

Graphical abstract
Keywords: Adefovir, Cidofovir, Antivirals, Prodrugs, Acyclic nucleoside phosphonate, Phosphonate ester, In vitro evaluation
Abstract
New Adefovir (PMEA) prodrugs with a pro-moiety consisting of decyl or decyloxyethyl chain bearing hydroxyl function(s), hexaethyleneglycol or a (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl unit were prepared starting from the tetrabutylammonium salt of the phosphonate drug and an appropriate alkyl bromide or tosylate. Analogously, two esters of Cidofovir [(S)-HPMPC] bearing a hexaethyleneglycol moiety were prepared. The activity of the prodrugs was evaluated in vitro against different virus families. A loss in the antiviral activities of the hydroxylated decyl or decyloxyethyl esters and hexaethyleneglycol esters of PMEA against human immunodeficiency virus (HIV) and herpesviruses [including herpes simplex virus (HSV), varicella-zoster virus (VZV), and human cytomegalovirus (CMV)] occurred in comparison with the parent compound. On the other hand, the (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl ester of PMEA showed significant activities against HIV and herpesviruses. (S)-HPMPC prodrugs exhibited anti-cytomegalovirus activities in the same range as the parent drug, whereas the anti-HSV and anti-VZV activities were one- to seven-fold lower than that of Cidofovir.
1. Introduction
Acyclic nucleoside phosphonates (ANPs) are a group of compounds with remarkable antiviral activities. Their development has resulted in three approved drugs, and research on ANPs continues to provide new active compounds.1, 2 The presence of the phosphonate group is responsible for their ionic character when subjected to physiological pH. The ionized molecule is not easily permeable through the gastrointestinal wall and biological membranes. In order to achieve better oral bioavailability, the phosphonate group of the drug can be transformed to a phosphonic ester or amidate, which is enzymatically cleaved to the parent drug after passing the intestinal barrier, or later inside the cells. A considerable number of ANP prodrugs have been evaluated, but only a few of them passed the preclinical studies. This likely arises from the complexity in achieving the right balance between the suitable chemical, physical, and pharmacological prodrug properties.
The basic aim of this work is to apply several less common pro-moieties to 9-[(2-phosphonylmethoxy)ethyl)]adenine (1) (PMEA, Adefovir). PMEA has demonstrated a broad spectrum of antiviral activity1 against human immunodeficiency virus (HIV) and other retroviruses and is active against various DNA viruses, including the hepatitis B virus and seventeen herpesviruses. The bis(pivaoyloxymethyl)ester of PMEA [Adefovir Dipivoxyl, bis(POM)–PMEA] (Fig. 1) has been approved as an oral prodrug for the treatment of hepatitis B. The orally administered prodrug is absorbed and then cleaved by carboxyesterases in the serum generating pivalate and formaldehyde as its byproducts. Formaldehyde is the significant contributor to the overall cytotoxic effects of the prodrug.3
Figure 1.
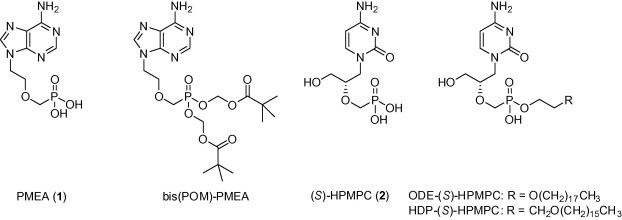
The structures of Adefovir (1), Cidofovir (2) and some of their prodrugs.
Another masking group of the pharmacochemistry of antibiotics—the dioxolenone unit—has been previously utilized to mask the carboxylic function analogously to the pivaoyloxymethyl variety. However, rare examples of dioxolenone esters on phosphate or phophonate have been published.4 The bioavailability and stability of (5-substituted 2-oxo-1,3-dioxolen-4-yl)methyl esters have been explored. Methyl as a substituent in position 5 proved to be optimal with respect to oral absorbability as well as stability of the prodrug.5 We have aimed at the preparation of a conjugate of this particular dioxolenone unit and 1.
Lipophilic esters constitute another important class of phosphate and phosphonate prodrugs. Some alkyl6 and alkyloxyalkyl7 esters of nucleotides or acyclic nucleoside phosphonates have already been advanced into clinical studies. Their antiviral activities are superior when compared to the parent drugs. On the other hand, their physical properties are far from being optimal. They suffer from low solubility in water. Additionally, a simple alkyl chain is subjected to a ω-oxidation process resulting in inactivation of the prodrug. Therefore, we have focused on those promoieties consisting of the aliphatic chain modified by means of addition(s) of hydroxyl group(s) or insertions of oxygen atoms. A hydroxylated alkyl or alkyloxyalkyl unit or a hexaethyleneglycol unit was attached by ester linkage to the phosphonate group of Adefovir as a model drug. The influence of the above-mentioned modifications on the in vitro antiviral activities is reported here. We also studied the effects of the addition of the hexaethylene glycol unit in Cidofovir [(S)-HPMPC, 2] as prodrugs closely related to Cidofovir hexadecyloxypropyl and octadecyloxyethyl esters, that is, HDP (S)-HPMPC and ODE (S)-HPMPC, respectively7 (Fig. 1 ). HDP (S)-HPMPC (CMX001) is currently being developed for use in the prophylactic and preemptive therapy of dsDNA viral infections.
Dioxolenone prodrugs are expected to be cleaved by serum or tissue esterases yielding the parent phosphonate, carbon dioxide and relatively nontoxic acetoin5, according to Scheme 1 . The lipid phosphonate diesters are cleaved into appropriate monoesters after permeation through the wall of the small intestine.8 Unlike the dioxolenone ester of phosphonate, lipid monoesters are stable in plasma due to their hydrolytic stability and the absence of phospholipases. The remaining ester linkage is cleaved by phosphoesterases C inside the cells.7b It was believed that the only role of the lipid pro-moiety is the facilitation of the passive transport through the phospholipid membranes. Recently, it was shown that the attachment of the masking alkyloxyalkyl unit to the phosphonate pharmacophore affects pharmacokinetics more complexly.7b
Scheme 1.
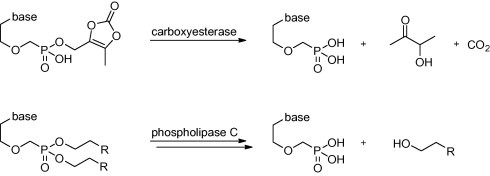
The activation of the considered prodrug types
2. Results
We have prepared several structural types of PMEA prodrugs which are outlined in Section 1 (see Fig. 2 ). The prodrugs of 1 were prepared in doublets of corresponding mono and diesters with the exception of compound 15. The presented pro-moieties are 10-hydroxydecyl unit (compounds 3 and 5), 9,10-dihydroxydecyl unit (compounds 4 and 6), 2-[(10-hydroxydecyl)oxy]ethyl unit (compounds 7 and 9), 2-[(9,10-dihydroxydecyl)oxy]ethyl unit (compounds 8 and 10), hexaethyleneglycol units (compounds 11–14) or (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl unit (compound 15). The length of the hydroxylated alkyl chain in compounds 3–10 was limited by the accessibility of the precursors (9-decen-1-ol, 10-undecen-1-ol). Hexaethyleneglycol pro-moiety was coupled also to Cidofovir (2), and thus prodrugs 16 and 17 were prepared.
Figure 2.
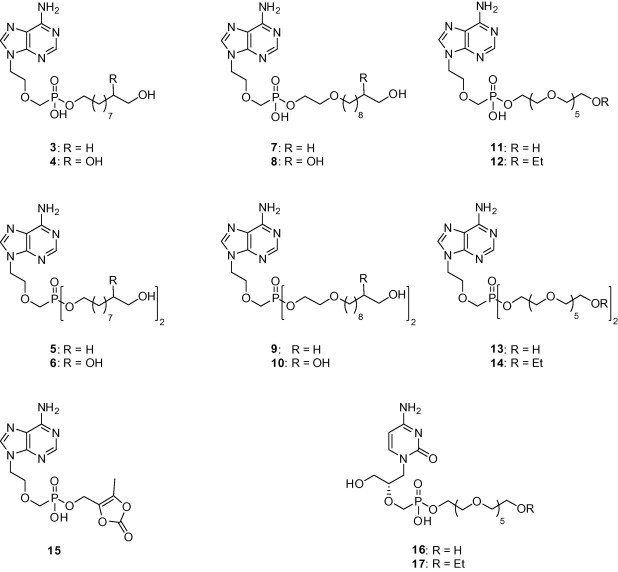
The structures of the synthesized prodrugs.
2.1. Chemistry
2.1.1. Alkylation of the phosphonate function
The synthesis of the prodrugs is based on nucleophilic substitution reactions. The acidic P–OH group can be deprotonated and the negatively charged oxygen atom acts as a nucleophile in the reaction with a functionalized alkyl bromide or tosylate. Their preparation is depicted in Section 2.1.2 below. Tetrabutylammonium hydroxide was used as a base for the deprotonation of alkyl phosphonic acid 1. The resulting tetrabutylammonium salt is soluble in organic solvents and reacts with 2 equiv of an appropriately protected alkyl bromide or tosylate in moderate yields. The acid labile protecting groups of the prepared phosphonate diesters 18–22 were cleaved in a subsequent step by conventional methods (AcOH, Dowex 50). The monoesters 3, 4, 7, 8, 11, 12 were prepared from appropriate diesters by means of the published procedure9 involving a treatment with excess of lithium azide. Resulting monoesters are sufficiently stable towards the additional nucleophilic attack of the azide ion due to the presence of charged oxygen atom of the phosphonate function. Thus, the appropriate monoester was isolated as a sole product.
Diisopropylethylamine was utilized first as a base for the preparation of the (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl ester of PMEA. Monitoring of the reaction mixture by TLC showed the formation of a relatively nonpolar product which was thought to be the appropriate diester. However, it disappeared during the working up the reaction mixture and monoester 15 was isolated as the sole product (see Scheme 3 ). Although the preparation procedure involving one equivalent of tetrabutylammonium hydroxide as a base seemed to be more suitable, the yield was still low (16%).
Scheme 3.
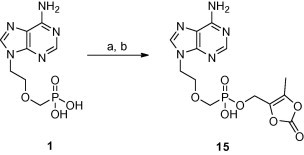
The preparation of prodrug 15. Reagents and conditions: (a) Bu4NOH, MeOH; (b) (5-Methyl-2-oxo-1,3-dioxolen-4-yl)methyl bromide, DMF.
Cidofovir was alkylated starting from its cyclic form (23).7a The tetrabutylammonium salt of 23 was treated with tosylate 28 or 29 (see Section 2.1.2). The resulting cyclic phosphonate esters yielded the monoesters (16 or 17) after the final working up (see Scheme 4 ).
Scheme 4.
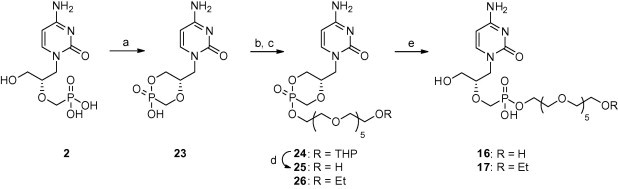
The preparation of prodrugs 16 and 17. Reagents and conditions: (a) DCC, N,N′-dicyclohexyl-4-morpholinecarboxamidine, DMF, 100 °C; (b) tetrabutylammonium hydroxide, MeOH; (c) 28 or 29, DMF, 100 °C; (d) AcOH, H2O; (e) NH3, H2O.
2.1.2. The preparation of functionalized alkyl bromides and tosylates
The (5-Methyl-2-oxo-1,3-dioxolen-4-yl)methyl bromide for the preparation of 15 was synthesized according to the published procedure.5 Bromides for the preparation of 18–21 were prepared from commercially available 9-decen-1-ol or 10-undecen-1-ol. Their hydroxyl groups were replaced with bromine and the double bond was oxidized to 1,2-diol by the treatment with osmium tetroxide and N-methylmorfoline-N-oxide (Scheme 5 ). The diol system in 22 was protected with an isopropylidene group, and the prepared bromide 23 was reacted in a subsequent step with ethylene glycol. The arising alcohol was converted to bromide 24. The diol system in 25 was oxidized by the action of sodium periodate followed by reductive treatment and MOM-protection procedure. The resulting bromide 26 was treated in the same two-step-way as compound 23 yielding bromide 27. Bromides 23, 24, 26, 27 were used for the alkylation of PMEA according to Scheme 2 . The acid labile protecting groups (isopropyliden, MOM) of the prepared phosphonate esters 18–21 were cleaved in a subsequent step by conventional methods (AcOH, Dowex 50) (see Scheme 2).
Scheme 5.
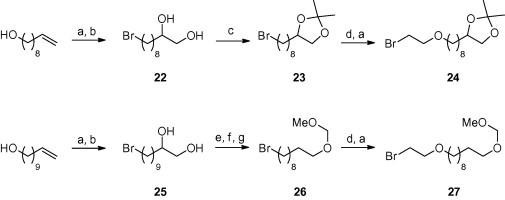
The preparation of protected bromides for the alkylation of 1 or 23. Reagents and conditions: (a) CBr4, PPh3, CH2Cl2, 0 °C→rt; (b) N-methylmorpholine-N-oxide, OsO4, acetone, H2O, rt; (c) 2,2-dimethoxypropane, TsOH, rt; (d) NaH, ethyleneglycol, DMF, rt; (e) NaIO4, H2O, THF, rt; (f) NaBH4, H2O, THF, rt; (g) CH3OCH2Br, CH2Cl2, iPr2EtN, 0 °C.
Scheme 2.
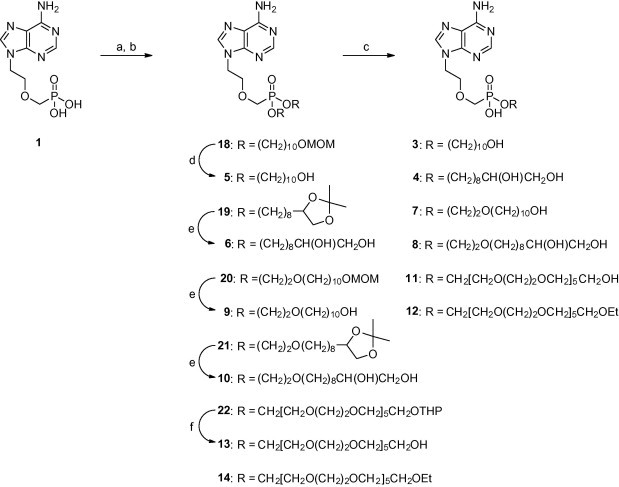
The preparation of prodrugs 3–14. Reagents and conditions: (a) Bu4NOH, MeOH; (b) 23, 24, 26–29, DMF, 100 °C; (c) LiN3, DMF, 100 °C; (d) HCl, MeOH, 65 °C; (e) Dowex 50WX8-400 [H+], MeOH, H2O, 60 °C; (f) AcOH, H2O, 50 °C.
The hexaethyleneglycol moiety was coupled to phosphonates 1 and 2 via its monotosylate. Hexaethyleneglycol was tosylated with one equivalent of tosylchloride with silver oxide as a base.10 The remaining hydroxyl group was protected as THP ether. The reaction of the prepared 28 with sodium ethoxide was followed by THP deprotection and tosylation. The three-step procedure yielded compound 29 (see Scheme 6 ). Both tosylates 28 and 29 were utilized for PMEA (1) or cHPMPC (23) alkylation (see Scheme 2). The THP protecting group of synthesized phosphonate esters 22 and 24 was cleaved in the presence of Dowex 50.
Scheme 6.
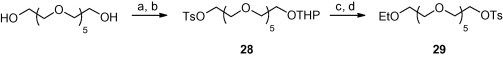
The preparation of protected hexaethylene glycol monotosylates for the alkylation of 1 and 23. Reagents and conditions: (a) TsCl, Ag2O, KI, CH2Cl2, 0 °C; (b) 3,4-dihydro-2H-pyran, PPTS, CH2Cl2, 40 °C; (c) EtOH, NaH, THF, r.t.; (d) Dowex 50WX8-400 [H+], MeOH, rt; (e) TsCl, Et3N, CH2Cl2, rt.
3. Biological activities
The antiviral activity of the different compounds was evaluated against various DNA viruses, including poxviruses [i.e., vaccinia virus (VACV)], herpesviruses [i.e., herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2), thymidine kinase-deficient HSV-1 (acyclovir-resistant, ACVr), varicella-zoster virus (VZV) and human cytomegalovirus (HCMV)] (Table 1 ). The compounds were also evaluated against retroviruses [(i.e., human immunodeficiency virus type 1 (HIV-1) and type 2 (HIV-2)] (Table 2 ). All of the compounds were also examined against several RNA viruses, including vesicular stomatitis virus (VSV), Coxsackie B4, respiratory syncytial virus (RSV), parainfluenza virus type 3, reovirus-1, Sindbis virus and Punta Toro virus (Table 2). None of the prodrugs showed activity against the different RNA viruses, except for HIV-1 and HIV-2. However, a loss in the antiviral activities of the hydroxylated decyl or decyloxyethyl and hexaethyleneglycol esters of PMEA (compounds 3–14) against HIV was observed since compound 7 {2-[(10-Hydroxydecyl)oxy]ethyl ester of PMEA} was the only compound among this type of prodrugs showing some activity against HIV at nontoxic concentrations. Thus, compound 7 was able to inhibit HIV replication with 50% effective concentrations (EC50) which were ∼10-fold higher than those observed for PMEA. In contrast, the (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl ester of PMEA (compound 15) had anti-HIV activities (EC50 values of 10.6 μM and 9.3 μM for HIV-1 and HIV-2, respectively) equivalent to that of PMEA (EC50 values of 10.4 and 8.6 μM for HIV-1 and HIV-2, respectively).
Table 1.
Antiviral and cytotoxic activity of the compounds against herpes- and poxviruses in cell culture
| Compound | Antiviral activity: EC50 (μM)a |
Cytotoxicity (μM) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HSV-1 |
HSV-2 | VZV |
HCMV |
Feline | Vaccinia virus | Cell morphology | Cell growth | |||||
| KOS | KOS ACVr | G strain | OKA | 07/1. | AD-169 | Davis | herpesvirus | (MCC)b | (CC50)c |
|||
| (HEL) | (HEL) | (HEL) | (HEL) | (HEL) | (HEL) | (HEL) | (CRFK) | (HEL) | (HEL) | (HEL) | CRFK | |
| 3 | >233 | >233 | >233 | 114 | 64.3 | >233 | >233 | >233 | >233 | >233 | >233 | >233 |
| 4 | >224 | >224 | >224 | >224 | >224 | >224 | >224 | >9 | >224 | >224 | >224 | 9.4 |
| 5 | >171 | >171 | >171 | >34 | >34 | >34 | >34 | >171 | >171 | 171 | >171 | >171 |
| 6 | >162 | >162 | >162 | 99 | 60.7 | >162 | >162 | >162 | >162 | >162 | >162 | >162 |
| 7 | 106 | 122 | 122 | 26 | 18.4 | >211 | >211 | >211 | >211 | >211 | >211 | >211 |
| 8 | >204 | >204 | >204 | 61.4 ± 57.4 | ⩾72.7 ± 41.5 | >204 | >204 | >204 | >204 | >204 | >204 | >204 |
| 9 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | 148 | 62.5 | >148 |
| 10 | >142 | >142 | >142 | >142 | >142 | >28.3 | >142 | >142 | >142 | ⩾142 | >142 | >142 |
| 11 | >186 | >186 | >186 | 18.6 ± 5.2 | 40.9 ± 18.4 | >186 | >186 | >186 | >186 | >186 | >186 | >186 |
| 12 | >177 | >177 | >177 | 74.3 | 69 | >177 | >177 | >177 | >177 | >177 | 146 | >177 |
| 13 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 |
| 14 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 |
| 15 | 51.9 | 51.9 | 98.6 | 1.92 ± 0.8 | 7.6 ± 2.4 | 259 | 259 | >259 | >259 | >259 | 117.2 ± 71.8 | >259 |
| 16 | 16.5 | 12.9 | 7.4 | 1.91 ± 0.6 | 0.60 ± 0.01 | 0.80 ± 0.20 | 0.70 ± 0.05 | 14.5 | 107 | >184 | >184 | >184 |
| 17 | 1.75 | 3.5 | 1.4 | 1.78 ± 1.09 | 0.64 ± 0.04 | 0.95 ± 0.18 | 0.61 ± 0.13 | 2.8 | 15.7 | >175 | >175 | >175 |
| Acyclovir | 0.27 ± 0.15 | 27.1 | 0.23 ± 0.15 | 3.0 ± 2.2 | 69.3 ± 23.5 | N.D.d | N.D. | N.D. | >250 | >222 | ⩾1421 ± 422 | N.D. |
| Brivudin | 0.028 ± 0.021 | 274.3 | 45.7 ± 41.7 | 0.013 ± 0.01 | ⩾105.4 ± 36.9 | N.D. | N.D. | N.D. | 4.0 ± 2.3 | >150 | 470 ± 165 | N.D. |
| Ganciclovir | 0.03 ± 0.02 | 15.7 | 0.027 ± 0.0058 | N.D. | N.D. | 6.0 ± 3.4 | 6.54 ± 4.0 | 8.1 | >250 | >392 | 435 ± 447 | >394 |
| (S)-HPMPC | 1.6 ± 1.0 | 2.0 ± 1.4 | 1.43 ± 0.60 | 0.29 ± 0.25 | 0.10 ± 0.06 | 0.92 ± 0.82 | 0.94 ± 0.67 | N.D. | 7.0 ± 3.8 | >317 | 234 ± 182 | N.D. |
| PMEA | 42.5 ± 13.2 | 41.4 ± 6.6 | 24.9 ± 14.6 | 6.7 ± 4.1 | 5.0 ± 0.8 | 117.6 ± 48.0 | 114.6 ± 28.2 | N.D. | >183 | >183 | ||
Effective concentration required to reduce virus-induced cytopathicity by 50%.
Minimum cytotoxic concentration that causes a microscopically detectable alteration of the cell morphology.
Cytotoxic concentration required to reduce cell growth by 50%.
Not determined.
Table 2.
Antiviral and cytotoxic activity of the compounds against RNA and retroviruses in cell culture
| Compound | Antiviral activity: EC50 (μM)a |
Cytotoxicity (μM) |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HIV-1 | HIV-2 | VSV | RSV | Coxackie B4 | Para | Reovirus-1 | Sindbis | Punta Toro | Feline | Influenza A | Influenza A | Influenza B | Cell morphology | Cell growth | |||||
| influenza-3 | coronavirus | H1N1 | H3N2 | (MCC)b | (CC50)c | ||||||||||||||
| (CEM) | (CEM) | (HEL) | (HeLa) | (HeLa) | (HeLa) | (Vero) | (Vero) | (Vero) | (Vero) | (Vero) | (CRFK) | (MDCK) | (MDCK) | (MDCK) | Vero | MDCK | HeLa | (CEM) | |
| 3 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 | >233 |
| 4 | >224 | >224 | >224 | >224 | >224 | >224 | >224 | >224 | >224 | >224 | >224 | >9 | >224 | >224 | >224 | >224 | >224 | >224 | >224 |
| 5 | >171 | >171 | >171 | >171 | 99 ± 0 | >171 | >34 | >34 | >34 | >34 | >34 | >171 | >171 | >171 | >171 | 171 | >171 | >171 | >171 |
| 6 | >162 | >162 | >162 | >162 | >162 | >162 | >162 | >162 | >162 | >162 | >162 | >162 | >32,4 | >32,4 | >32,4 | >162 | >162 | >162 | >162 |
| 7 | 106/122 | 122/95 | >211 | >211 | >211 | >211 | >211 | >211 | >211 | >211 | >211 | >211 | >42 | >42 | >42 | >211 | >211 | >211 | >211 |
| 8 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 | >204 |
| 9 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >29.7 | >5.9 | >5.9 | >5.9 | 148 | 29.7 | 148 | 13.5 |
| 10 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 | >142 |
| 11 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 | >186 |
| 12 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 | >177 |
| 13 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 | >125 |
| 14 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 | >117 |
| 15 | 10.6 ± 8.3 | 9.3 ± 1.7 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | >259 | 88 ± 9.1 |
| 16 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 | >184 |
| 17 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 | >175 |
| Oseltamivir | N.D.d | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 12 ± 6.5 | 90 ± 21 | 23.5 ± 15.2 | N.D. | >100 | N.D. | N.D. |
| Ribavirin | N.D. | N.D. | N.D. | 14.8 ± 10.7 | 3.0 ± 10.7 | 69 ± 50 | >250 | 121 ± 99 | ⩾174 ± 96 | >250 | 125 ± 98 | N.D. | 9.8 ± 1.5 | 12.5 ± 5.2 | 7.3 ± 2.4 | N.D. | >100 | >250 | N.D. |
| PMEA | 10.4 ± 4.9 | 8.6 ± 3.4 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | >250 | 164 ± 37 |
Effective concentration required to reduce virus-induced cytopathicity by 50%.
Minimum cytotoxic concentration that causes a microscopically detectable alteration of cell morphology.
Cytotoxic concentration required to reduce cell growth by 50%
Not determined.
The 2-[(10-Hydroxydecyl)oxy]ethyl ester of PMEA (7) exhibited also activities against HSV-1, HSV-2 and VZV. Similarly to HIV, the anti-herpesvirus activities of the prodrugs 3–14 decreased when compared to the parent compound, with compound 7 being the most active one among these prodrugs. In the case of HSV and VZV, the drop in antiviral activities was inferior to that observed for HIV since the EC50 values for compound 7 against HSV and VZV were three- to four-fold lower than those for PMEA. However, compound 7 was totally inactive against CMV. The (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl ester 15 had anti-herpesvirus activities. The activity of compound 15 against herpesviruses was equivalent (HSV-1 and VZV) or two- (CMV) to four-fold (HSV-2) lower than that observed for the parent compound PMEA. Other esters of PMEA, that is, compounds 6, 8, 11 and 12, showed weak activities only against VZV, with EC50 values in the range of 18–114 μM, which is 3- to 15-fold higher than the EC50 values for the parent compound. None of the PMEA prodrugs showed activity against vaccinia virus or feline herpesvirus, like the parent compound PMEA.
The two prodrugs of (S)-HPMPC, that is, compounds 16 {14-hydroxy-3,6,9,12-tetraoxaheptadecyl ester of (S)-1-[3-hydroxy-2-(phophonomethoxy)propyl]cytosine} and 17 {3,6,9,12,15-pentaoxaeicosyl ester of (S)-1-[3-hydroxy-2-(phophonomethoxy)propyl]cytosine} were able to inhibit the replication of the different herpesviruses tested and the poxvirus vaccinia virus. Both prodrugs had activities equivalent to that of (S)-HPMPC against CMV, with EC50 values in the range of 0.6–0.95 μM as compared to 0.9 μM for (S)-HPMPC. However, compounds 16 and 17 had six- to seven-fold lower activities than (S)-HPMPC against VZV. Compound 17 had activities against HSV, feline herpesvirus, and vaccinia virus similar to those found for (S)-HPMPC while the activities of compound 16 against these viruses were 5- to 15-fold lower.
As already mentioned, prodrugs 16 and 17 represent the penta oxy-analogs of alkyloxyalkyl prodrugs developed by Hostetler7b Table 3 compares some antiviral activities of 12 (HIV), 16 and 17 (CMV) with analogous simple alkoxyalkyl ester.11, 12 The comparison demonstrates that insertion of oxygen atoms into the chain in 16 and 17 caused 400- to 1000-fold drop of activity against CMV. The loss of anti-HIV activity of 12 is even more significant (>107-fold compared to HDP–PMEA). Comparison of activities of compounds 3–11 with HDP–PMEA is not possible because different chain length plays a role. We can only speculate based on the relationship between activity and chain length, as observed with Cidofovir prodrugs.11 The adequate chain shortening from 20 atoms (HDP) to 12 atoms (octyloxypropyl) caused circa 1000-fold drop in anti-CMV activity. However, the decrease in antiviral activity of compounds 3–8 is more significant (∼107-fold compared to HDP–PMEA) and is hardly accountable solely to the chain shortening.
Table 3.
Antiviral activity of compounds 16 and 17 compared to TDP-(S)-HPMPC and HDP-(S)-HPMPC and of compound 12 compared to HDP–PMEA
| Compound | Number of atoms in alkyloxyalkyl chain | Type of alkyloxyalkyl chain | CMV (AD-169)/EC50 (μM) |
|---|---|---|---|
| 16 | 18 | Hydroxytetraoxaheptadecyl- | 0.80 ± 0.20 |
| TDP (S)-HPMPC | 18 | Tetradecyloxypropyl- | 0.002 ± 0.002a |
| 17 | 20 | Pentaoxaeicosyl- | 0.95 ± 0.18 |
| HDP (S)-HPMPC | 20 | Hexadecyloxypropyl- | 0.0009 ± 0.0001a |
| (S)-HPMPC | 0.92 ± 0.82 | ||
| (S)-HPMPC | 1.20 ± 0.43a | ||
| HIV-1/EC50 (μM) | |||
| 12 | 20 | Pentaoxaeicosyl- | >177 |
| HDP–PMEA | 20 | Hexadecyloxypropyl- | 0.000015 ± 0.00003b |
| PMEA | 10.4 ± 4.9 | ||
| PMEA | 1.1 ± 0.6b | ||
The superior antiviral activity of HDP esters is explained by the interplay of increased permeation through the cell membrane and the by-pass of the phosphorylation step leading to the rapid formation of bioactive Cidofovir diphosphate.7b Since the prodrugs 3–14, 16 and 17 are less active than the parent drug in some cases, decreased cellular uptake or slow cleavage of the promoiety by intracellular phospholipase C may be the reason for their poor antiviral activities.
4. Conclusions
A set of new amphiphilic prodrugs of PMEA (Adefovir) and (S)-HPMPC (Cidofovir) has been prepared and evaluated in vitro. The phosphonate group of PMEA was masked with a hexaethyleneglycol unit or hydroxylated decyl or decyloxyethyl chain. The first prodrug type was applied also to (S)-HPMPC (Cidofovir). As a less toxic analog of Adefovir Dipivoxyl, the (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl ester of PMEA was prepared. However, complete masking of the phosphonate group with the dioxolenone unit was not accomplished. A loss in the antiviral activities of hexaethyleneglycol esters and hydroxylated decyl or decyloxyethyl esters of PMEA was noted while the (5-methyl-2-oxo-1,3-dioxolen-4-yl)methyl ester of PMEA showed significant activities against HIV and herpesviruses. (S)-HPMPC prodrugs bearing hexaethyleneglycol pro-moiety exhibited significant activities against cytomegalovirus, which were in the same range as (S)-HPMPC. Activities against herpesviruses and poxviruses were one- to fifteen-fold lower than that of (S)-HPMPC. Considering these results, we assume that the prodrugs 3–14 and 16–17 are taken up less efficiently or are not suitable substrates for phospholipase C.
5. Experimental
Unless stated otherwise, solvents were evaporated at 40 °C and compounds were dried at 100 Pa. The purification of the products by reverse phase HPLC technique was performed on a Waters Delta 600 instrument with a Waters 2487 Dual λ Absorbance Detector using Luna Phenomenex® C-18 preparative columns (10 μM, 21 × 250 mm, flow 12 ml/min); the elution conditions are given in the text. The column chromatography was performed on 60 μM silica gel (Fluka). The 1H and 13C NMR spectra were measured in CDCl3, D2O or DMSO-d 6 on a Bruker Avance II 600 spectrometer (1H at 600 MHz, 13C at 151 MHz) or Bruker Avance II 500 spectrometer (1H at 500 MHz, 13C at 125.7 MHz). The spectra were referenced to TMS or dioxane (δ 3.75 and 67.19) as internal standards or to the residual solvent signal (δ 2.50 and 39.7 for DMSO). The 31P NMR spectra were measured on Bruker Avance II 500 spectrometer (202.3 MHz) in CDCl3 using H3PO4 as the external standard. The general numbering for the assignment of the NMR signals is depicted in Figure 3 . Mass spectra were measured by the ESI technique using LCQ Fleet or LTQ Orbitrap XL (Thermo Fisher Scientific). Most of the chemicals and ion-exchange resins were purchased from Sigma-Aldrich (Czech Republic).
Figure 3.
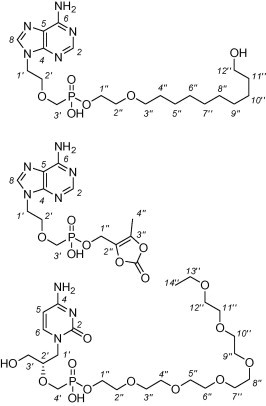
The general numbering for the assignment of the NMR signals.
5.1. General procedure for the preparation of PMEA diesters
PMEA (1) (273 mg, 1.0 mmol) was suspended in MeOH (5 ml) and dissolved by a solution of tetrabutylamomonium hydroxide (2.0 ml, 1 M solution in MeOH). The solution was then evaporated and co-distilled with isopropanol (5 ml) and toluene (3 × 5 ml). The syrupy residue was dissolved in DMF (4 ml) and appropriate bromide (23, 24, 26 or 27) or tosylate (28 or 29) (2.1 mmol) was added. The mixture was stirred at 100 °C (8–12 h), evaporated, after which the residue was chromatographed on a silica gel column (80 g) in 10% MeOH/CHCl3 (for compounds 12 and 13) or 6% MeOH/CHCl3 for all of the others.
5.1.1. Bis(10-Hydroxydecyl) ester of 9-[2-(phosphonomethoxy)ethyl]adenine (5)
Compound 18, prepared by a general procedure from 1 and 26, was refluxed in methanolic hydrochloric acid (5 ml, pH ∼2) for 6 h. The mixture was evaporated, co-distilled with ethanol and crystallized from EtOH–Et2O. Yield: 270 mg (46%) of crystals; mp 101–104 °C. 1H NMR (DMSO-d 6, ppm) δ: 1.20–1.28 (m, 24H, H-3′′–H-8′′), 1.39 (m, 4H, H-9′′), 1.48 (m, 4H, H-2′′), 3.36 (t, 4H, J 10′′–9′′ = 6.6, H-10′′), 3.80–3.88 (m, 6H, H-1′′, H-3′), 3.91 (t, 2H, J 2′–1′ = 5.0, H-2′), 4.43 (t, 2H, J 1′–2′ = 5.0, H-1′), 8.44 (s, 1H, H-8), 8.51 (s, 1H, H-2), 8.80 (b s, NH), 9.46 (b s, NH). 13C NMR (DMSO-d 6, ppm) δ: 25.10 (C-3′′), 25.71 (C-8′′), 28.71, 29.13, 29.15, 29.24 (C-4′′, C-5′′, C-6′′, C-7′′), 30.11 (d, J 2′′–P = 5.5, C-2′′), 32.73 (C-9′′), 43.42 (C-1′), 60.89 (C-10′′), 63.83 (d, J 3′–P = 162.7, C-3′), 65.76 (d, J 1′′–P = 6.5, C-1′′), 70.19 (d, J 2′–P = 11.2, C-2′), 118.03 (C-5), 144.24 (C-8), 145.39 (C-2), 148.76 (C-4), 150.72 (C-6). 31P NMR (CDCl3, ppm) δ: 21.38 (m). ESI–MS, m/z: 584.2 (100) [M−H]−, 585.2 (32) [M−H]−, 620.0 (18), 652.2 (18). ESI–HRMS calcd for C28H51N5O6P 584.3582, found: 584.3578 [M−H]−.
5.1.2. Bis(9,10-Dihydroxydecyl) ester of 9-[2-(phosphonomethoxy)ethyl]adenine (6)
Compound 19, prepared by a general procedure from 1 and 23, was treated with Dowex 50WX8-400 [H+] in methanol/water (1:1, 20 ml) at 60 °C for 1 h. The resin was filtered off, washed with conc. ammonia/methanol (1:4). Yield: 225 mg (46%) of crystallizing syrup. Crystallization from EtOH–Et2O; mp 72–73 °C. 1H NMR (DMSO-d 6, ppm) δ: 1.16–1.27 (m, 20H, H-3′′–H-6′′, H-7′′a, H-8′′a), 1.35–1.41 (m, 4H, H-7′′b, H-8′′b), 1.47 (m, 4H, H-2′′), 3.19–3.27 (m, 4H, H-10′′), 3.37 (m, 2H, H-9′′), 3.80–3.89 (m, 8H, H-1′′, H-3′, H-2′), 4.32 (t, 2H, J 1′–2′ = 5.1, H-1′), 4.35 (d, 2H, J OH–9′′ = 5.0, OH), 4.43 (t, 2H, J OH–10′′ = 5.7, OH), 7.19 (b s, 2H, NH2), 8.06 (s, 1H, H-8), 8.13 (s, 1H, H-2). 13C NMR (DMSO-d 6, ppm) δ: 25.13 (C-3′′), 25.38 (C-7′′), 28.74, 29.23, 29.44 (C-4′′, C-5′′, C-6′′), 30.13 (d, J 2′′–P = 5.5, C-2′′), 33.62 (C-8′′), 42.58 (C-1′), 63.86 (d, J 3′–P = 162.7, C-3′), 65.79 (d, J 1′′–P = 6.5, C-1′′), 66.21 (C-10′′), 70.48 (d, J 2′–P = 11.5, C-2′), 71.29 (C-9′′), 118.77 (C-5), 141.18 (C-8), 149.71 (C-4), 152.55 (C-2), 156.14 (C-6). 31P NMR (CDCl3, ppm) δ: 21.37 (m). ESI–MS, m/z: 616.2 (100) [M−H]−, 617.2 (30) [M−H]−, 662.0 (27), 684.2 (10), 900.1 (12). ESI–HRMS calcd for C28H51N5O8P 616.3481, found: 616.3455 [M−H]−.
5.1.3. Bis{2-[(10-Hydroxydecyl)oxy]ethyl} ester of 9-[2-(phosphonomethoxy)ethyl]adenine (9)
Compound 20, prepared by a general procedure from 1 and 27, was treated with Dowex 50WX8-400 [H+] in methanol/water (1:1, 20 ml) at 60 °C for 10 h. The resin was filtered off, washed with conc. ammonia/methanol (1:4). Yield: 276 mg (41%) of solid; mp 52 °C. 1H NMR (DMSO-d 6, ppm) δ: 1.20–1.27 (m, 24H, H-5′′–H-10′′), 1.38 (m, 4H, H-11′′), 1.44 (m, 4H, H-4′′), 3.30–3.38 (m, 8H, H-3′′, H-12′′), 3.45 (t, 4H, H-2′′, J 2′′–1′′ = 4.7, H-2′′), 3.87–3.89 (m, 4H, H-3′, H-2′), 3.99 (m, 4H, H-1′′), 4.30–4.33 (m, 2H, H-1′′, OH), 7.18 (b s, 2H, NH2), 8.07 (s, 1H, H-8), 8.12 (s, 1H, H-2). 13C NMR (DMSO-d 6, ppm) δ: 25.70 (C-10′′), 25.80 (C-5′′), 29.10, 29.16, 29.22, 29.28, 29.34 (C-4′′, C-6′′, C-7′′, C-8′′, C-9′′), 32.74 (C-11′′), 42.58 (C-1′), 60.91 (C-12′′), 63.99 (d, J 3′–P = 163.2, C-3′), 65.03 (d, J 1′′–P = 6.5, C-1′′), 69.29 (d, J 2′′–P = 5.6, C-2′′), 70.44 (C-3′′), 70.49 (d, J 2′–P = 11.6, C-2′), 118.75 (C-5), 141.19 (C-8), 149.68 (C-4), 152.53 (C-2), 156.13 (C-6). 31P NMR (CDCl3, ppm) δ: 22.17 (m). ESI–MS, m/z: 255.3 (11), 283.3 (21), 672.2 (100) [M−H]−, 673.3 (32) [M−H]−, 694.2 (24), 740.3 (13), 956.1 (11). ESI–HRMS calcd for C32H59N5O8P 672.4107, found: 672.4085 [M−H]−.
5.1.4. Bis{2-[(9,10-Dihydroxydecyl)oxy]ethyl ester of 9-[2-(phosphonomethoxy)ethyl]adenine (10)
Compound 21, prepared by a general procedure from 1 and 24, was treated with Dowex 50WX8-400 [H+] in methanol/water (1:1, 20 ml) at 60 °C for 1 h. The resin was filtered off, washed with conc. ammonia/methanol (1:4). Yield: 279 mg (40%) of thick syrup. 1H NMR (DMSO-d 6, ppm) δ: 1.17–1.28 (m, 20H, H-5′′–H-8′′, H-9′′a, H-10′′a), 1.34–1.41 (m, 4H, H-9′′b, H-10′′b), 1.45 (m, 4H, H-4′′), 3.23 (m, 4H, H-12′′), 3.31–3.38 (m, 8H, H-3′′, H-11′′), 3.45 (t, 4H, J 2′′–1′′ = 4.6, H-2′), 3.87–3.89 (m, 4H, H-2′, H-3′), 3.99 (m, 4H, H-1′′), 4.31–4.33 (m, 4H, C-1′, OH), 4.41 (t, 2H, J OH–12′′ = 5.7, OH), 7.18 (b s, 2H, NH2), 8.08 (s, 1H, H-8), 8.12 (s, 1H, H-2). 13C NMR (DMSO-d 6, ppm) δ: 25.37 (C-9′′), 25.81 (C-5′′), 29.09, 29.29, 29.35, 29.46 (C-4′′, C-6′′, C-7′′, C-8′′), 33.60 (C-10′′), 42.60 (C-1′), 63.99 (d, J 3′–P = 163.3, C-3′), 65.04 (d, J 1′′–P = 6.4, C-1′′), 66.19 (C-12′), 69.29 (d, J 2′′–P = 5.7, C-2′′), 70.45 (C-3′′), 70.50 (d, J 2′–P = 11.2, C-2′), 71.28 (C-11′′), 118.75 (C-5), 141.21 (C-8), 149.69 (C-4), 152.54 (C-2), 156.13 (C-6). 31P NMR (CDCl3, ppm) δ: 22.84 (m). ESI–MS, m/z: 704.2 (100) [M−H]−, 705.5 (33) [M−H]−, 772.2 (11). ESI–HRMS calcd for C32H59N5O10P 704.4005, found: 704.3981 [M−H]−.
5.1.5. Bis(14-Hydroxy-3,6,9,12-tetraoxooctadecyl) ester of 9-[2-(phosphonomethoxy)ethyl]adenine (13)
The ester 22, prepared by a general procedure from 1 and 28, was contaminated with tetrabutylammonium salts. The mixture was treated with Dowex 50WX8-400 [H+] in methanol at 40 °C. The suspension was put on a Dowex 50WX8-400 [H+] column. Rinsing with water was followed by 1% ammonia. The UV-adsorbing eluate was evaporated. Yield: 375 mg (47%) of syrup. 1H NMR (DMSO-d 6, ppm) δ: 3.40 (m, 4H, H-11′′), 3.46–3.51 (m, 40H, H-2′′–H-10′′, H-12′′), 3.87–3.90 (m, 4H, H-2′, H-3′), 4.00 (m, 4H, H-1′′), 4.32 (t, 2H, J 1′–2′ = 5.1, H-1′), 4.59 (t, J OH–12′′ = 5.5, OH), 7.20 (b s, 2H, NH2), 8.08 (s, H-8), 8.13 (s, H-2). 13C NMR (DMSO-d 6, ppm) δ: 42.64 (C-1′), 60.40 (C-12′′), 63.95 (d, J 3′–P = 162.7, C-3′), 65.04 (d, J 1′′–P = 6.5, C-1′′), 69.66–70.00 (m, C-2′′–C-10′′), 70.46 (d, J 2′–P = 11.2, C-2′), 72.54 (C-11′′), 118.76 (C-5), 141.29 (C-8), 149.70 (C-4), 152.57 (C-2), 156.14 (C-6). 31P NMR (CDCl3, ppm) δ: 22.84 (m). ESI–MS, m/z: 413.0 (18), 423.9 (31), 802.3 (14) [M+H]+, 824.4 (100) [M+Na]+, 825.4 (37) [M+Na]+, 826.4 (10) [M+Na]+. ESI–HRMS calcd for C32H61N5O16P 802.3845, found: 802.3844 [M+H]+.
5.1.6. Bis(3,6,9,12,15,18-Hexaoxaeicosyl) ester of 9-[2-(phosphonomethoxy)ethyl]adenine (14)
The product obtained according to the general procedure using tosylate 29 was contaminated with tetrabutylammonium bromide. It was further purified on a Dowex 50WX8-400 [H+] column (5 ml). Rinsing with water was followed by an elution of 1% ammonia. The UV-absorbing fractions were evaporated and co-distilled with ethanol and toluene. Yield: 430 mg (50%) of syrup. 1H NMR (DMSO-d 6, ppm) δ: 1.08 (t, 6H, J 13′′–14′′ = 7.0, H-14′′), 3.41 (q, 4H, J 14′′–13′′ = 7.0, H-13′′), 3.43–3.52 (m, 44H, H-2′′–H-12′′), 3.87–3.90 (m, 4H, H-2′, H-3′), 4.00 (m, 4H, H-1′′), 4.32 (t, 2H, J 1′–2′ = 5.2, H-1′), 7.18 (b s, 2H, NH2), 8.08 (s, 1H, H-8), 8.13 (s, 1H, H-2). 13C NMR (DMSO-d 6, ppm) δ: 15.32 (C-14′′), 42.63 (C-1′), 63.96 (d, J 3′–P = 162.8, C-3′), 65.04 (d, J 1′′–P = 6.5, C-1′′), 65.74 (C-13′′), 69.42–70.03 (m, C-2′′–C-12′′), 70.46 (d, J 2′–P = 11.0, C-2′), 118.77 (C-5), 141.28 (C-8), 149.70 (C-4), 152.56 (C-2), 156.15 (C-6). 31P NMR (CDCl3, ppm) δ: 22.84 (m). ESI–MS, m/z: 333.3 (15), 440.9 (22), 451.9 (42), 858.3 (17) [M+H]+, 880.4 (100) [M+Na]+. ESI–HRMS calcd for C36H69N5O16P 858.4471, found: 858.4468 [M+H]+.
5.2. General procedure for the preparation of PMEA monoesters
The appropriate diester (0.16 mmol) and LiN3 (78 mg, 1.6 mmol) in DMF (1 ml) was heated to 100 °C. The conversion was monitored by means of TLC (CHCl3/MeOH, 9:1). The conversion was complete in 10–16 h.
5.2.1. 10-Hydroxydecyl ester of 9-[2-(phosphonomethoxy)ethyl]adenine (3)
The product was crystallized overnight from the reaction mixture. Recrystallization from H2O–EtOH afforded 50 mg (73%); mp 181–183 °C. 1H NMR (D2O, ppm) δ: 0.85–0.95 (m, 6H), 1.03 (m, 2H), 1.11 (m, 2H, H-3′′–H-7′′), 1.18–1.24 (m, 4H, H-2′′, H-8′′), 1.48 (m, 2H, H-9′′), 3.55–3.62 (m, 6H, H-3′, H-1′′, H-10′′), 3.86 (t, 2H, J 2′–1′ = 4.9, H-2′), 4.40 (t, 2H, J 1′–2′ = 4.9, H-1′), 8.15 (s, 1H, H-2), 8.18 (s, 1H, H-8). 13C NMR (D2O, ppm) δ: 25.27 (C-3′′), 25.64 (C-8′′), 28.75, 29.10, 29.15, 29.17 (C-4′′–C-7′′), 30.33 (d, J 2′′–P = 6.1, C-2′′), 31.95 (C-9′′), 44.34 (C-1′), 62.49 (C-10′′), 66.04 (d, J 1′′–P = 5.8, C-1′′), 66.54 (d, J 3′–P = 157.9, C-3′), 71.12 (d, J 2′–P = 14.1, C-2′), 118.79 (C-5), 143.58 (C-8), 149.25 (C-4), 152.80 (C-2), 155.94 (C-6). 31P NMR (D2O, ppm) δ: 18.03 (m). ESI–MS, m/z: 428.2 (100) [M−H]−, 429.3 (21) [M−H]−, 856.8 (15) [2M−H]−, 879.3 (11); ESI–HRMS calcd for C18H31N5O5P 428.2068, found: 428.2072 [M−H]−.
5.2.2. 9,10-Dihydroxydecyl ester of 9-[2-(phosphonomethoxy)ethyl]adenine (4)
The product was crystallized overnight from the reaction mixture. The recrystallization from H2O–EtOH afforded 52 mg (73%); mp 257–258 °C. 1H NMR (D2O, ppm) δ: 0.85–1.44 (m, 14H, H-2′′–H-8′′), 3.44 (dd, J gem. = 11.7, J 10a′′–9′′ = 7.0, H-10′′a), 3.54–3.58 (m, 3H, H-1′′, H-10′′b), 3.61 (d, 2H, J 3′–P = 9.2), 3.65 (m, 1H, H-9′′), 3.87 (m, 2H, H-2′), 4.42 (m, 2H, H-1′), 8.18 (s, 1H, H-2), 8.20 (s, H-8). 13C NMR (D2O, ppm) δ: 25.28, 25.30 (C-3′′, C-7′′), 28.74, 29.09, 29.21 (C-4′′, C-5′′, C-6′′), 30.34 (d, J 2′′–P = 5.7, C-2′′), 32.85 (C-8′′), 44.36 (C-1′), 66.05 (d, J 1′′–P = 6.1, C-1′′), 66.09 (C-10′′), 66.53 (d, J 3′–P = 157.8, C-3′), 71.09 (d, J 2′–P = 14.0, C-2′), 72.49 (C-9′′), 118.88 (C-5), 143.68 (C-8), 149.38 (C-4), 152.87 (C-2), 156.03 (C-6). 31P NMR (D2O, ppm) δ: 18.02 (m). ESI–MS, m/z: 444.3 (100) [M−H]−, 445.3 (19) [M−H]−. ESI–HRMS calcd for C18H31N5O6P 444.2026, found: 444.2017 [M−H]−.
5.2.3. 2-[(10-Hydroxydecyl)oxy]ethyl ester of 9-[2-(phosphonomethoxy)ethyl]adenine (7)
The reaction mixture was evaporated, the residue was deionized on Dowex 50WX8-400 column ([H+], 4 ml, gradient water→2% aqueous ammonia) and then on a Dowex 1 column ([AcO−], 4 ml, gradient water→1% aqueous acetic acid). Yield: 67 mg (88%), mp 169–171 °C. 1H NMR (D2O, ppm) δ: 1.02–1.21 (m, 10H, H-5′′–H-9′′), 1.25 (m, 2H, H-10′′), 1.30 (m, 2H, H-4′′), 1.49 (m, 2H, H-11′′), 3.21 (t, 2H, J 3′′–4′′ = 6.6, H-3′′), 3.35 (m, 2H, H-2′′), 3.57 (t, 2H, J 12′–11′ = 6.7, H-12′′), 3.64 (d, 2H, J 3′–P = 9.1, H-3′), 3.78 (m, 2H, H-1′′), 3.89 (m, 2H, H-2′), 4.42 (m, 2H, H-1′), 8.17 (s, 1H, H-2), 8.21 (s, 1H, H-8). 13C NMR (D2O, ppm) δ: 25.65 (C-10′′), 25.74 (C-5′), 29.02, 29.06, 29.12, 29.23 (C-4′′, C-6′′–C-9′′), 31.94 (C-11′′), 44.37 (C-1′), 62.50 (C-12′′), 64.34 (d, J 1′′–P = 5.7, C-1′′), 66.82 (d, J 3′–P = 158.8, C-3′), 70.23 (d, J 2′′–P = 6.9, C-2′′), 71.17 (d, J 2′–P = 13.7, C-2′), 71.30 (C-3′′), 118.84 (C-5), 143.57 (C-8), 149.33 (C-4), 152.89 (C-2), 156.01 (C-6). 31P NMR (D2O, ppm) δ: 18.30 (m). ESI–MS, m/z: 472.3 (100) [M−H]−, 473.3 (22) [M−H]−, 944.8 (44), 945.9 (16) [2M−H]−. ESI–HRMS calcd for C20H35N5O6P 472.2330, found: 472.2333 [M−H]−.
5.2.4. 2-[(9,10-Dihydroxydecyl)oxy]ethyl ester of 9-[2-(phosphonomethoxy)ethyl]adenine (8)
The reaction mixture was evaporated, the residue was deionized on a Dowex 50WX8-400 column ([H+], 4 ml, gradient water→2% aqueous ammonia) and then on Dowex 1 column ([AcO−], 4 ml, gradient water→1% aqueous acetic acid). Yield: 72 mg (92%), mp 156 °C. 1H NMR (D2O, ppm) δ: 1.02–1.46 (m, 14H, H-4′′–H-10′′), 3.20 (t, 2H, J 3′′–4′′ = 6.7, H-3′′), 3.33 (m, 2H, H-2′′), 3.44 (dd, 1H, J gem = 11.7, J 12b′′–11′′ = 7.0, H-12b′′), 3.57 (dd, 1H, J gem = 11.7, J 12a′′–11′′ = 3.8, H-12a′′), 3.62–3.68 (m, 4H, H-3′, H-11′′), 3.76 (m, 2H, H-1′′), 3.89 (m, 2H, H-2′), 4.43 (m, 2H, H-1′), 8.19 (s, 1H, H-2), 8.22 (s, 1H, H-8). 13C NMR (D2O, ppm) δ: 25.30 (C-9′′), 25.72 (C-5′′), 28.98, 29.05, 29.15, 29.24 (C-4′′, C-6′′–C-8′′), 32.86 (C-10′′), 44.37 (C-1′), 64.33 (d, J 1′′–P = 5.7, C-1′′), 66.09 (C-12′′), 66.82 (d, J 3′–P = 158.4, C-3′), 70.22 (d, J 2′′–P = 6.6, C-2′′), 71.18 (d, J 2′–P = 13.6, C-2′), 71.30 (C-3′′), 72.50 (C-11′′), 118.87 (C-5), 143.61 (C-8), 149.38 (C-4), 152.92 (C-2), 156.04 (C-6). 31P NMR (D2O, ppm) δ: 18.28 (m). ESI MS: 488.2 (100) [M−H]−, 489.3 (25) [M−H]−. ESI–HRMS calcd for C20H35N5O7P 488.2280, found: 488.2279 [M−H]−.
5.2.5. 17-Hydroxy-3,6,9,12,15-pentaoxaheptadecyl ester of 9-[2-(phosphonomethoxy)ethyl]adenine (11)
The reaction mixture was evaporated, the residue was deionized on a Dowex 50WX8-400 column ([H+], 4 ml, water) and then on Dowex 1 column ([AcO−], 4 ml, gradient water→0.5% aqueous acetic acid). Yield: 70 mg (81%) of thick syrup. 1H NMR (DMSO, ppm) δ: 3.40 (m, 2H, H-11′′), 3.45–3.49 (m, 20H, H-2′′–H-10′′, H-12′′), 3.70 (d, 2H, J 3′–P = 8.4, H-3′), 3.86–3.90 (m, 4H, H-2′, H-1′′), 4.32 (t, 2H, J 1′–2′ = 5.2, H-1′), 7.48 (b s, 2H, NH2), 8.15, 8.16 (s, s, H-2, H-8). 13C NMR (DMSO, ppm) δ: 42.82 (C-1′), 60.44 (C-12′′), 64.32 (d, J 1′′–P = 5.7, C-1′′), 65.30 (d, J 3′–P = 160.3, C-3′), 69.82–70.13 (m, C-2′′–C-10′′), 70.36 (d, J 2′–P = 10.6, C-2′), 72.56 (C-11′′), 118.68 (C-5), 141.75 (C-8), 149.52 (C-4), 151.69 (C-2), 155.53 (C-6). 31P NMR (D2O, ppm) δ: 18.38 (m). ESI–MS, m/z: 255.5 (14), 283.4 (19), 536.3 (100) [M−H]−, 618.0 (27). ESI–HRMS calcd for C20H35N5O10P 536.2127, found: 536.2129 [M−H]−.
5.2.6. 3,6,9,12,15,18-Hexaoxaeicosyl ester of 9-[2-(phosphonomethoxy)ethyl]adenine (12)
The reaction mixture was evaporated, the residue was deionized on a Dowex 50WX8-400 column ([H+], 4 ml, water) and then on a Dowex 1 column ([AcO−], 4 ml, gradient water→0.5% aqueous acetic acid). Yield: 72 mg (80%) of thick syrup. 1H NMR (DMSO, ppm) δ: 1.08 (t, 3H, J 14′′–13′′ = 7.0, H-14′′), 3.40 (q, 2H, J 13′′–14′′ = 7.0, H-13′′), 3.43–3.49 (m, 22H, H-2′′–H-12′′), 3.70 (d, 2H, J 3′–P = 8.4, H-3′), 3.86–3.90 (m, 4H, H-2′, H-1′′), 4.32 (t, 2H, J 1′–2′ = 5.2, H-1′), 7.51 (b s, 2H, NH2), 8.15, 8.16 (s, s, H-2, H-8). 13C NMR (DMSO, ppm) δ: 15.40 (C-14′′), 42.90 (C-1′), 64.36 (d, J 1′′–P = 5.6, C-1′′), 65.35 (d, J 3′–P = 160.1, C-3′), 65.83 (C-13′′), 69.49–70.12 (m, C-2′′–C-12′′), 70.39 (d, J 2′–P = 11.0, C-2′), 118.68 (C-5), 141.91 (C-8), 149.52 (C-4), 151.51 (C-2), 155.39 (C-6). 31P NMR (D2O, ppm) δ: 18.39 (m). ESI–MS, m/z: 564.2 (100) [M−H]−, 565.2 (25) [M−H]−, 586.2 (13), 645.9 (11), 1150.6 (23), 1151.6 (10). ESI–HRMS calcd for C22H39N5O10P 564.2440, found: 564.2440 [M−H]−.
5.2.7. (5-Methyl-2-oxo-1,3-dioxolen-4-yl)methyl ester of 9-[2-(phosphonomethoxy)ethyl]adenine (15)
A suspension of 1 (450 mg, 1.65 mmol) in methanol (5 ml) was neutralized with a 1 M solution of methanolic tetrabutylammonium bromide (1.65 ml, 1.65 mmol). PMEA (1) was dissolved, the solution was evaporated and co-distilled with ethanol and toluene. The residue was dissolved in anhydrous DMF (10 ml) and (5-methyl-2-oxo-1,3-dioxolen-4-yl)methylbromide5 (350 mg, 1.81 mmol) was added. The mixture was stirred for four days at room temperature. The resulting precipitate (400 mg) was purified on a Dowex 1 column ([AcO−], 50 ml, gradient water→1% aqueous acetic acid). Unreacted 1 was eluted at first, then 15. The fractions containing the product were lyophilized. Yield: 100 mg (16%).; mp 196 °C. 1H NMR (DMSO-d 6, ppm) δ: 1.99 (s, 3H, H-4′′), 3.67 (d, 2H, J 3′–P = 9.2, H-3′), 3.88 (m, 2H, H-2′), 4.29 (bd, 2H, J 1′′–P = 7.0, H-1′′), 4.41 (m, 2H, H-1′), 8.13 (s, H-2), 8.14 (s, H-8). 13C NMR (DMSO-d 6, ppm) δ: 8.74 (C-4′′), 44.29 (C-1′), 55.26 (d, J 1′′–P = 4.4, C-1′′), 66.94 (d, J 3′–P = 157.5, C-3′), 71.18 (d, J 2′–P = 14.3, C-2′), 118.53 (C-5), 135.25 (d, J 2′′–P = 6.8, C-2′′), 140.35 (C-3′′), 143.52 (C-8), 149.23 (C-4), 152.79 (C-2), 154.63 (CO), 155.75 (C-6). 31P NMR (D2O, ppm) δ: 18.20 (m). ESI–MS, m/z: 384.1 (100) [M−H]−, 385.1 (13) [M−H]−. ESI–HRMS: calcd for C13H15N5O7P 384.0715, found: 384.0717 [M−H]−.
5.3. General procedure for the preparation of (S)-HPMPC esters
Cyclic (S)-HPMPC (23) (261 mg, 1.0 mmol, prepared according to lit.7a) was suspended in MeOH (5 ml) and dissolved by a solution of tetrabutylamomonium hydroxide (1.0 ml, 1 M solution in MeOH). The solution was evaporated and co-distilled with isopropanol (5 ml) and toluene (3 × 5 ml). The syrupy residue was dissolved in DMF (4 ml) and the appropriate tosylate (28 or 29) (1.05 mmol) was added. The mixture was stirred at 100 °C (8 h), evaporated and the residue was chromatographed on a silica gel column (80 g) in 16% MeOH/CHCl3. The obtained cyclic ester of HPMPC was contaminated with tetrabutylammonium salts.
5.3.1. 14-Hydroxy-3,6,9,12-tetraoxaheptadecyl ester of (S)-1-[3-hydroxy-2-(phophonomethoxy)propyl]cytosine (16)
The crude ester 24, prepared by general procedure from 23 and 28, was heated in 50% aqueous acetic acid (10 ml) at 50 °C for 2 h. The mixture was then evaporated, co-distilled with H2O. The residue was deionized on a Dowex 50WX8-400 [H+] column (12 ml). Rinsing with water was followed with 0.5% ammonia. The UV-adsorbing eluate was concentrated and put on a Dowex 1 [AcO−] column (12 ml). The column was washed with water and the product was eluted with 0.25% aqueous AcOH. The UV-adsorbing fractions were evaporated, co-distilled with water, ethanol and toluene. Yield: 205 mg (38%) of syrup. 1H NMR (DMSO, ppm) δ: 3.40–3.65 (m, 27H, H-1′b, H-3′, H-4′, H-2′′–H-12′′), 3.77 (m, 1H, H-2′), 3.86 (m, 2H, H-1′′), 4.02 (dd, 1H, J gem = 13.9, J 1′a–2′ = 2.9, H-1′a), 5.82 (d, 1H, J 5–6 = 7.5, H-5), 7.70 (d, 1H, J 6–5 = 7.5, H-6), 8.73, 9.09 (b s, b s, 2H, NH2). 13C NMR (DMSO, ppm) δ: 50.17 (C-1′), 60.39 (C-3′, C-12′′), 63.43 (d, J 1′′–P = 5.4, C-1′′), 64.77 (d, J 4′–P = 168.0, C-4′), 69.89–70.0 (m, C-3′′–C-10′′), 70.46 (d, J 2′′–P = 5.8, C-2′′), 72.53 (C-11′′), 79.05 (C-2′), 93.09 (C-5), 149.42 (C-6), 150.89 (C-2), 162.04 (C-4). 31P NMR (D2O, ppm) δ: 18.49 (m). ESI–MS, m/z: 255.5 (14), 283.4 (19), 536.3 (100) [M−H]−, 618.0 (27). ESI–HRMS calcd for C20H35N5O10P 536.2127, found: 536.2129 [M−H]−.
5.3.2. 3,6,9,12,15-Pentaoxaeicosyl ester of (S)-1-[3-hydroxy-2-(phophonomethoxy)propyl]cytosine (17)
The crude ester 26, prepared by a general procedure from 23 and 29, was deionized on a Dowex 50WX8-400 [H+] column (12 ml), gradient water→0.5% ammonia, after which concentrated UV-adsorbing eluate was put on a Dowex 1 [AcO−] (12 ml), gradient water→0.25% acetic acid. Yield: 255 mg (46%) of syrup. 1H NMR (DMSO, ppm) δ: 1.09 (t, 3H, J 14′′–13′′ = 7.0, H-14′′), 3.41 (q, 2H, J 13′′–14′′ = 7.0, H-13′′), 3.43–3.64 (m, 27H, H-1′b, H-3′, H-4′, H-2′′–H-12′′), 3.76 (m, 1H, H-2′), 3.86 (m, 2H, H-1′′), 4.01 (dm, 1H, J gem = 13.6, H-1′a), 5.79 (m, 1H, H-5), 7.67 (m, 1H, H-6), 8.51, 8.91 (b s, b s, 2H, NH2). 13C NMR (DMSO, ppm) δ: 15.33 (C-14′′), 50.11 (C-1′), 60.42 (C-3′), 63.50 (C-1′′), 64.73 (d, J 4′–P = 158.8, C-4′), 65.74 (C-13′′), 69.43–70.02 (m, C-3′′–C-12′′), 70.45 (d, J 2′′–P = 6.0, C-2′′), 79.14 (C-2′), 93.09 (C-5), 149.16 (C-6), 151.2 (C-2), 162.3 (C-4). 31P NMR (D2O, ppm) δ: 18.48 (m). ESI–MS, m/z: 570.2 (100) [M−H]−, 571.2 (25) [M−H]−, 592.1 (13), 1162.9 (22), 1163.9 (10). ESI–HRMS calcd for C22H41N3O12P 570.2433, found: 570.2435 [M−H]−.
5.4. Synthesis of functionalized alkyl bromides and tosylates
5.4.1. General procedure for the preparation of diols 22 and 25
Triphenylphosphine (9.23 g, 35.2 mmol) was added in several portions to a cooled (0 °C) solution of 9-decen-1-ol or 10-undecen-1-ol (32.0 mmol) and CBr4 (33.6 mmol, 11.1 g) in dichloromethane (50 ml). The reaction mixture was stirred for 1 h at 0 °C and 1 h at room temperature and subsequently evaporated. The residue was filtered through a short silica gel column (5 × 10 cm) in hexane as a mobile phase. The eluate was evaporated and the crude bromide was dissolved in acetone (150 ml) and N-methyl-morpholine N-oxide (10 ml of 50% aqueous solution, 48.0 mmol) and osmium tetroxide (2.0 g of 4% aqueous solution) were added. The reaction mixture was stirred for 3 h at room temperature, then concentrated (∼ 50 ml) and partitioned between ether (100 ml) and an aqueous solution of sodium thiosulfate (100 ml). The aqueous layer was washed with ether again (50 ml). The combined ether portions were dried over MgSO4, filtered and evaporated. The residue was purified on a short silica gel column (5 × 10 cm) in ethyl acetate/hexane (3:2). The fractions were evaporated and the product was crystallized from toluene–hexane. Yield: 6.85 g of 10-bromo-1,2-decandiol or 7.31 g of 11-bromo-1,2-dodecanediol (85%).
5.4.1.1. 10-Bromodecane-1,2-diol (22)
Yield 6.85 g (85%). Anal. Calcd for C10H21BrO2: C, 47.44; H, 8.36; Br, 31.56. Found: C, 47.59; H, 8.52; Br, 31.43. ESI MS, m/z: 275.1 (100) [M+Na]+, 277.1 (94) [M+Na]+, 526.1 (19), 528.7 (24). ESI–HRMS calcd for C10H21O2BrNa 275.06171, found: 275.06182 and 277.05968 [M+Na]+.
5.4.1.2. 11-Bromododecane-1,2-diol (25)
Yield 7.31 g (85%). Anal. Calcd for C11H23BrO2: C, 49.45; H, 8.68; Br, 29.90. Found: C, 49.65; H, 8.96; Br, 29.57. ESI MS, m/z: 289.1 (100) [M+Na]+, 291.1 (94) [M+Na]+, 554.7 (27), 556.6 (37). ESI–HRMS calcd for C11H23O2BrNa 289.07736, found: 289.07755 and 291.07537 [M+Na]+.
5.4.2. 4-(8-Bromooctyl)-2,2-Dimethyl-1,3-dioxolane (23)
Diol 22 (6.80 g, 26.9 mmol) in 2,2-dimethoxypropane (50 ml) was treated with toluenesulfonic acid (100 mg, 0.53 mmol) at room temperature for 30 min. The solution was then extracted with an aqueous solution of NaHCO3 (50 ml). The aqueous solution was washed with ether (50 ml) again and the combined organic fractions were dried (MgSO4) and evaporated. The crude product (7.79 g, 99%) was used in the next step without further purification. 1H NMR (CDCl3, ppm) δ: 1.23–1.45 (m, 10H, H-3–H-7), 1.36 (s, 3H, CH3), 1.41 (s, 3H, CH3), 1.50 (m, 1H, H-8), 1.64 (m, 1H, H-8), 1.85 (m, 2H, H-2), 3.41 (t, 2H, J 2–1 = 6.9), 3.50 (t, 1H, J 10b–9 = 7.4, H-10b), 4.02–4.10 (m, 2H, H-10a, H-9). 13C NMR (CDCl3, ppm) δ: 25.70 (C-7), 25.74 (CH3), 26.95 (CH3), 28.10 (C-3), 28.63, 29.27, 29.51 (C-4, C-5, C-6), 32.77 (C-2), 33.56 (C-8), 33.99 (C-1), 69.50 (C-10), 76.11 (C-9), 108.56 (O–C–O). ESI MS, APCI MS: no mol. peak.
5.4.3. 4-[8-(2-Bromoethoxy)octyl]-2,2-dimethyl-1,3-dioxolane (24)
Ethyleneglycol (900 l, 16 mmol) was slowly added to a cooled (0 °C) suspension of NaH (400 mg of 60% suspension in mineral oil, 10 mmol) in dry DMF (3 ml). The suspension was stirred overnight at room temperature, after which bromide 23 (2.35 g, 8.0 mmol) in DMF (3 ml) was added. The mixture was stirred for 24 h. The excess of NaH was decomposed with several drops of MeOH. The solvents were evaporated and the residue was partitioned between chloroform (40 ml) and water (40 ml). The aqueous layer was treated with water again. The combined organic extracts were dried (MgSO4) and evaporated. The residue was chromatographed on a silica gel column (60 g) in 10%→20% aceton/hexane. The fractions containing the main product were evaporated. The syrupy residue (1.14 g) was dissolved in dichloromethane (10 ml) and CBr4 (1.46 g, 4.4 mmol) was added. The solution was cooled to 0 °C and triphenylphosphine (1.21 g, 4.6 mmol) was added. The reaction mixture was stirred for 1 h at 0 °C, 1 h at room temperature and subsequently evaporated. The residue was chromatographed on a silica gel column in 5→10% acetone/hexane. Yield: 1.35 g (50%) of liquid. 1H NMR (CDCl3, ppm) δ: 1.23–1.42 (m, 10H, H-5–H-9), 1.35 (q, 3H, J Me–Me = 0.7, CH3), 1.41 (q, 3H, J Me–Me = 0.7, CH3), 1.50 (m, 1H, H-10), 1.58 (m, 2H, H-4), 1.63 (m, 1H, H-10), 3.45–3.51 (m, 5H, H-12a, H-3, H-1), 3.74 (t, 2H, J 2–1 = 6.3, H-2), 4.03 (m, 1H, H-12b), 4.07 (m, 1H, H-11). 13C NMR (CDCl3, ppm) δ: 25.71 (C-9), 25.74 (CH3), 25.99 (C-5), 26.93 (CH3), 29.28, 29.39, 29.55 (C-6, C-7,C-8), 30.48 (C-1), 33.56 (C-10), 69.50 (C-12), 70.57 (C-2), 71.27 (C-3), 76.12 (C-11), 108.54 (O-C-O). ESI–MS: no mol. peak.
5.4.4. 1-Bromo-10-(methoxymethoxy)decane (26)
A solution of diol 25 (4.13 g, 16.3 mmol) in THF/H2O (1:1, 200 ml) was cooled to 0 °C and NaIO4 (5.3 g, 24.8 mmol) was added while stirring. NaBH4 (1.1 g, 29.1 mmol) was added 1 h later at 0 °C. The reaction mixture was stirred overnight at room temperature and then concentrated to ∼ 100 ml. The aqueous residue was extracted with EtOAc (3 × 50 ml), combined organic layers were dried (MgSO4) and evaporated. The residue was dissolved in CH2Cl2 (50 ml), the solution was cooled (0 °C) and treated with iPr2EtN (3.8 ml, 21.8 mmol) and methoxymethyl bromide (1.7 ml, 20.8 mmol). The solution was allowed to heat to room temperature and stirred overnight. The mixture was then washed with an aqueous solution of NaHCO3 (50 ml), the dichloromethane portion was dried (MgSO4) and evaporated. The residue was chromatographed on a silica gel column (200 g) in acetone/hexane (6:100). Yield: 3.55 g (77%) of liquid. 1H NMR (CDCl3, ppm) δ: 1.27–1.88 (m, 16H, H-2–H-9), 3.36 (s, 3H, CH3), 3.41 (t, 2H, J 1–2 = 6.9, H-1), 3.52 (m, 2H, H-10), 4.62 (s, 2H, O-CH2-O). 13C NMR (CDCl3, ppm) δ: 26.17–32.80 (m, C-2–C-9), 34.02 (C-1), 55.07 (CH3), 67.81 (C-10), 96.37 (O-C-O). ESI MS, m/z: 288.3 (47), 289.3 (18), 301.2 (50), 303.2 (26) [M+Na]+, 305.2 (25) [M+Na]+, 310.3 (95), 335.1 (21), 413.3 (25), 492.4 (44), 509.2 (40), 597.0 (64), 663.2 (100), 685.5 (47), 722.1 (34). ESI–HRMS calcd for C12H25O2BrNa 303.09301, found: 303.09293 and 305.09089 [M+Na]+.
5.4.5. 1-(2-Bromoethoxy)-10-(methoxymethoxy)decane (27)
Ethyleneglycol (900 l, 16 mmol) was slowly added to a cooled (0 °C) suspension of NaH (400 mg of 60% suspension in mineral oil, 10 mmol) in dry DMF (3 ml). The suspension was stirred for 1 h at room temperature, then bromide 26 (2.28 g, 8.1 mmol) in DMF (3 ml) was added. The mixture was stirred for 24 h. The excess of NaH was decomposed with several drops of MeOH. The solvents were evaporated and the residue was partitioned between chloroform and water. The aqueous layer was treated with water again. The combined organic extracts were dried (MgSO4) and evaporated. The residue was chromatographed on a silica gel column (60 g) in 10%→20% acetone/hexane. Fractions containing the main product were evaporated. The syrupy residue (1.07 g, 4.01 mmol) was dissolved in dichloromethane (10 ml) and CBr4 (1.46 g, 4.4 mmol) was added. The solution was cooled to 0 °C and triphenylphosphine (1.21 g, 4.6 mmol) was added. The reaction mixture was stirred for 1 h at 0 °C, 1 h at room temperature and then evaporated. The residue was chromatographed on a short silica gel column in 10% acetone/hexane. Yield: 1.24 g (47%) of liquid. 1H NMR (CDCl3, ppm) δ: 1.26–1.61 (m, 16H, H-4–H-11), 3.46 (t, 2H, J 1–2 = 6.3, H-1), 3.48 (t, 2H, J 3–4 = 6.7, H-3), 3.52 (t, 2H, J 12–11= 6.7, H-12), 3.74 (t, 2H, J 2–1 = 6.3, H-2), 4.62 (s, 2H, O-CH2-O); 13C NMR (CDCl3, ppm) δ: 26.02–29.73 (m, C-4–C-11), 30.49 (C-1), 55.07 (CH3), 67.86 (C-12), 70.59 (C-2), 71.32 (C-3), 96.37 (O-C-O). ESI MS, m/z: 342.1 (100), 344.1 (87), 347.2 (70) [M+Na]+, 349.2 (65) [M+Na]+, 590.1 (54), 592.1 (90), 594.1 (52), 597.2 (49), 599.2 (28). ESI–HRMS calcd for C14H29O3BrNa 347.11923, found: 347.11938 and 349.11714 [M+Na]+.
5.4.6. 17-[(Tetrahydro-2H-pyran-2-yl)oxy]-1-tosyloxy-3,6,9,12,15-pentaoxaheptadecan (28)
A mixture of hexaethylene glycol (7.13 g, 25.3 mmol), Ag2O (8.78 g, 37.9 mmol) and KI (0.84 g, 5.05 mmol) in CH2Cl2 (80 ml) was sonicated (15 min). The suspension was cooled down to −30 °C and a solution of tosylchloride (4.91 g, 25.8 mmol) in dichloromethane (100 ml) was added dropwise while stirring. The mixture was then gradually heated up to 0 °C and kept for 15 min at this temperature. The mixture was dried (MgSO4) and salts were filtered off. The filtrate was evaporated and the syrupy residue chromatographed on a silica gel column (300 g). The appropriate ditosylate was eluted with EtOAc (0.75 g), the desired monotosylate was eluted with 10% MeOH/EtOAc (8.70 g). The fractions containing monotosylate were evaporated, then co-distilled with toluene (2 × 50 ml). The residue was dissolved in dichloromethane (80 ml), 2,3-dihydro-2H-pyran (3.0 ml, 28.9 mmol) and pyridinium p-toluensulphonate (200 mg) were added and the mixture was refluxed (6 h). The reaction mixture was cooled down and washed with water (50 ml). The aqueous layer was extracted with EtOAc (4 × 40 ml). The combined organic portions were dried (MgSO4) and chromatographed on a silica gel column 300 g in gradient EtOAc→5% MeOH/EtOAc. Yield: 7.75 g (59%). 1H NMR (CDCl3, ppm) δ: 1.48–1.63 (m, 4H, H-3′a, H-4′a, H-5′, THP), 1.72 (m, 1H, H-3′b, THP), 1.83 (m, 1H, H-4′b, THP), 2.45 (s, 3H, CH3), 3.48–4.17 (m, 22H, H-6′, THP, H-1–H-10), 4.63 (m, 1H, H-2′, THP), 7.34 (m, 2H, H-3′′, Ts)), 7.80 (m, 2H, H-2′′, Ts); 13C NMR (CDCl3, ppm) δ: 19.46 (C-4′, THP), 21.62 (CH3), 25.40 (C-5′, THP), 30.53 (C-3′, THP), 61.67–72.58 (m, C-6′, THP, C-1–C-10), 98.93 (C-2′, THP), 127.96 (C-2′′, Ts), 129.79 (C-3′′, Ts), 132.99 (C-1′′, Ts), 144.76 (C-4′′, Ts). ESI MS, m/z: 538.1 (100), 539.1 (36), 540.1 (10), 543.2 (35) [M+Na]+.
5.4.7. 1-Tosyloxy-3,6,9,12,15,18-hexaoxaeicosan (29)
Absolute EtOH (0.5 ml, 8.5 mmol) was added dropwise to a stirred suspension of 60% NaH (0.36 g, 9.0 mmol) in anhydrous THF (10 ml). The mixture was stirred for 24 h at room temperature. Subsequently, a solution of tosylate 28 (2.63 g, 5.05 mmol) in THF (20 ml) was added. The mixture was stirred for three additional days at r.t. The excess of NaH was carefully decomposed with MeOH and the solution was concentrated in vacuo. The residue was partitioned between EtOAc (20 ml) and H2O (20 ml), and the aqueous layer was extracted with EtOAc (3 × 20 ml) again. The combined organic portions were dried (MgSO4), evaporated and purified on a silica gel column (50 g) in EtOAc. The fractions containing the main product (1.69 g) were evaporated and treated with Dowex 50WX8-400 [H+] in methanol (50 ml) overnight. The resin was then filtered off, washed with methanol, and combined filtrates were evaporated and co-distilled with toluene (3 × 10 ml). The residue (1.25 g) was tosylated with tosylchloride (1.54 g, 8.08 mmol) in a mixture of CH2Cl2 (20 ml) and Et3N (1.3 ml, 9.3 mmol) at room temperature for 24 h. The mixture was washed with water (20 ml), and the aqueous layer was extracted with EtOAc (3 × 20 ml) again. Combined organic fractions were dried (MgSO4) and evaporated. The syrupy residue was purified on a silica gel column (70 g) in 20% hexane/EtOAc→EtOAc. Yield: 1.70 g (72%). 1H NMR (CDCl3, ppm) δ: 1.21 (t, 3H, J 12–11 = 7.0, H-12), 2.54 (s, 3H, CH3, Ts), 3.52 (q, 2H, J 11–12 = 7.0, H-11), 3.57–3.66, 3.69, 4.16 (m, m, m, 16H, 2H, 2H, H-1–H-10), 7.34 (m, 2H, H-3′, Ts), 7.80 (m, 2H, H-2′, Ts); 13C NMR (CDCl3, ppm) δ: 15.12 (C-12), 21.62 (CH3, Ts), 66.60 (C-11), 68.65, 69.21, 69.78, 70.48, 70.52, 70.53, 70.55, 70.58, 70.62, 70.72 (C-1–C-10), 127.96 (C-2′, Ts), 129.79 (C-3′, Ts), 132.98 (C-1′, Ts), 144.76 (C-4′, Ts). ESI MS, m/z: 482.1 (100), 483.1 (24), 487.2 (42), 488.2 (10) [M+Na]+. ESI–HRMS calcd for C21H36O9SNa 487.19722, found: 487.19737 [M+Na]+.
5.5. Antiviral activity assays
The compounds were evaluated against the following viruses: herpes simplex virus type 1 (HSV-1) strain KOS, thymidine kinase-deficient (TK-) HSV-1 KOS strain resistant to ACV (ACVr), herpes simplex virus type 2 (HSV-2) strains Lyons and G, varicella-zoster virus (VZV) strain Oka, TK- VZV strain 07-1, human cytomegalovirus (HCMV) strains AD-169 and Davis, feline herpesvirus, vaccinia virus Lederle strain, human immunodeficiency virus (HIV) type 1 (IIIB) and type 2 (ROD), respiratory syncytial virus (RSV) strain Long, vesicular stomatitis virus (VSV), Coxsackie B4, Parainfluenza 3, Reovirus-1, Sindbis, Punta Toro, feline coronavirus, influenza A virus subtypes H1N1 and H3N2, and influenza B virus. The antiviral assays, other than HIV, were based on the inhibition of virus-induced cytopathicity or plaque formation in human embryonic lung (HEL) fibroblasts, African green monkey cells (Vero), human epithelial cells (HeLa), Crandell-Rees feline kidney cells (CRFK), or Madin Darby canine kidney cells (MDCK). Confluent cell cultures in microtiter 96-well plates were inoculated with 100 CCID50 of virus (1 CCID50 being the virus dose to infect 50% of the cell cultures) or with 20 plaque forming units (PFU). After 1–2 h adsorption period, the residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations of the test compounds. Viral cytopathicity or plaque formation (VZV) was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the test compounds. Antiviral activity was expressed as the EC50 or concentration required reducing virus-induced cytopathogenicity or viral plaque formation by 50%. The methodology of the anti-HIV assays was as follows: human CEM (∼3 × 105 cells/cm3) were infected with 100 CCID50 of HIV-1 (IIIB) or HIV-2 (ROD)/ml and seeded in 200-μL wells of a microtiter plate containing appropriate dilutions of the test compounds. After four days of incubation at 37 °C, HIV-induced CEM giant cell formation was examined microscopically.
5.6. Cytotoxicity assays
Cytotoxicity measurements were based on the inhibition of cell growth. HEL cells were seeded at a rate of 5 × 103 cells/well into 96-well microtiter plates and allowed to proliferate for 24 h. Then, medium containing different concentrations of the test compounds was added. After three days of incubation at 37 °C, the cell number was determined with a Coulter counter. The cytostatic concentration was calculated as the CC50, or the compound concentration required reducing cell proliferation by 50% relative to the number of cells in the untreated controls. CC50 values were estimated from graphic plots of the number of cells (percentage of control) as a function of the concentration of the test compounds. Alternatively, the cytotoxicity of the test compounds was expressed as the minimum cytotoxic concentration (MCC) or the compound concentration that caused a microscopically detectable alteration of cell morphology.
Acknowledgments
This work is a part of the Research Project of the Institute AV0Z40550506 and Centre of New Antivirals and Antineoplastics 1M0508 supported by the Ministry of Education, Youth and Sports of the Czech Republic. It was also supported by Gilead Sciences Inc. (Foster City, CA, USA) and the ‘Geconcerteerde Onderzoeksacties (GOA), Krediet nr. 10/014’ of the K.U. Leuven. The authors would like to thank to the staff of the Mass Spectrometry (Dr. J. Cvačka, Head), the Analytical Department of the institute (Dr. S. Matějková, Head) and Mrs. Julie Křelinová for technical assistance. We also thank Anita Camps, Steven Carmans, Frieda De Meyer, Leentje Persoons, Leen Ingels, Wim Van Dam and Lies Van den Heurck for their excellent technical assistance with the antiviral assays.
References and notes
- 1.De Clercq E. Antiviral Res. 2007;75:1. doi: 10.1016/j.antiviral.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Lee W.A., Martin J.S. Antiviral Res. 2006;71:254. doi: 10.1016/j.antiviral.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 3.Zídek Z., Kmoníčková E., Holý A. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 2005;149:315. doi: 10.5507/bp.2005.049. [DOI] [PubMed] [Google Scholar]
- 4.Sun C.Q., Cheng P.T.W., Stevenson J., Dejneka T., Brown B., Wang T.C., Robl J.A., Poss M.A. Tetrahedron Lett. 2002;43:1161. [Google Scholar]
- 5.Sakamoto F., Ikeda S., Tsukamoto G. Chem. Pharm. Bull. 1984;32:2241. doi: 10.1248/cpb.32.2241. [DOI] [PubMed] [Google Scholar]
- 6.Saneyoshi M., Morozumi M., Kodama K., Machida H., Kuninaka A., Yoshino H. Chem. Pharm. Bull. 1980;28:2915. doi: 10.1248/cpb.28.2915. [DOI] [PubMed] [Google Scholar]
- 7.(a) Beadle J.R., Hartline C., Aldern K.A., Rodriguez N., Harden E., Kern E.R., Hostetler K.Y. Antimicrob. Agents Chemother. 2002;46:2381. doi: 10.1128/AAC.46.8.2381-2386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hostetler K.Y. Antiviral Res. 2009;82:A84. doi: 10.1016/j.antiviral.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Serafinowska H.T., Ashton R.J., Bailey S., Harnden M.R., Jackson S.M., Sutton D. J. Med. Chem. 1995;38:1372. doi: 10.1021/jm00008a015. [DOI] [PubMed] [Google Scholar]; (b) Starrett J.E., Tortolani D.R., Russell J., Hitchcock M.J.M., Whiterock V., Martin J.C., Mansuri M.M. J. Med. Chem. 1994;37:1857. doi: 10.1021/jm00038a015. [DOI] [PubMed] [Google Scholar]
- 9.Holý A. Synthesis. 1998;29:381. [Google Scholar]
- 10.Loiseau F.A., Hii K.K., Hill A.M. J. Org. Chem. 2004;69:639. doi: 10.1021/jo035042v. [DOI] [PubMed] [Google Scholar]
- 11.Wan W.B., Beadle J.R., Hartline C., Kern E.R., Ciesla S.L., Valiaeva N., Hostetler K.Y. Antimicrob. Agents Chemother. 2005;49:656. doi: 10.1128/AAC.49.2.656-662.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valiaeva N., Beadle J.R., Aldern K.A., Trahan J., Hostetler K.Y. Antiviral Res. 2006;72:10. doi: 10.1016/j.antiviral.2006.03.007. [DOI] [PubMed] [Google Scholar]