.0 Hz, CHaHbOTBDPS), 3.85 (dd, 1H, J = 6.0, 10.5 Hz, CHaHbOTBDPS), 3.68–3.60 (m, 1H, 5-H), 3.19 (irregular t, 1H, J = 9.5, 11.0 Hz, SCHaCHb), 3.11 (dd, 1H, J = 8.0, 11.5 Hz, SCHaCHb), 1.09 (s, 9H, t-Bu); 13C NMR (50 MHz, CDCl3) δ 168.6, 161.9, 149.9, 141.7, 136.0, 135.9, 135.6, 133.3, 133.2, 131.7, 130.8, 130.3, 129.6, 128.2, 103.5, 94.9 (d, J = 189.7 Hz), 65.8 (d, J = 25.6 Hz), 65.0 (d, J = 1.7 Hz), 48.9 (d, J = 19.5 Hz), 28.2 (d, J = 7.1 Hz), 27.2, 19.7; LRMS(FAB) m/z 589 (M+H)+; Anal. Calcd for C32H33FN2O4SSi: C, 65.28; H, 5.65; N, 4.76; S, 5.45. Found: C, 64.97; H, 5.70; N, 4.97; S, 5.66.
4.9. (−)-3-Benzoyl-1-[(3S,4S,5R)-5-(tert-butyl-diphenyl-silanyloxymethyl)-4-fluoro-tetrahydro-thiophen-3-yl]-5-methyl-1H-pyrimidine-2,4-dione (14)
Compound 12 (100 mg, 0.26 mmol) was converted to 14 (102 mg, 66%) as a colorless sticky oil according to the similar procedure used in the preparation of compound 13: UV (CHCl3) λ max 253 nm; −0.7 (c 1.45, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.95–7.36 (m, 15H, 3× Ar), 7.15 (d, 1H, J = 1.0 Hz, H-6), 5.35 (ddd, 1H, J = 6.5, 8.0, 53.0 Hz, 4-H), 5.04–4.96 (m, 1H, 3-H), 3.91 (dd, 1H, J = 6.0, 11.0 Hz, TBDPSOCHaHb), 3.84 (dd, 1H, J = 6.0, 10.5 Hz, TBDPSOCHaHb), 3.64 (qd, 1H, J = 6.0, 19.0 Hz, 5-H), 3.22 (irregular t, 1H, J = 10.0, 11.0 Hz, SCHaHb), 3.08 (dd, 1 H, J = 7.5, 11.0 Hz, SCHaHb), 1.98 (d, 3H, J = 1.0 Hz, CH3), 1.08 (s, 9H, t-Bu); 13C NMR (125 MHz, CDCl3) δ 168.6, 162.4, 149.6, 137.1, 135.6, 135.6, 135.1, 132.9, 132.8, 131.5, 130.4, 129.9, 129.2, 127.8, 127.8, 111.9, 94.4 (d, J = 190.0 Hz), 64.6, 62.2, 48.2 (d, J = 19.1 Hz), 27.4 (d, J = 7.8 Hz), 26.8, 19.3, 12.6; 19F NMR (470 MHz, CDCl3) δ −187.58; LRMS(FAB) m/z 625 (M+Na)+; Anal. Calcd for C33H35FN2O4SSi: C, 65.75; H, 5.85; N, 4.65; S, 5.32. Found: C, 65.40; H, 5.83; N, 4.75; S, 5.17.
4.10. (−)-1-[(3S,4S,5R)-5-(tert-Butyl-diphenyl-silanyloxymethyl)-4-fluoro-tetrahydro-thiophen-3-yl]-1H-pyrimidine-2,4-dione (15)
To a stirred solution of 13 (110 mg, 0.19 mmol) in methanol (4.5 mL) and CH2Cl2 (1.5 mL) was added 1 M NaOMe (0.40 mL, 0.40 mmol, in MeOH) at 0 °C and the reaction mixture was stirred for 6 h at room temperature. The mixture was partitioned between EtOAc and water, dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography using hexane and ethyl acetate (1.5:1) as the eluent to give the uracil nucleoside 15 (74 mg, 82%) as a colorless sticky oil: −2.9 (c 0.90, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.68 (br s, 1H, NH), 7.69–7.40 (m, 10H, 2× Ar), 7.22 (d, 1H, J = 8.0 Hz, H-6), 5.73 (d, 1H, J = 7.5 Hz, H-5), 5.29 (td, 1H, J = 7.0, 53.0 Hz, 4-H), 5.06–4.98 (m, 1H, 3-H), 3.87 (d, 1H, J = 1.0 Hz, CHaHbOTBDPS), 3.86 (s, 1H, CHaHbOTBDPS), 3.68–3.61 (m, 1H, 5-H), 3.15–3.10 (m, 2H, SCH2), 1.10 (s, 9H, t-Bu); 13C NMR (125 MHz, CDCl3) δ 162.7, 150.5, 141.3, 135.6, 135.6, 132.9, 132.8, 129.9, 129.9, 127.8, 103.1, 94.5 (d, J = 190.0 Hz), 64.3 (d, J = 26.0 Hz), 62.2, 48.6 (d, J = 19.5 Hz), 27.8 (d, J = 7.3 Hz), 26.8, 19.3; Anal. Calcd for C25H29FN2O3SSi: C, 61.95; H, 6.03; N, 5.78; S, 6.62. Found: C, 62.08; H, 5.74; N, 5.99; S, 6.45.
4.11. (−)-1-[(3S,4S,5R)-5-(tert-Butyl-diphenyl-silanyloxymethyl)-4-fluoro-tetrahydro-thiophen-3-yl]-5-methyl-1H-pyrimidine-2,4-dione (16)
Compound 14 (84 mg, 0.14 mmol) was converted to 16 (65 mg, 94%) as a colorless sticky oil according to the similar procedure used in the preparation of compound 15: UV (CHCl3) λ max 266 nm; −6.4 (c 2.44, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.57 (s, 1H, imide-H), 7.71–7.41 (m, 10 H, 2× Ar), 7.05 (s, 1H, H-6), 5.31 (ddd, 1H, J = 7.0, 8.0, 53.0 Hz, 4-H), 5.08–5.00 (m, 1H, 3-H), 3.90 (dd, 1H, J = 6.0, 10.0 Hz, TBDPSOCHaHb), 3.87 (dd, 1H, J = 5.5, 10.5 Hz, TBDPSOCHaHb), 3.64 (qd, 1H, J = 6.0, 19.0 Hz, 5-H), 3.16 (t, 1H, J = 10.5 Hz, SCHaHb), 3.07 (dd, 1H, J = 7.5, 11.5 Hz, SCHaHb), 1.94 (s, 3H, CH3), 1.10 (s, 9H, t-Bu); 13C NMR (125 MHz, CDCl3) δ 163.1, 150.4, 137.0, 135.6, 135.6, 133.0, 132.8, 129.9, 129.9, 127.8, 127.8, 111.8, 94.3 (d, J = 190.0 Hz), 64.5, 64.0 (d, J = 25.1 Hz), 48.3 (d, J = 19.6 Hz), 27.5 (d, J = 7.4 Hz), 26.8, 19.3, 12.5; 19F NMR (470 MHz, CDCl3) δ −188.06; LRMS(FAB) m/z 499 (M+H)+; Anal. Calcd for C26H31FN2O3SSi: C, 62.62; H, 6.27; N, 5.62; S, 6.43. Found: C, 62.47; H, 6.60; N, 5.39; S, 6.68.
4.12. (−)-1-((3S,4S,5R)-4-Fluoro-5-hydroxymethyl-tetrahydrothiophen-3-yl)-1H-pyridine-2,4-dione (1)
To a stirred solution of uracil nucleoside 15 (57 mg, 0.12 mmol) in THF (4 mL) was added 1 M tetrabutylammonium fluoride solution (0.34 mL, 0.34 mmol, TBAF, in THF) at 0 °C, and the reaction mixture was stirred at room temperature for 2 h. After the reaction mixture was concentrated in vacuo, the resulting residue was purified by silica gel column chromatography using methylene chloride and methanol (15:1) as the eluent to give the final uracil nucleoside 1 (28 mg, 97%) as a colorless oil, which was crystallized in ether and a small amount of methanol to produce a white solid: mp 143.4–145.2 °C; UV (MeOH) λ max 262 nm; −23.6 (c 0.63, MeOH); 1H NMR (500 MHz, CD3OD) δ 7.72 (d, 1H, J = 7.5 Hz, H-6), 5.70 (d, 1H, J = 8.5 Hz, H-5), 5.33 (ddd, 1H, J = 6.0, 8.0, 53.0 Hz, 4-H), 5.10–5.01 (m, 1H, 3-H), 3.81 (dd, 1H, J = 6.0, 11.0 Hz, HOCHaHb), 3.70 (dd, 1H, J = 6.5, 12.0 Hz, HOCHaHb), 3.54 (qd, 1H, J = 6.0, 19.5 Hz, 5-H), 3.23 (t, 1H, J = 11.0 Hz, SCHaHb), 3.03 (dd, 1H, J = 8.0, 11.0 Hz, SCHaHb); 13C NMR (125 MHz, CD3OD) δ 166.2, 152.7, 144.7, 103.2, 96.0 (d, J = 187.3 Hz), 66.0 (d, J = 24.6 Hz), 64.4 (d, J = 1.4 Hz), 49.7 (d, J = 19.1 Hz), 27.8 (d, J = 7.8 Hz); 19F NMR (470 MHz, CD3OD) δ −186.21; LRMS(FAB) m/z 247 (M+H)+; Anal. Calcd for C9H11FN2O3S: C, 43.90; H, 4.50; N, 11.38; S, 13.02. Found: C, 43.65; H, 4.61; N, 11.59; S, 13.30.
4.13. (−)-1-((3S,4S,5R)-4-Fluoro-5-hydroxymethyl-tetrahydro-thiophen-3-yl)-5-methyl-1H-pyrimidine-2,4-dione (2)
Compound 16 (66 mg, 0.13 mmol) was converted to 2 (34 mg, 98%) as a white solid according to the similar procedure used in the preparation of compound 1: mp = 92.3–94.5 °C; UV (MeOH) λ max 266 nm; −23.3 (c 0.77, MeOH); 1H NMR (500 MHz, CD3OD) δ 7.56 (d, 1H, J = 1.0 Hz, H-6), 5.32 (ddd, 1H, J = 7.0, 8.5, 54.0 Hz, 4-H), 5.10-5.01 (m, 1H, 3-H), 3.82 (dd, 1H, J = 5.5, 11.5 Hz, HOCHaHb), 3.70 (dd, 1H, J = 6.5, 11.0 Hz, HOCHaHb), 3.53 (qd, 1H, J = 6.0, 18.5 Hz, 5-H), 3.22 (t, 1H, J = 11.0 Hz, SCHaHb), 2.99 (ddd, 1H, J = 1.0, 8.0, 11.0 Hz, SCHaHb), 1.88 (d, 3H, J = 1.0 Hz, 5-CH3); 13C NMR (125 MHz, CD3OD) δ 166.4, 152.9, 140.3, 112.2, 95.9 (d, J = 187.3 Hz), 65.5 (d, J = 24.6 Hz), 64.5, 49.5 (d, J = 19.3 Hz), 27.6 (d, J = 8.3 Hz), 12.4; 19F NMR (470 MHz, CD3OD) δ −186.80; LRMS(FAB) m/z 261 (M+H)+; Anal. Calcd for C10H13FN2O3S: C, 46.14; H, 5.03; N, 10.76; S, 12.32. Found: C, 45.82; H, 5.39; N, 10.62; S, 12.05.
4.14. (−)-4-Amino-1-((3S,4S,5R)-4-fluoro-5-hydroxymethyl-tetrahydro-thiophen-3-yl)-1H-pyrimidin-2-one (3)
To a stirred solution of uracil nucleoside 1 (18 mg, 0.07 mmol) in anhydrous pyridine (1.5 mL) was added acetic anhydride (0.02 mL, 0.21 mmol) at room temperature, and the reaction mixture was stirred at the same temperature overnight. The reaction mixture was evaporated in vacuo and partitioned between ethyl acetate and dilute HCl aqueous solution. The organic layer was washed with saturated NaHCO3 aqueous solution, dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting residue was used to the next reaction without further purification. To a stirred suspension of the acetated residue 17 and 1,2,4-triazole (86 mg, 1.25 mmol) in anhydrous pyridine (2.5 mL) was added phosphorus oxychloride (0.08 mL, 0.83 mmol) at room temperature, and the reaction mixture was stirred at the same temperature for overnight. 1,4-Dioxane (2 mL) and 28% ammonium hydroxide (2 mL) were added to the reaction mixture at 0 °C and stirred at room temperature overnight. After the volatiles were evaporated under reduced pressure, methanolic ammonia (3 mL) was added to the resulting residue, and the reaction mixture was stirred at room temperature overnight. After the volatiles were removed in vacuo, the resulting residue was purified by silica gel column chromatography using methylene chloride and methanol (7:1) as the eluent to give the final cytosine nucleoside 3 (9 mg, 54%) as a light brownish solid, which was recrystallized in ether and a small amount of methanol: hygroscopic; UV (MeOH) λ max 271 nm; −26.1 (c 0.57, MeOH); 1H NMR (500 MHz, CD3OD) δ 7.72 (d, 1H, J = 7.5 Hz, H-6), 5.92 (d, 1H, J = 7.5 Hz, H-5), 5.42 (ddd, 1H, J = 6.0, 8.0, 54.0 Hz, 4-H), 5.08–5.00 (m, 1H, 3-H), 3.83 (dd, 1H, J = 6.5, 12.0 Hz, HOCHaHb), 3.71 (dd, 1H, J = 6.5, 11.5 Hz, HOCHaHb), 3.60–3.52 (m, 1H, 5-H), 3.30 (t, 1H, J = 11.0 Hz, SCHaHb), 3.04 (ddd, 1H, J = 1.0, 8.0, 11.0 Hz, SCHaHb); 13C NMR (125 MHz, CD3OD) δ 166.4, 157.4, 144.1, 95.3, 94.9 (d, J = 186.3 Hz), 66.3 (d, J = 25.0 Hz), 63.3 (d, J = 1.9 Hz), 48.8 (d, J = 19.5 Hz), 26.9 (d, J = 7.8 Hz); 19F NMR (470 MHz, CD3OD) δ −38.20; LRMS(FAB) m/z 246 (M+H)+; Anal. Calcd for C9H12FN3O2S: C, 44.07; H, 4.93; N, 17.13; S, 13.07. Found: C, 44.32; H, 5.03; N, 16.87; S, 12.99.
4.15. (−)-9-[(3S,4S,5R)-5-(tert-Butyl-diphenyl-silanyloxymethyl)-4-fluoro-tetrahydro-thiophen-3-yl]-6-chloro-9H-purine (18)
Compound 12 (65 mg, 0.17 mmol) was converted to 18 (78 mg, 88%) as a colorless sticky oil according to the similar procedure used in the preparation of compound 13: UV (CHCl3) λ max 264 nm; −12.4 (c 0.56, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.75 (s, 1H, H-8), 8.23 (s, 1H, H-2), 8.68–7.39 (m, 10H, 2× Ar), 5.74 (ddd, 1H, J = 1.5, 8.0, 52.5 Hz, 4-H), 5.32–5.24 (m, 1H, 3-H), 3.92 (dd, 1H, J = 5.5, 11.0 Hz, TBDPSOCHaHb), 3.84 (dd, 1H, J = 6.0, 10.5 Hz, TBDPSOCHaHb), 3.75 (qd, 1H, J = 6.0, 20.0 Hz, 5-H), 3.71 (t, 1H, J = 11.5 Hz, SCHaHb), 3.28 (dd, 1H, J = 7.5, 11.0 Hz, SCHaHb), 1.10 (s, 9H, t-Bu); 13C NMR (50 MHz, CDCl3) δ 151.9, 151.5, 151.4, 141.8, 135.5, 135.4, 132.5, 132.7, 132.0, 129.8, 127.7, 127.6, 94.8 (d, J = 191.0 Hz), 64.7 (d, J = 1.7 Hz), 62.9 (d, J = 24.3 Hz), 48.3 (d, J = 19.5 Hz), 28.4 (d, J = 7.1 Hz), 26.6, 19.2; 19F NMR (470 MHz, CDCl3) δ −187.66; LRMS(FAB) m/z 527 (M+H)+; Anal. Calcd for C26H28ClFN4OSSi: C, 59.24; H, 5.35; N, 10.63; S, 6.08. Found: C, 59.51; H, 5.40; N, 10.44; S, 5.91.
4.16. (−)-[(2R,3S,4S)-4-(6-Amino-purin-9-yl)-3-fluoro-tetrahydro-thiophen-2-yl]-methanol (4)
A solution of 6-chloropurine nucleoside 18 (78 mg, 0.15 mmol) in methanolic ammonia (6 mL) was heated to 80 °C in a glass bomb overnight. After cooling, the volatiles were removed in vacuo and the resulting residue was purified by silica gel column chromatography using methylene chloride and methanol (10:1) to give adenine nucleoside 4 (27 mg, 68%) as a colorless sticky oil, which was crystallized in ether and a small amount of methanol to afford a white solid: mp = 159.4-160.7 °C; UV (MeOH) λ max 259 nm; −32.5 (c 1.49, MeOH); 1H NMR (500 MHz, CD3OD) δ 8.30 (s, 1H, H-8), 8.24 (s, 1H, H-2), 5.61 (ddd, 1H, J = 6.5, 8.5, 52.5 Hz, 3-H), 5.39–5.31 (m, 1H, 4-H), 3.79 (dd, 1H, J = 6.0, 11.0 Hz, HOCHaHb), 3.72 (dd, 1 H, J = 6.0 11.0 Hz, HOCHaHb), 3.72–3.62 (m, 1H, 2-H), 3.61 (t, 1H, J = 10.5 Hz, SCHaHb), 3.24 (ddd, 1H, J = 1.5, 7.5, 11.0 Hz, SCHaHb); 13C NMR (125 MHz, CD3OD) δ 156.7, 152.7, 150.9, 142.1, 120.7, 96.9 (d, J = 189.1 Hz), 64.5 (d, J = 1.4 Hz), 63.5 (d, J = 24.1 Hz), 49.9 (d, J = 19.1 Hz), 29.0 (d, J = 7.4 Hz); 19F NMR (470 MHz, CD3OD) δ −186.41; LRMS(FAB) m/z 270 (M+H)+; Anal. Calcd for C10H12FN5OS: C, 44.60; H, 4.49; N, 26.01; S, 11.91. Found: C, 44.84; H, 4.51; N, 25.77; S, 12.18.
Acknowledgments
This work was supported by Pusan National University Research Grant 2005 (to H. R. Moon) and a grant from the Korea Science and Engineering Foundation (R01-2005-000-10162-0, to L. S. Jeong).
References and notes
- 1.(a) Furman P.A., Fyfe J.A., St. Clair M.H., Weinhold K., Rideout J.L., Freeman G.A., Lehrman S.N., Bolognesi D.P., Broder S., Mitsuya H., Barry D.W. Proc. Natl. Acad. Sci. U.S.A. 1986;83:8333. doi: 10.1073/pnas.83.21.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sarin P.S. Annu. Rev. Pharmacol. 1988;28:411. doi: 10.1146/annurev.pa.28.040188.002211. [DOI] [PubMed] [Google Scholar]; (c) De Clercq E. Anticancer Res. 1987;7:1023. [PubMed] [Google Scholar]; (d) Mitsuya H., Weinhold K.L., Furman P.A., St. Clair M.H., Lehrman S.N., Gallo R.C., Bolognesi D., Barry D.W., Broder S. Proc. Natl. Acad. Sci. U.S.A. 1985;82:7096. doi: 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yarchoan R., Klecker R.W., Weinhold K.J., Markham P.D., Lyerly H.K., Durack D.T., Gelmann E., Lehrman S.N., Blum R.M., Barry D.W. Lancet. 1986;1:575. doi: 10.1016/s0140-6736(86)92808-4. [DOI] [PubMed] [Google Scholar]
- 2.Faulds D., Brogden R.N. Drugs. 1992;44:94. doi: 10.2165/00003495-199244010-00008. [DOI] [PubMed] [Google Scholar]
- 3.Whittington R., Brogden R.N. Drugs. 1992;44:656. doi: 10.2165/00003495-199244040-00009. [DOI] [PubMed] [Google Scholar]
- 4.Baba M., Pauwels R., Herdewinjn P., De Clercq E., Desmyter J., Vandeputte M. Biochem. Biophys. Res. Commun. 1987;142:128. doi: 10.1016/0006-291x(87)90460-8. [DOI] [PubMed] [Google Scholar]
- 5.(a) Soudeyns H., Yao S.-J., Gao W., Belleau B., Kraus J.-L., Nguyen-Ba N., Spira B., Wainberg M.A. Antimicrob. Agents Chemother. 1991;35:1386. doi: 10.1128/aac.35.7.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Coates J.A.V., Cammack N., Jenkinson H.J., Jowett A.J., Jowett M.I., Pearson B.A., Penn C.R., Rouse P., Viner K.C., Cameron J.M. Antimicrob. Agents Chemother. 1992;36:733. doi: 10.1128/aac.36.4.733. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schinazi R.F., Chu C.K., Peck A., McMillan A., Mathis R., Cannon D., Jeong L.S., Beach J.W., Choi W.-B., Yeola S., Liotta D.C. Antimicrob. Agents Chemother. 1992;36:672. doi: 10.1128/aac.36.3.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Terao Y., Akamatsu M., Achiwa K. Chem. Pharm. Bull. 1991;39:823. doi: 10.1248/cpb.39.823. [DOI] [PubMed] [Google Scholar]; (b) Bamford M.J., Humber C.C., Storer R. Tetrahedron Lett. 1991;32:271. [Google Scholar]
- 7.Sells T.B., Nair V. Tetrahedron Lett. 1993;34:3527. [Google Scholar]
- 8.Marquez V.E., Tseng C.K.-H., Kelley J.A., Mitsuya H., Broder S., Roth J.S., Driscoll J.S. Biochem. Pharmacol. 1987;36:2719. doi: 10.1016/0006-2952(87)90254-1. [DOI] [PubMed] [Google Scholar]
- 9.(a) Huang J.T., Chen L.C., Wang L., Kim M.H., Warshaw J.A., Zhu Q.Y., Chou T.C., Watanabe K.A., Matulic-Adamic J., Su T.L., Fox J.J., Polsky B., Baron P.A., Gold J.W.M., Hardy W.D., Zuckerman E. J. Med. Chem. 1991;34:1640. doi: 10.1021/jm00109a017. [DOI] [PubMed] [Google Scholar]; (b) Watanabe K.A., Harada K., Zeidler J., Matulic-Adamic J., Takahashi K., Ren W.Y., Cheng L.C., Fox J.J., Chou T.C., Zhu Q.Y., Polsky B., Gold J.W.M., Armstrong D. J. Med. Chem. 1990;33:2145. doi: 10.1021/jm00170a016. [DOI] [PubMed] [Google Scholar]; (c) Martin J.A., Bushnell D.J., Duncan I.B., Dunsdon S.J., Hall M.J., Machin P.J., Merrett J.H., Parkes K.E.B., Roberts N.A., Thomas G.J., Galpin S.A., Kinchington D. J. Med. Chem. 1990;33:2137. doi: 10.1021/jm00170a015. [DOI] [PubMed] [Google Scholar]
- 10.Matthes E., Lehmann C., Scholz D., Rosenthal H.A., Langen P. Biochem. Biophys. Res. Commun. 1988;153:825. doi: 10.1016/s0006-291x(88)81170-7. [DOI] [PubMed] [Google Scholar]
- 11.Moravcova J., Capkova J., Stanek J. Carbohydr. Res. 1994;263:61. [Google Scholar]
- 12.Yoshimura Y., Kitano K., Yamada K., Satoh H., Watanabe M., Miura S., Sakata S., Sasaki T., Matsuda A. J. Org. Chem. 1997;62:3140. doi: 10.1021/jo9700540. [DOI] [PubMed] [Google Scholar]
- 13.(a) Jeong L.S., Moon H.R., Yoo S.J., Lee S.N., Chun M.W., Lim Y.-H. Tetrahedron Lett. 1998;39:5201. [Google Scholar]; (b) Jeong L.S., Nicklaus M.C., George C., Marquez V.E. Tetrahedron Lett. 1994;35:7569. [Google Scholar]; (c) Jeong L.S., Yoo S.J., Moon H.R., Kim Y.H., Chun M.W. J. Chem. Soc., Perkin Trans. 1. 1998:3325. [Google Scholar]
- 14.Yamada K., Sakata S., Yoshimura Y. J. Org. Chem. 1998;63:6891. doi: 10.1021/jo980667s. [DOI] [PubMed] [Google Scholar]
- 15.Rodriquez J.B., Marquez V.E., Nicklaus M.C., Mitsuya H., Barchi J.J., Jr. J. Med. Chem. 1994;37:3389. doi: 10.1021/jm00046a024. [DOI] [PubMed] [Google Scholar]
- 16.Shi J., Du J., Ma T., Pankiewicz K.W., Patterson S.E., Tharnish P.M., McBrayer T.R., Stuyver L.J., Otto M.J., Chu C.K., Schinazi R.F., Watanabe K.A. Bioorg. Med. Chem. 2005;13:1641. doi: 10.1016/j.bmc.2004.12.011. [DOI] [PubMed] [Google Scholar]