

Abstract
Tumor-associated macrophages (TAMs) are important tumor-promoting cells. However, the mechanisms underlying how the tumor and its microenvironment reprogram these cells remain elusive. Here we report that lipids play a crucial role in generating TAMs in the tumor microenvironment. Macrophages from both human and murine tumor tissues were enriched with lipids due to increased lipid uptake by macrophages. TAMs expressed elevated levels of the scavenger receptor CD36, accumulated lipids, and used fatty acid oxidation (FAO) instead of glycolysis for energy. High levels of FAO promoted mitochondrial oxidative phosphorylation, production of reactive oxygen species, phosphorylation of JAK1 and dephosphorylation of SHP1, leading to STAT6 activation and transcription of genes that regulate TAM generation and function. These processes were critical for TAM polarization and activity both in vitro and in vivo. In summary, we highlight the importance of lipid metabolism in the differentiation and function of pro-tumor TAMs in the TME.
Introduction
Heterogeneity of macrophages (MΦs) has long been recognized as the result of the plasticity and versatility of these cells to different microenvironmental stimuli (1). In tumors, tumor-associated macrophages (TAMs) have been shown to regulate many critical processes associated with malignancy including promotion of tumor cell growth (2,3), drug resistance (4–6) and metastasis (7), induction of angiogenesis (8), and immune suppression (9,10).
Although it is known that tumor and tumor microenvironment (TME) play an important role in polarizing MΦs into tumor-promoting TAMs (11), the underlying mechanisms remain partially elucidated. Previous studies showed that prostate cancer-derived cathelicidin-related antimicrobial peptide reeducated MΦs into TAMs (12), and hypoxic cancer cell-derived oncostatin M and eotaxin differentiated MΦs into TAM (13). Moreover, myeloma-derived MIF drives MΦs into TAM differentiation (6). Additionally, as an important factor of TME, hypoxia only maintains the phenotype of M2-like MHC-IIlow TAM (14), although strong tumor hypoxia and a high density of hypoxic TAMs have been shown to correlate with a decreased survival rate (15). However, there is very little information regarding the biological or clinical role of lipid metabolism in TAMs in cancer.
In this study, we report an interesting finding that lipids in TME are crucial for TAM differentiation and function in different tumor settings. We found that, compared with control MΦs, TAMs from different human and murine tumor tissues were enriched with lipids. Similar phenomenon was observed with in vitro differentiated human and murine TAMs. Thus, we hypothesized that accumulation of lipid in TAMs is crucial for their differentiation and function in promoting tumor growth. In this study we elucidated the mechanisms underlying TAM accumulation of lipids and their function on TAM differentiation and function.
Materials and Methods
Human samples
The frozen sections of human breast, colon, and prostate tumor tissues were purchased from OriGene. Sections of normal tissues from these cancer patients were used as controls. Tumor tissue samples were obtained from newly diagnosed patients with melanoma or colon cancer; bone marrow (BM) aspirates were from newly diagnosed patients with multiple myeloma (MM) or monoclonal gammopathy of undetermined significance (MGUS). The same patients also provided normal tissue samples as controls. BM aspirates from healthy donors were used as controls for myeloma. All the human participants provided written informed consent, and this study was conducted in accordance with the U.S. Common Rule and approved by the Institutional Review Board at Cleveland Clinic and Houston Methodist Research Institute.
Mice
C57BL/6J, CD36−/−, and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from the Jackson Laboratory. C57BL/KaLwRij mice were purchased from Envigo (Horst, Netherland). Mice were maintained in a temperature- and humidity-controlled environment and given unrestricted access to chow diet and acidified water. Detailed methods about EL4 cell-induced lymphoma mouse model, VK*MYC and 5TGM1 cell-induced myeloma mouse model are described in extended experimental procedures.
Cells
Murine myeloma 5TGM1 and 5TGM1-luc cell lines were a kind gift from Dr. Frederic J Reu (16) and in culture with IMDM medium supplemented with 10% FBS (Gibco). Other human and murine cell lines were purchased from ATCC and maintained in the indicated medium. TAMs were generated by culturing MΦs with tumor cells or their tumor-conditioning medium (TCM) for 3 days. All the cell lines (except 5TGM1 and 5TGM1-luc cells) used in this study were purchased from ATCC. The mycoplasma testing and cell authentication were performed by The Cytogenetics and Cell Authentication Core of MD Anderson Cancer Center (Houston, TX). All cells used in this study were mycoplasma negative. Detailed method is described in extended experimental procedures.
Isolation and generation of MΦs
Murine and human MΦs were generated as described previously (4). For mouse MΦs generation, murine BM cells were collected by flushing the femurs of 8 to 10 weeks old C57BL/6J or CD36 KO mice with ice-cold RPMI 1640 medium to generate MΦs. Red blood cells were lysed with ACK Lysing Buffer (Gibco) and washed twice with RPMI 1640 medium. After 2 hour incubation in RPMI 1640 medium at 37 °C with 5% CO2, culture supernatant was changed with fresh RPMI 1640 supplemented with 10% FBS (Gibco), 100 U/ml penicillin, and 100 μg/ml streptomycin. Human M-CSF (10 ng/mL) was used to generate BM-derived MΦs.
For human MΦs, as described previously (17), peripheral blood mononuclear cells (PBMCs) are isolated from the whole blood by a density gradient centrifugation method using Ficoll. The cells were incubated in a 24-well or 6-well plate for 2 hours in FBS-free RPMI 1640 medium at 37 °C to remove non-adherent cells. After 2-hour incubation, the adherent cells were incubated in RPMI 1640 medium supplemented with 10% FBS (Gibco) and 10 ng/mL M-CSF (216-MC-500, R&D) to induce MΦ differentiation. Medium was replaced with fresh RPMI 1640 supplemented with 10% FBS and 10 ng/mL M-CSF every 3 days. On day 7, MΦs were collected and analyzed by flow cytometry.
In vitro generation of TAMs
In vitro differentiated TAMs were generated as described previously (18). Detailed method is described in extended experimental procedures.
Lipid content measurement
Cellular lipid content was measured using BODIPY 493/503. Briefly, after wash with PBS, cells were stained for 15 minutes with BODIPY 493/503 at room temperature in PBS (FBS or fatty acid free) and examined immediately using both confocal microscopy and flow cytometry.
Quantitative RT-PCR
Total RNA from MΦs was extracted with TRIzol RNA isolation reagents (Invitrogen) or a RNeasy Mini Kit (QIAGEN), followed by cDNA synthesis with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time quantitative PCR was conducted with SYBR Select Master Mix (Applied Biosystems). Expression was normalized to the mouse or human housekeeping gene HPRT. Primers used are listed in the Supplementary Table 1.
Western blot analysis
Cell lysates and immunoblot were performed as previously described (19).
Microarray analysis
The datasets during and/or analyzed during the current study are available on the GEO database (GSE143025 and GSE143583). Detailed method is described in extended experimental procedures.
Measurement of oxygen-consumption rates
Briefly, control MΦs and TAMs after indicated treatment were transferred on to XF microplates (15, 000 cells per well) in complete culture medium for 1 day. MΦs were washed and equilibrated for 1 hour. To determine fatty acid oxidation in MΦs, Mito Stress Tests were carried out using sequential injections of oligomycin (1.5 μM), FCCP (1.5 μM), and Rotenone plus Antimycin A (1.5 μM). To determine glycolysis in MΦs, Glycolysis Stress Test was carried out using sequential injections of glucose (10 mM), oligomycin (1.0 μM), and 2-DG (50 mM). OCR and ECAR were recorded by an Extracellular Flux Analyzer XF24 (Seahorse Bioscience).
Lipid uptake assay
Fatty acid uptake was measured using a Free Fatty Acid Uptake Assay kit (ab176768, Abcam, USA), following the manufacturers’ protocol. Briefly, cells were seeded at a density of 50,000 cells per well in a 96-well plate 1 day before measurement, and these cells were deprived of serum for 1 hour. Next, cells were incubated with TF2-C12 fatty acid at room temperature, and fluorescence (excitation: 485 nm, emission: 515 nm) was measured using a Wallac 1420 Victor2 Microplate Reader (Perkin Elmer, USA).
For the fluorescent FA pulse-chase assay, cells were deprived of serum for 1 hour and pulsed with Red C12 overnight in order to allow the Red C12 to accumulate in lipid droplets. After washing with PBS, cells were measured with confocal microscopy.
Statistical analysis
The ImageJ software (National Institutes of Health) was used for protein band quantification. The Student t test (two-tailed) was used to compare various experimental groups. When more than 2 groups were included in an analysis, Bonferroni’s corrected significance level was used. Survival was evaluated using Kaplan-Meier estimates and log-rank tests. One-way ANOVA was performed to analyze the difference of tumor volumes. All results are shown as mean ± SEM, unless otherwise indicated. A P value less than 0.05 was considered statistically significant.
Ethics approval and consent to participate
This study was approved by the Institutional Animal Care and Use Committee (IACUC) and Institutional Review Board (IRB) of Cleveland Clinic (Protocol NO. 2016–0158; IRB # 13–791) and Houston Methodist Research Institute (Protocol NO. 0918–0053; IRB # Pro00019970). All animals received humane care in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the “Guide for the Care and Use of Laboratory Animals” prepared by the Institute of Laboratory Animal Resources and published by the National Institutes of Health (NIH Publication NO. 86–23, revised 1996).
Results
Increased lipid accumulation in TAMs from human and murine tumor tissues
We have been investigating the role of TAMs in MM drug resistance (4,6,20) and sorted out TAMs from MM patient’s BM samples for functional studies. Under light microscope, TAMs were morphologically different from control MΦs (Fig. 1A and B). One of the striking differences was that TAMs were enriched with lipids (Fig. 1A and B). Additionally, we observed obvious increase of lipids, which co-localized with CD68 staining in tissue sections of patients with breast (Fig. 1C and Supplementary Fig. S1A), colon (Fig. 1D and Supplementary Fig. S1B), and prostate cancers (Fig. 1E and Supplementary Fig. S1C). To confirm the results, we used a lipophilic fluorescent dye BODIPY 493/503 to semi-quantify the amounts of lipids in TAMs. As shown in Fig. 1F and G, CD11b+CD68+ MΦs isolated from human melanoma (Fig. 1F) and colon cancer (Fig. 1G) tissues displayed significantly more lipid accumulation compared to MΦs isolated from normal tissues. Additionally, an increased lipid accumulation in TAMs from patient’s BM was positively and significantly associated with the progression of MGUS to MM (Fig. 1H).
Fig. 1. Lipid accumulation in TAMs.
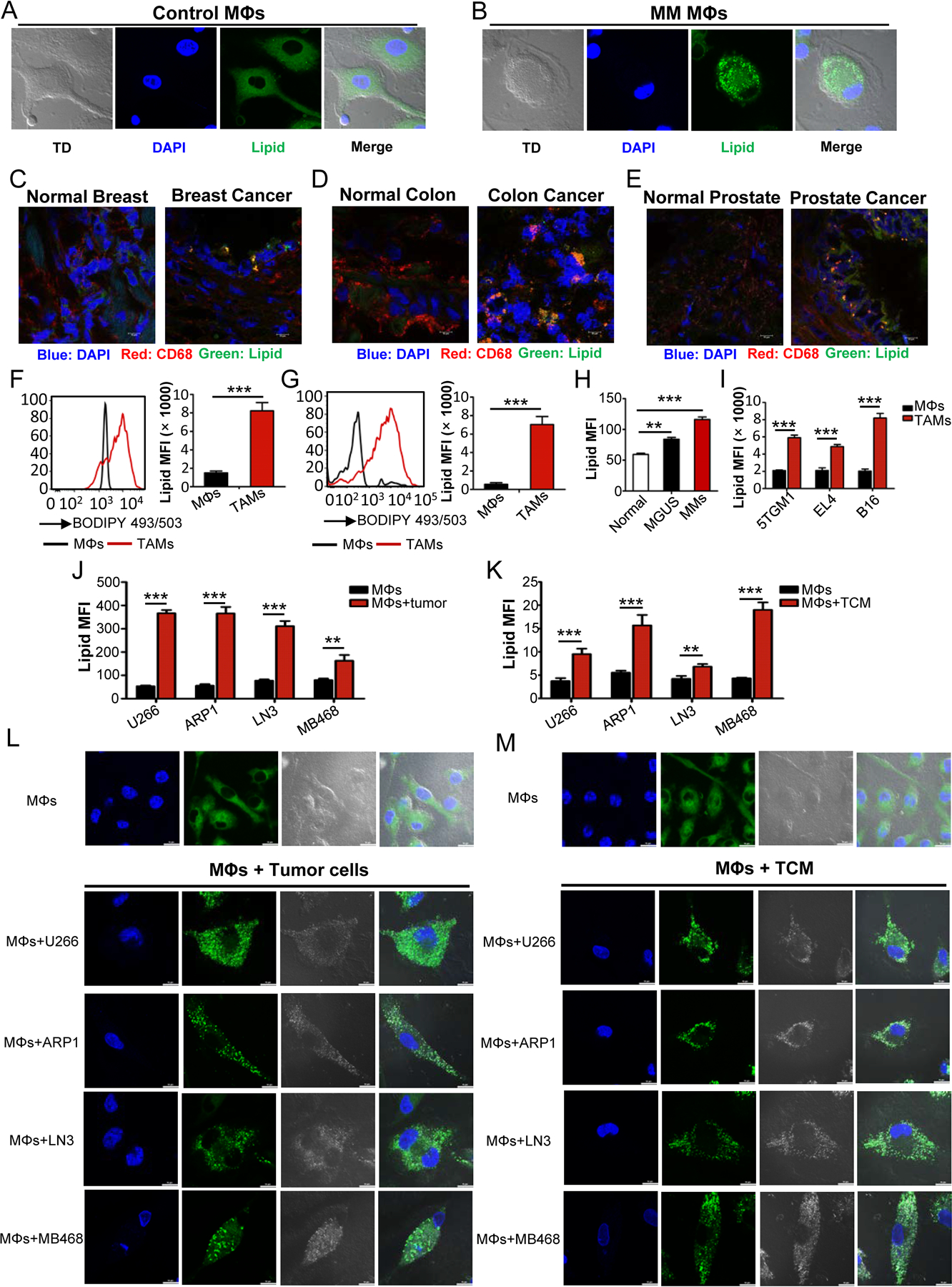
A-B, Representative fluorescent confocal images showing the morphology and lipid levels of human MΦs sorted from BM samples of normal donors (A) and MM patients (B). TD represents contrast image by transmitted light differential interference.
C-E, Representative images of lipid levels and CD68 expression in human frozen sections of breast, colon, and prostate cancer and normal tissues.
F, Representative histogram showing the levels of lipids in CD11b+CD68+ MΦs from human colon normal and cancer tissues (left panel). Right panel showing the statistic result of the mean fluorescence intensity (MFI) of lipids in MΦs from normal and tumor tissues. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. ***, P < 0.001.
G, Representative histograms (left panel) and quantitative result (right panel) of the lipid levels in MΦs from human normal and melanoma tissues. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. ***, P < 0.001.
H, Bar graph representing the lipid levels in CD11b+CD68+ MΦs from normal, MGUS, and MM BM samples. Statistical significance was determined by two-tailed Student’s t test between normal and disease group. **, P < 0.01; ***, P < 0.001.
I, Statistic results of lipid levels in F4/80+CD11b+ MΦs of tumor tissues and their corresponding normal tissues from indicated tumor-bearing (TB) or normal mice. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. ***, P < 0.001.
J-K, Bar graphs depicting the MFI of lipids in MΦs cultured with or without indicated tumor cells (J) or TCM (K). Statistical significance was determined by two-tailed Student’s t test between control MΦs and different TAMs. **, P < 0.01; ***, P < 0.001.
L-M, Representative confocal images of lipid droplet in MΦs cultured with tumor cells (L) or TCM (M).
We examined lipid level in TAMs in tumor-bearing mice. CD11b+/F4/80+ MΦs isolated from 5TGM1 murine myeloma-bearing C57BL/KalwRij mice, EL4 murine lymphoma-bearing or B16 murine melanoma-bearing C57BL/6J mice all contained significantly more lipid than their normal counterparts of tumor-free mice (Fig. 1I).
Next, we examined whether in vitro generated TAMs also accumulate lipids. Human PBMC-derived MΦs (MΦs) were generated and incubated with human tumor cells (Fig. 1J) or TCM (Fig. 1K) for 72 hours. These tumor-cocultured MΦs (TAMs) displayed more than two-fold increase in lipid compared with control MΦs (Fig. 1J and K). Using confocal microscopy with BODIPY 493/503 staining of neutral lipids, we showed that TAMs contained large bright dots which were lipid droplets, while control MΦs contained small, even-distributed green smear (Fig. 1L and M). Similar results were also obtained with mouse BM cell-derived MΦs after (co)cultured with different murine tumor cells or their TCM (Supplementary Fig. S1D–H). However, no significant difference was observed in lipid accumulation in TAMs under hypoxia or normoxia conditions (Supplementary S1I and J), indicating that hypoxia does not significantly influence TAM accumulation of lipids. These results clearly showed that both human and murine TAMs accumulate lipids, which may be important and required for TAM function.
TAMs accumulate lipids by enhanced lipid uptake through surface scavenger receptor CD36
To elucidate the mechanisms underlying lipid accumulation in TAMs, in vitro-generated human TAMs and control MΦs were washed, cultured in serum-free medium for 1 hour, and then treated with fluorescent fatty acid analogue (FA-Red C12). TAMs showed markedly higher uptake of fatty acid than control MΦs (Fig. 2A and B). These results suggested that an increased uptake of lipids may be a reason for the increased lipid accumulation in TAMs.
Fig. 2. Blocking CD36 abolishes lipid accumulation in TAMs.
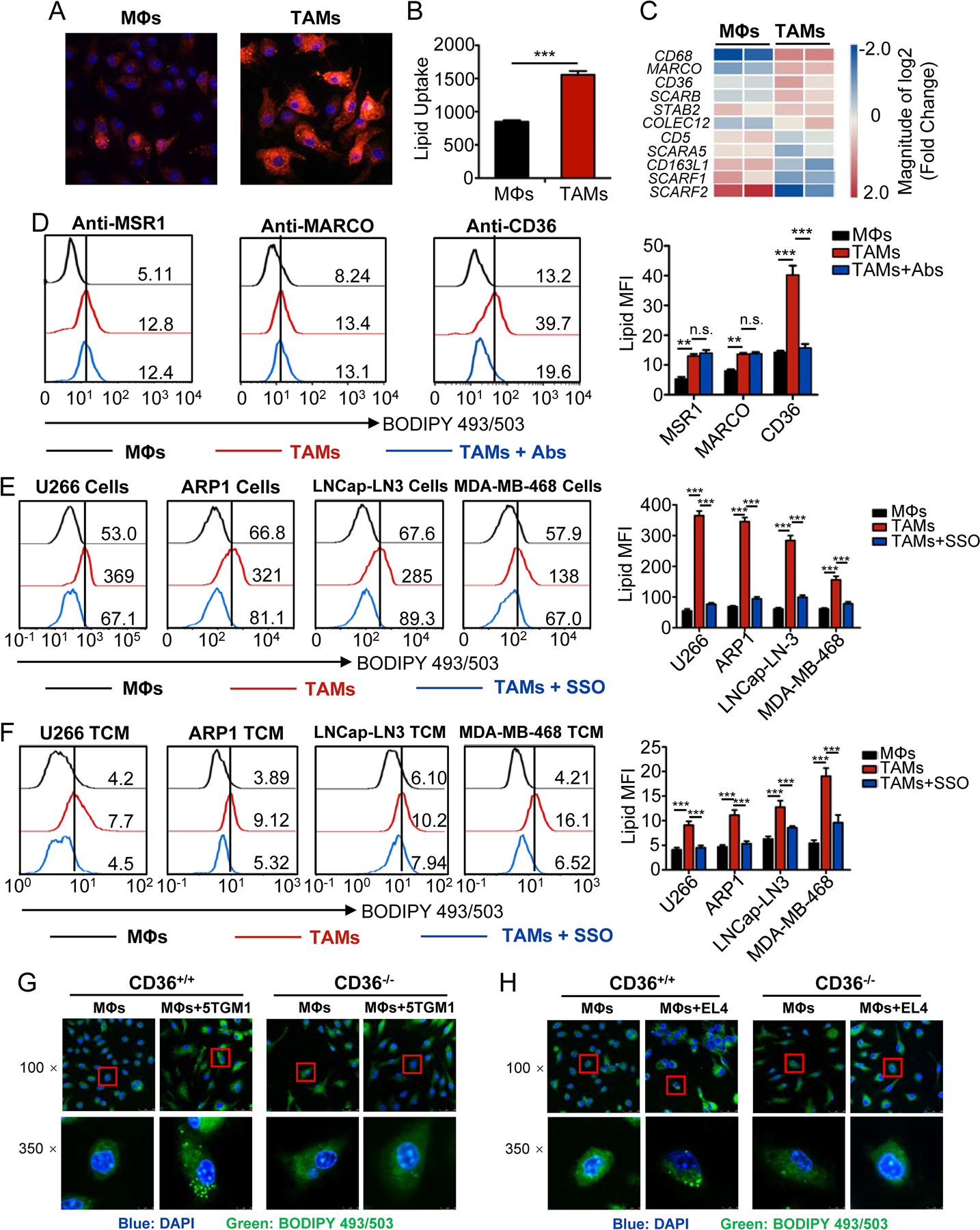
A, Representative fluorescent confocal images showing Fatty Acid-Red C12 uptake by human control MΦs and TAMs.
B, Bar graph depicting ELISA results of fatty acid uptake by control MΦs and TAMs using a Free Fatty Acid Uptake Assay kit. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. ***, P < 0.001.
C, Heat map showing the relative expression of human scavenger receptors in TAMs compared with control MΦs.
D, Representative histograms (left) and statistic results (right) showing lipids in MΦs and TAMs with indicated blocking antibody treatments. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. **, P < 0.01; ***, P < 0.001; n.s., not significant.
E-F, Representative histograms (left) and statistic results (right) showing lipid levels in control MΦs and indicated TAMs cultured with or without SSO. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. ***, P < 0.001.
G-H, Representative confocal images of lipids of murine MΦs from CD36 WT and KO mice cultured with or without 5TGM1 (G) or EL4 (H) cells in vitro.
To investigate why TAMs have higher lipid uptake than control MΦs, we analyzed microarray data to determine the expression of scavenger receptors (SRs) in human control MΦs and TAMs. Compared with control MΦs, three of the four major SRs, including CD68, MARCO, and CD36, were significantly upregulated in TAMs, while MSR-1 was not detected (Fig. 2C), suggesting that tumor cells upregulate the expression of these SRs on MΦs. This result was confirmed by qPCR (Supplementary Fig. S2A) and flow cytometry (Supplementary Fig. S2B–F). Next, we studied the role of these 3 receptors in lipid accumulation in TAMs by using blocking antibodies to MSR1, MARCO and CD36. Among these receptors, only CD36-blocking antibody abolished lipid accumulation in TAMs (Fig. 2D). To confirm that lipid accumulation in TAMs was indeed mediated by CD36, we used a CD36-specific inhibitor, sulfo-N-succinimidyl oleate (SSO) to inhibit the enzyme activity of CD36 in human MΦs (Fig. 2E and F), or knocked down (KD) Cd36 expression in murine MΦs by shRNA (Supplementary Fig. S3A). SSO (Fig. 2E and F) or Cd36-KD (Supplementary Fig. S3B) significantly decreased lipid accumulation in TAMs (Fig. 2E, F and Supplementary Fig. S3A and B). Additionally, we determined lipid level of TAMs from CD36 knockout (KO) mice. Consistent with the above results, lipid accumulation was dampened in CD36-KO TAMs as compared to wildtype (WT) cells (Fig. 2G, H and Supplementary Fig. S3C and D). However, no significant effect of hypoxia on lipid uptake and accumulation in TAMs, or the expression of CD36 and M1/M2 polarization was observed (Supplementary Fig. S3E–H). Collectively, these results suggested that lipid uptake and accumulation in TAMs were mainly mediated via the SR CD36.
TAMs accumulate lipids as source of energy
As TAMs accumulate lipids, we hypothesized that TAMs use the accumulated lipids as the source of fuels to produce energy for their activity. To test our hypothesis, we first analyzed the expression of genes involved in fatty acid and glucose metabolism in human MΦs and TAMs. As shown in Fig. 3A and B, most of the genes involved in fatty acid β-oxidation were significantly upregulated in TAMs as compared with control MΦs (Fig. 3A and B), whereas no significant differences were observed in the genes involved in glucose metabolism (Supplementary Fig. S4A–E) between the cells. We confirmed by qPCR that TAMs displayed an elevated expression pattern of genes associated with FAO, including CPT1A, which is a FAO rate-limiting enzyme, ACADM, HADHA, and HADH, while no change of the expression of ACACA was observed in human (Fig. 3C) and mice (Supplementary Fig. S4F). Second, we measured the mitochondrial oxygen consumption rates (OCRs) in TAMs treated with or without etomoxir, which is the inhibitor of CPT-1, to determine whether the high level of fatty acid oxidation contributed to ATP production-linked oxidative phosphorylation in mitochondria. As shown in Fig. 3D, E, and Supplementary Fig. S4G, TAMs showed an enhanced mitochondrial OCRs, ATP production, and markedly increased spare respiratory capacity (the quantitative difference between the maximal OCR and the initial basal OCR) (Fig. 3D, E and Supplementary Fig. S4G), which are indicative of an increased commitment to oxidative phosphorylation in TAMs and suggest that fatty acid oxidation inhibition in TAMs suppresses oxidative phosphorylation and ATP production. No difference in extracellular acidification rate (ECAR) was observed between control MΦs and TAMs (Supplementary Fig. S4H and I).
Fig. 3. Culture with tumor cells increases the fatty acid oxidation in TAMs.
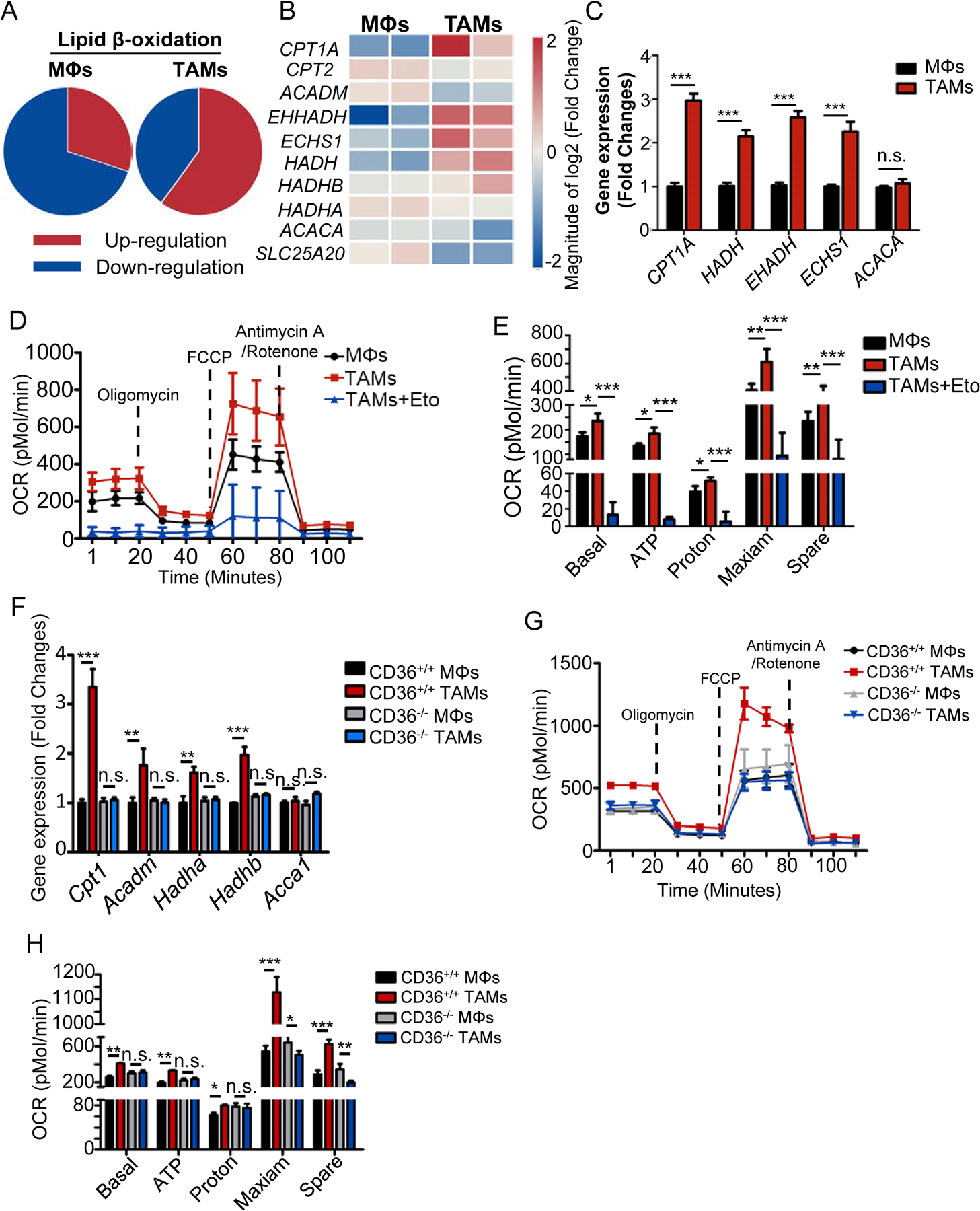
A-B, Microarray analysis of genes related to lipid β-oxidation in control MΦs and TAMs. Pie charts showing the percentage of total gene changes (A) in each indicated group of cells. Heat map showing the relative expression of lipid β-oxidation-associated genes (B) in control MΦs and TAMs.
C, Relative mRNA expression of indicated genes related to lipid β-oxidation in control MΦs and TAMs. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. ***, P < 0.001; n.s., not significant.
D-E, Oxygen consumption rates (OCRs) of control MΦs, TAMs and etomoxir treated TAMs by Seahorse XF Analyzer (D). Statistic results depicting the basal and maximal respiration and spare respiratory capacity in control MΦs and TAMs by analyzing the OCRs (E). Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
F, Relative mRNA expression of indicated genes in control MΦs and TAMs from CD36 WT and KO mice using qPCR. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. **, P < 0.01; ***, P < 0.001; n.s., not significant.
G-H, OCRs of control MΦs and TAMs from CD36 WT and KO mice (G) and statistic results (H) depicting the basal and maximal respiration and spare respiratory capacity. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. *, P < 0.05; **, P < 0.01; ***, P < 0.001, n.s., not significant.
Finally, to further determine the relationship between oxidative phosphorylation and lipid accumulation in TAMs, we measured the expression levels of fatty acid oxidation-related genes and OCRs in control MΦs and TAMs from CD36 WT and KO mice. Our data showed that the expression levels of these fatty acid oxidation-related genes in CD36-KO TAMs were similar to control MΦs but were significantly lower than the WT TAMs (Fig. 3F). We also observed that the basal respiratory rate, ATP production and oxidative phosphorylation were significantly attenuated in CD36-KO TAMs compared with WT TAMs (Fig. 3G and H). Taken together, these findings confirmed our hypothesis that high level of lipids uptaken by CD36 promotes TAM fatty acid oxidation and oxidative phosphorylation to generate more energy for TAMs.
Lipid accumulation and fatty acid oxidation are required for the differentiation and protumor function of TAMs
To determine whether lipid accumulation and enhanced fatty acid oxidation are required for TAM differentiation, the expression profiles of genes in murine control MΦs and TAMs were investigated. Gene array data showed that, compared with control MΦs, TAMs expressed higher levels of molecules favoring tumor proliferation and progression, such as Mrc1, Tgm2, Arg1, Vegf, Hif1a, Ccl2, and Pparg (Fig. 4A). Quantitative RT-PCR confirmed the results (Fig. 4B). Moreover, the elevated expression of these TAM-associated genes in MΦs was significantly reduced by fatty acid oxidation inhibition (Fig. 4C), suggesting that TAM differentiation is controlled by fatty acid oxidation. We then explored the role of CD36 in TAM polarization. As shown in Fig. 4D, 20 genes were upregulated in WT TAMs compared with control MΦs; 12 genes were downregulated in CD36-KO TAMs compared with WT TAMs; and 12 genes remained unchanged between CD36-KO and control MΦs. Only two genes, Arg1 and Ccl2, were present in the intersection of the three gene sets, suggesting that Arg1 and Ccl2 are regulated by CD36 in TAMs. This result was confirmed by examining the mRNA (Fig. 4E) and protein expression (Fig. 4F) of Arg-1 and Ccl2 in control MΦs and TAMs from CD36 WT and KO mice.
Fig. 4. Fatty acid oxidation promotes TAM differentiation.
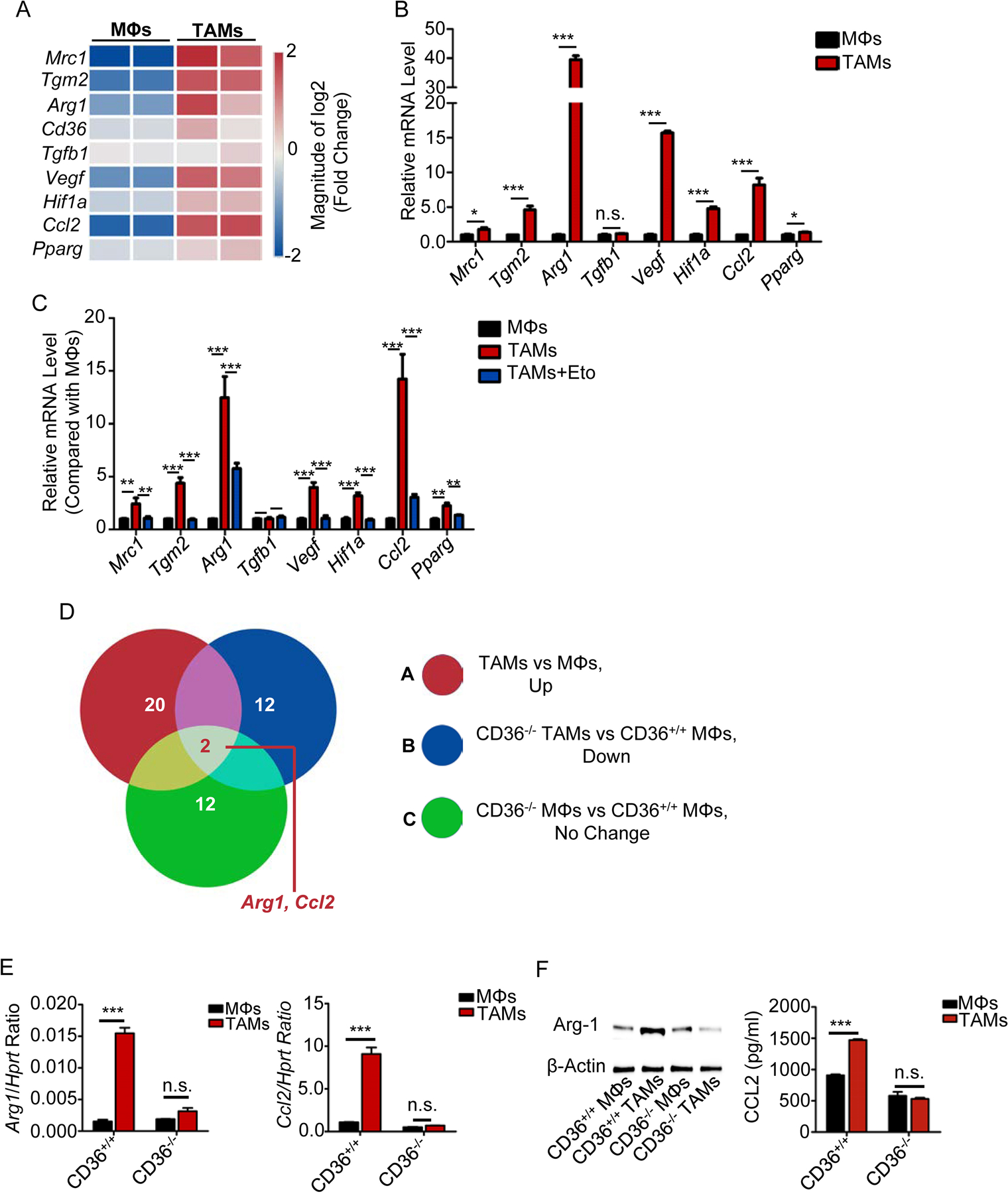
A, Heat map showing the relative expression of TAM signature genes in control MΦs and TAMs.
B, Relative mRNA expression of TAM signature genes in control MΦs and TAMs by qPCR. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. *, P < 0.05; ***, P < 0.001, n.s., not significant.
C, Relative mRNA expression of indicated genes in the MΦs with indicated treatment. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. **, P < 0.01; ***, P < 0.001, n.s., not significant.
D, Venn diagram showing the changes in gene expression in control MΦs and TAMs under various conditions. Arg1 and Ccl2 were in the intersection between these three gene sets.
E-F, Relative mRNA expression (e) and protein levels (f) of Ccl2 and Arg1 in control MΦs and TAMs from CD36 WT and KO mice. Statistical significance was determined by two-tailed Student’s t test between control MΦs and TAMs. ***, P < 0.001, n.s., not significant.
As TAMs play an important role in promoting tumor growth and progression (21), we next hypothesized that lipid accumulation and metabolism in TAMs are also required for their tumor-promoting function. We showed that, by determining the expression of Ki-67 and cell cycle progression in human myeloma cells U266 (Fig. 5A) and ARP-1 (Fig. 5B), human prostate carcinoma cells LNCaP-LN3 (Fig. 5C), human breast adenocarcinoma cells MDA-MB-468 (Fig. 5D), as well as murine myeloma cells 5TGM1 (Supplementary Fig. S5A) and murine lymphoma cells EL4 (Supplementary Fig. S5B), after coculture with control MΦs or TAMs in the presence or absence of etomoxir. TAMs significantly enhanced the expression of Ki-67 (Fig. 5A–D and Supplementary Fig. S5A and B) and increased the S-phase population in tumor cells (Fig. 5E, F and Supplementary Fig. S5C–E) as compared with control MΦs. Etomoxir-treated TAMs failed to promote tumor cell proliferation (Fig. 5A–F and Supplementary Fig. S5). CD36-KO TAMs lost their ability to promote tumor proliferation (Fig. 5G and H). Taken together, these results suggested that the inhibition of fatty acid oxidation in TAMs suppresses their function as a protumor cell.
Fig. 5. Inhibition of fatty acid oxidation impairs protumor function of TAMs.
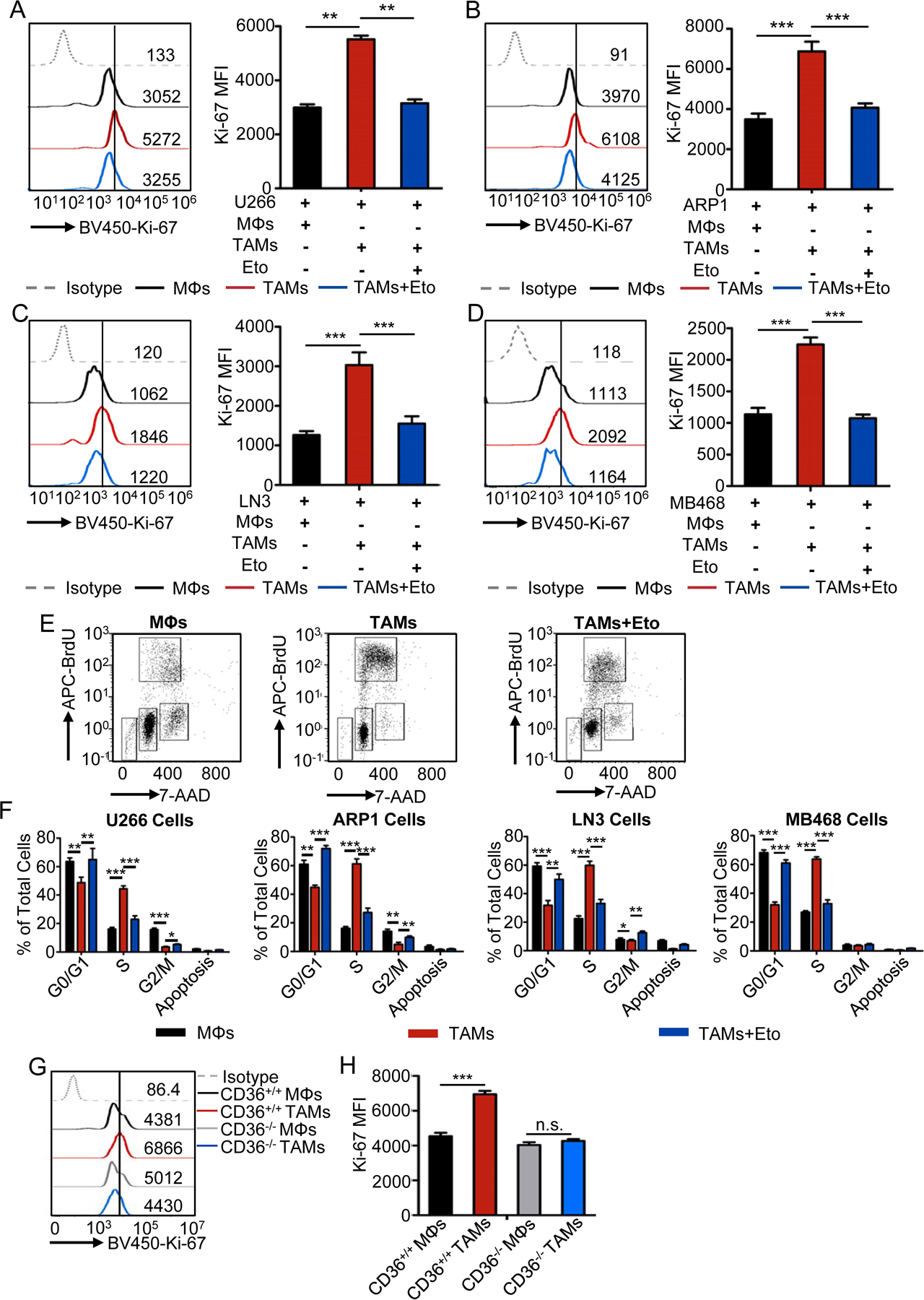
A-D, Representative histograms (left) and statistic results (right) of the expression of Ki-67 in human U266 (A) and ARP1 (B) myeloma cells, LNCap-LN3 prostate tumor cells (C), and MDA-MB-468 breast tumor cells (D), cultured with control MΦs or TAMs in the presence or absence of etomoxir. Statistical significance was determined by two-tailed Student’s t test between indicated groups. **, P < 0.01; ***, P < 0.001.
E, Representative dot plots showing cell cycle progression of tumor cells with indicated coculture conditions using APC-BrdU and 7-AAD.
F, Bar graphs showing the percentage of human U266, ARP1, LNCaP-LN3, and MDA-MB-468 cells in the indicated cell cycle stages under indicated coculture conditions. Statistical significance was determined by two-tailed Student’s t test between indicated groups. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
G-H, Representative histograms (G) and statistic results (H) showing the expression of Ki-67 in 5TGM1 cells cultured with control MΦs or TAMs from CD36 WT and KO mice. Statistical significance was determined by two-tailed Student’s t test between indicated groups. ***, P < 0.001; n.s., not significant.
High levels of fatty acid oxidation enhance the phosphorylation of STAT6
To elucidate the mechanism of fatty acid oxidation in regulating TAM polarization, we performed the ingenuity pathway analysis (IPA) to identify canonical signaling pathways that were activated in the cells. As shown in Fig. 6A, TAMs displayed, in comparison with control MΦs, activated signaling pathways including IL-4, IL-10, IL-13, PPAR-γ, fatty acid oxidation, CSF1R, and STAT6, which are highly related to immunosuppressive and tumor promoting function of MΦs (22). As STAT6 cross-talks with these signaling pathways that are activated in TAMs, we determined the phosphorylation status of STAT6 in the cells. Compared with control MΦs, STAT6 was phosphorylated at Tyr641 in TAMs (Fig. 6B and Supplementary Fig. S6A). We also observed high levels of oxidative stress in TAMs compared with control MΦs (Fig. 6C). As IL-4 and IL-13 directly activate the phosphorylation of STAT6 in MΦs (23), we measured the production of IL-4 and IL-13 in tumor cells used in our study. No production of IL-4/IL-13 was detected in the supernatants of the tumor cells (Supplementary Fig. S6B–E), indicating that the observed STAT6 phosphorylation in TAMs was not induced by the cytokines but by oxidative stress. To confirm our hypothesis, we first examined the phosphorylation levels of JAKs (JAK1, JAK2, and JAK3), STAT6 kinases, and SHP1, which is a STAT6-specific tyrosine phosphatase (24), in control MΦs and TAMs. As shown in Fig. 6D, E, and Supplementary Fig. S6F–I, the phosphorylation of JAK1 was significantly increased whereas the phosphorylation of SHP1 was inhibited in TAMs compared with MΦs. Second, to determine whether the phosphorylation of JAK1 and SHP1 in TAMs was regulated by oxidative stress, we treated MΦs with H2O2 and TAMs with antioxidant N-Acetyl-L-cysteine (NAC), and then examined the phosphorylation levels of JAK1, SHP1 and STAT6 in the cells. As shown in Fig. 6F and Supplementary Fig. S6J–M, H2O2 treatment significantly enhanced JAK1 and STAT6 phosphorylation but suppressed SHP1 phosphorylation in MΦs. In contrast, NAC treatment suppressed JAK1 and STAT6 phosphorylation but increased SHP1 phosphorylation in TAMs (Fig. 6F and Supplementary Fig. S6J–M). No significant changes in STAT6 or SHP1 phosphorylation were observed in MΦs and TAMs generated from CD36-KO mice (Fig. 6G). These results clearly indicated that higher fatty acid-oxidation-induced oxidative stress is responsible for STAT6 phosphorylation by dephosphorylating SHP1 in TAMs.
Fig. 6. High level of fatty acid oxidation enhances the phosphorylation of STAT6.
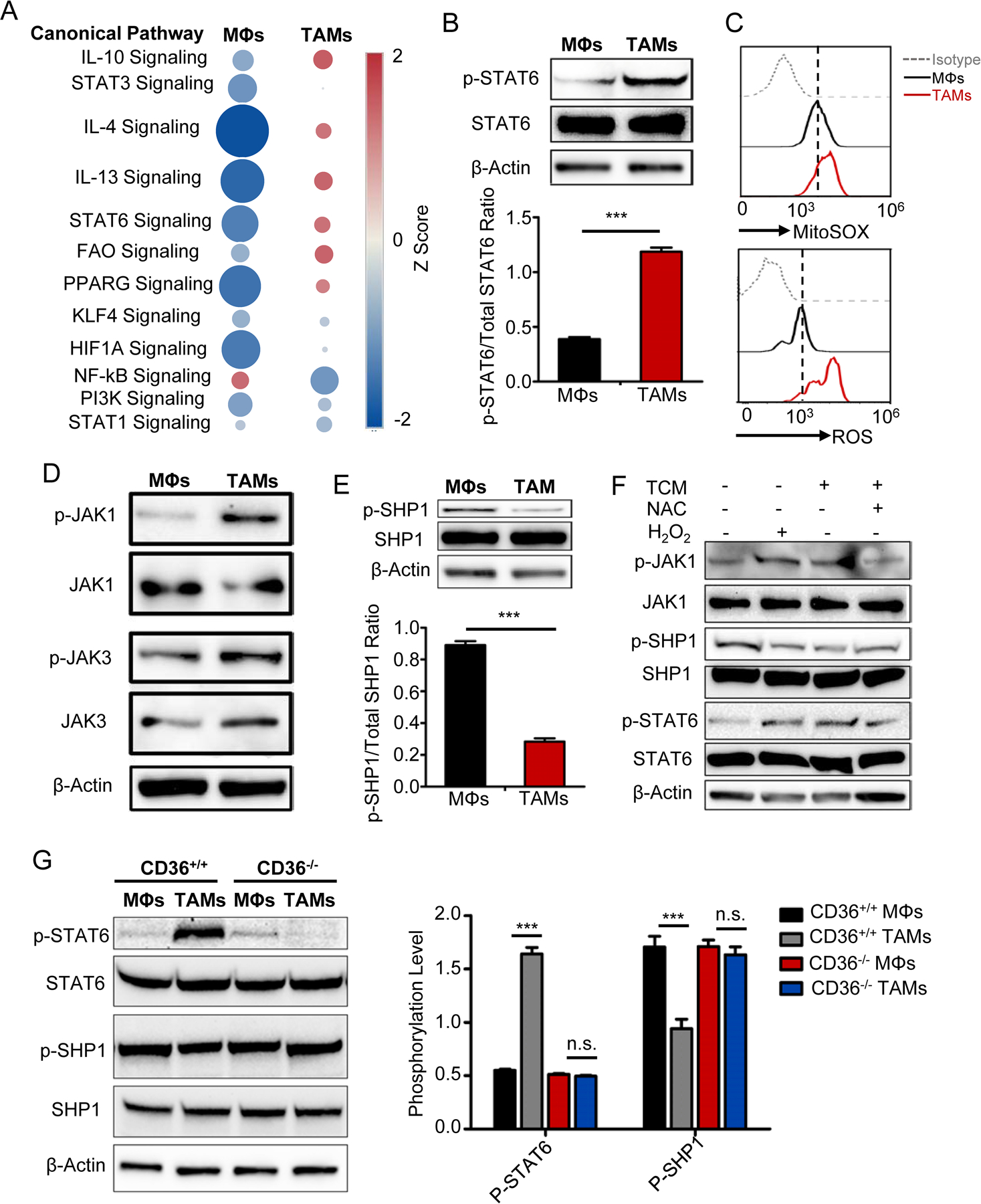
A, IPA analysis of canonical signaling pathways in control MΦs and TAMs. The circle surface area is proportional to −log (P-value) and the color intensity of circles indicates the Z score.
B, Representative western blots (upper) of 3 independent experiments and statistic result (lower) showing STAT6 phosphorylation in control MΦs and TAMs. Statistical significance was determined by two-tailed Student’s t test between indicated groups. ***, P < 0.001.
C, Representative histograms showing the levels of superoxide determined by Mitosox Red (upper) and cellular ROS by DCFDA (lower) in control MΦs and TAMs.
D, Representative immunoblots of JAK1 and JAK3 phosphorylation in control MΦs and TAMs.
E, Representative immunoblots (lower) and quantitative result (upper) of the SHP1 phosphorylation in control MΦs and TAMs. Statistical significance was determined by two-tailed Student’s t test between indicated groups. ***, P < 0.001.
F, immunoblots of the phosphorylated and total JAK1, STAT6, and SHP1 in MΦs with indicated treatment.
G, Western blots of STAT6 and SHP1 phosphorylation in control MΦs and TAMs from CD36 WT and KO mice. Bar graph showing means ± SEM of 3 independent experiments. Statistical significance was determined by two-tailed Student’s t test between indicated groups. ***, P < 0.001, n.s., not significant.
Clinical significance of CD36 expression in TAMs
To determine which cells express CD36 in tumor tissues, we analyzed 2 independent single-cell sequencing data from ArrayExpress and GEO (Supplementary Table 2, available online) on human lung (25) and breast cancers (26). Interestingly, CD36 was mainly expressed in CD14+CD11b+ myeloid cluster, which was one of 8 unbiased major clusters (Supplementary Fig. S7A). The majority (71.9%) of CD36-expressing cells were myeloid cells and 83% of these myeloid cells were MΦs (Supplementary Fig. S7B). Among CD36+ MΦs, 59.9% were tumor-derived MΦs (TAMs) and expressed a significantly higher level of CD36 expression compared with MΦs from normal tissues, which is consistent with the elevated levels of CD36 in total tumor tissues (Supplementary Fig. S7C). Similarly, the majority (49.3%) of CD36-expressing cells were MΦs and 99.9% of these MΦs were tumor-derived MΦs in human breast cancer (Supplementary Fig. S7D). Moreover, high expression of CD36 in CD68high TAM clusters was positively correlated with an upregulated expression of genes associated with TAM differentiation and fatty acid oxidation (Supplementary Fig. S7E). These data indicated that CD36 is mainly expressed by MΦs in tumor tissues and related with TAMs differentiation and fatty acid oxidation of the cells. Furthermore, we analyzed published array data from human renal carcinoma tissues (27,28) and found relatively higher levels of CD36 mRNA in tumor tissues than their normal counterparts and a strong correlation between CD36 expression and TAM signature gene expression (Supplementary Fig. S7F and G). In addition, we examined the published dataset from glioblastoma tissues (29) and observed a strong correlation between the expressions of CD36 and gene cluster involved in fatty acid oxidation pathway (Supplementary Fig. S7H), which is consistent with the results from our in vitro co-culture and in vivo studies. Taken together, these human cancer results demonstrated that CD36 is indeed highly expressed in human tumor tissues especially in MΦs and its expression is highly correlated with the expression of TAM signature genes and fatty acid oxidation-related genes.
Therapeutic efficacy of targeting TAM lipid metabolism in cancer treatment
Finally we determined whether targeting TAM lipid metabolic pathways can be used to treat cancer. First, the VK*MYC myeloma mouse model (30,31) was used and established in WT and CD36-KO C57BL/6J mice. Interestingly, while most of the WT B6 mice developed myeloma and succumbed to the disease, all CD36-KO mice survived without developing tumor (Fig. 7A and B). Second, we subcutaneously injected EL4 lymphoma cells into B6 mice together with or without TAMs generated from WT or CD36-KO mice and monitored tumor growth. Mice bearing EL4 cells and co-injection of WT TAMs had significantly lager tumor burdens than mice injected with EL4 alone or co-injected with EL4 and CD36-KO TAMs, indicating that exogenously injected CD36-KO TAMs did not promote EL4 growth in vivo (Fig. 7C). We then examined the infiltration of MΦs into tumor tissues in tumor-bearing CD36 WT or KO mice, and found that CD36 deficiency impaired the infiltration of MΦs into tumor bed (Supplementary Fig. S8A–D). In addition, we determined MΦ polarization status in tumor-bearing CD36 WT and KO mice using gene microarray analysis and observed that, compared to CD36 WT MΦs, CD36-deficient MΦs expressed significantly higher M1-signature genes (Supplementary Fig. S8E) and lower M2-signature genes (Supplementary Figure S8F). qPCR (Supplementary Fig. S8G and H) and flow cytometry (Supplementary Fig. S8I and J) confirmed these results, indicating that CD36 deficiency polarizes MΦs to M1-type cells.
Fig. 7. Effect of fatty acid oxidation inhibition on protumor function of TAMs.
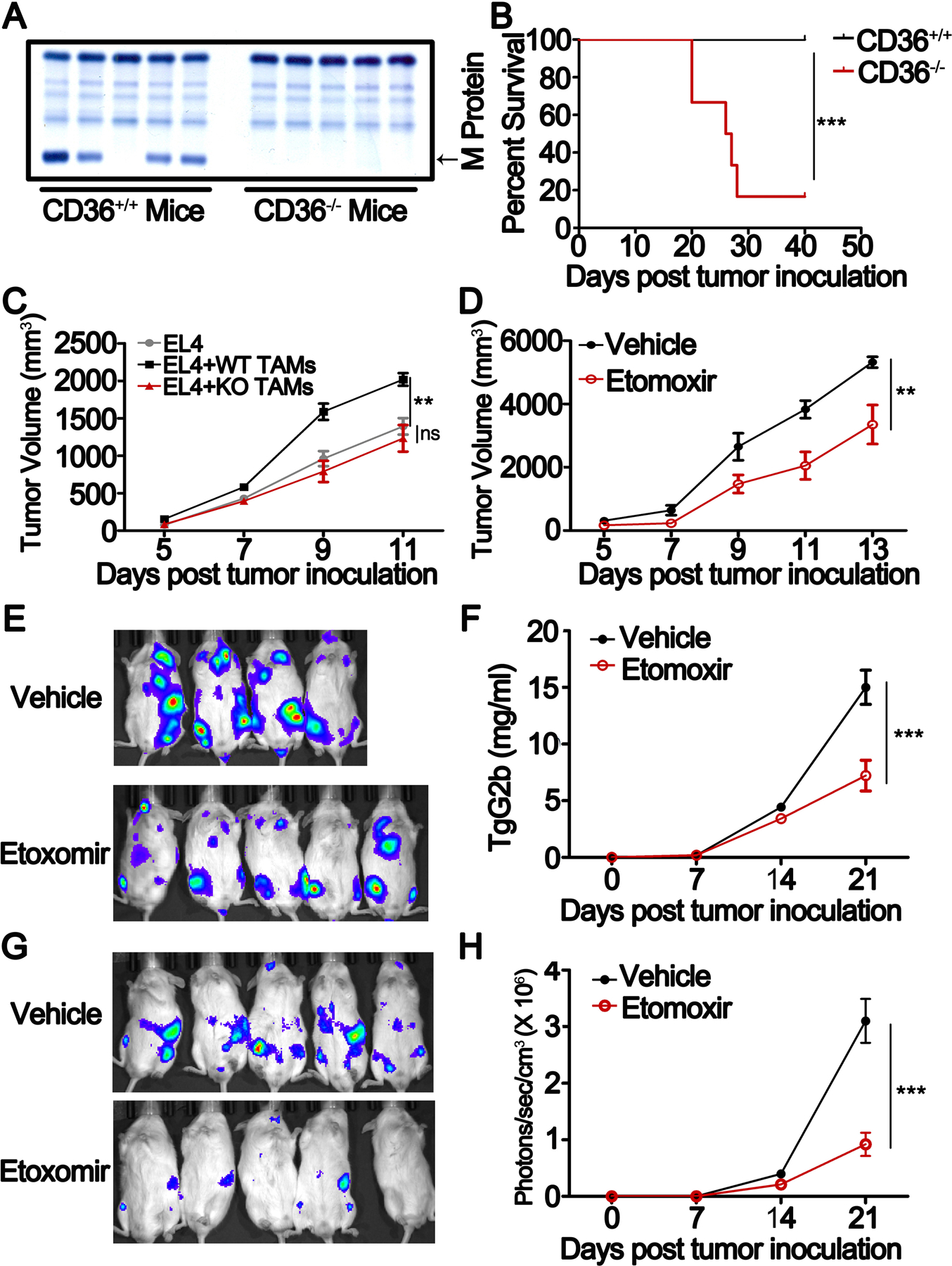
A, SPEP was performed on a representative Vk*MYC-bearing CD36 WT and KO mice. Arrows indicate M protein in Vk*MYC-bearing mice.
B, Survival curves of CD36 WT and KO mice bearing Vk*MYC myeloma cells. The results were analyzed by Log-rank (Mantel-Cox) test (**, P < 0.01).
C, EL4 lymphoma cells were mixed with or without CD36 WT or KO TAMs at a ratio of 5:1, and then injected subcutaneously into C57BL/6J mice. Tumor growth was monitored for 7 days. Results of tumor volumes are shown as means ± SEM. (* P < 0.05)
D, EL4-bearing NSG mice were treated with 50 mg/kg etomoxir intraperitoneally every two days after tumor injection. Tumor growth was followed for 9 days. Results of tumor volumes were shown as means ± SEM. (* P < 0.05)
E, Bioluminescent images of 5TGM1-luc myeloma cell-bearing NSG mice treated with 50 mg/kg etomoxir or vehicle on day 21.
F, Tumor burden measured as serum concentration of IgG2b in 5TGM1-bearing mice treated with etomoxir or vehicle. Results are shown as means ± SEM. **, P < 0.01.
G-H, Bioluminescent images (G) and statistic results of bioluminescent signals (H) of human MM.1S-luc cell-bearing NSG mice on day 21. Results are presented as means ± SEM. **, P < 0.01.
To explored whether inhibiting fatty acid oxidation can be used to treat cancer in vivo. we used etomoxir to inhibit fatty acid oxidation. Etomoxir treatment significantly suppressed tumor growth in EL4 lymphoma- (Fig. 7D) and 5TGM1 myeloma-bearing mice (Fig. 7E and F). Finally, in human myeloma-xenografted mouse model, etomoxir was also able to inhibit human myeloma growth in vivo (Fig. 7G and H). These results provide direct evidence that inhibiting TAM lipid metabolism could suppress the growth and progression of tumors in vivo.
Discussion
We and others have shown that in the TME, TAMs can promote tumor growth and induce drug resistance (4,5). Depleting TAMs exerted promising antitumor responses in vivo (4). It is unclear, however, how TAMs acquire a tumor-promoting phenotype in TME. In this study, we observed that TAMs from both human and murine tumor tissues were enriched with high levels of lipid. Inhibition of lipid uptake by blocking CD36 or knocking down Cd36, or inhibiting fatty acid oxidation in MΦs can block the generation of TAMs abolished their protumor functions in vitro and in vivo. These results indicated that lipid metabolism may play an important role in the differentiation and function of TAMs. In line with our results, a number of studies showed that lipid is a crucial factor regulating tumor-associated dendritic cells (32) and myeloid-derived suppressor cell (MDSC) function (33).
Generally, activated MΦs show changes in their metabolism that are associated with their cellular function. For example, classically in vitro LPS/IFN-γ-activated, pro-inflammatory M1 MΦs switch their metabolism toward enhance anaerobic glycolysis (34), while alternatively activated M2 MΦs induced by IL-4/IL-13 are associated with enhanced FAO and OXPHOS without changes in glycolysis (35). However, little is known about the metabolic characteristics of TAMs and the role of lipid metabolism in modulating their phonotype and function in the context of cancer development and progression. A recent study (36) reported that aerobic glycolysis is increased in TAMs and closely related with the promotive functions of TAMs on tumors. However, another study (37) demonstrated that monocyte-derived TAMs from human gliomas, compared to tissue-resident TAMs, exhibited a decreased glycolytic metabolism and an increased immunosuppression in the TME that were associated with poor patient survival. These studies suggest that reduced glycolytic activity in TAMs favors tumor progression via both nutritional and immunological circuitries. Interestingly, a recent study (38) also found that increased FA metabolism in TAMs was linked to their antitumor/protumor functions dependent on the type of FA enriched in cells. Compared to cells enriched with saturated FA that have protumor activity, TAMs enriched with unsaturated FA have antitumor function (39).
TAMs are highly heterogeneous (1), which presents a major challenge in identifying a treatment modality to target protumor TAMs. It has been shown that tumors can polarize MΦs into TAMs independent in IL-4/IL-13, such as prostate cancer-derived cathelicidin-related antimicrobial peptide (12), Hypoxic cancer cell-derived oncostatin M and eotaxin (13), and Myeloma-derived MIF (6). In this study we discovered that enhanced lipid accumulation and metabolism can also reprogram MΦs into TAMs in the TME. Therefore, our study suggests that targeting lipid metabolism in TAMs may be a promising therapy method for cancer patients.
CD36 plays a key role in the occurrence and development of tumors and has been repeatedly proposed as a prognostic marker in various cancers, mostly of epithelial origin such as breast (40), prostate (41), ovary(42), and colon(43), and also for hepatic carcinoma (44) and gliomas (45). By analyzing large, published datasets of human cancers, we also confirmed that CD36 is highly expressed by human tumor tissues especially the MΦs, and its expression is positively correlated with the expressions of TAMs and fatty acid oxidation gene signatures. However, there is little information about the role of CD36 in regulating TAM development. Our results showed that CD36 is a key receptor for lipid uptake and TAM generation in MΦs. Interestingly, CD36 also plays an important role in MDSCs for increasing fatty acid uptake and fatty acid oxidation to support their immunosuppressive functions (46). These findings indicate that CD36 may be a critical factor for regulating the lipid metabolism in tumor-surrounding cells to promote tumor growth and metastasis and inhibit the immune system, suggesting CD36 as a potential biomarker or target for diagnosis, prognosis, and therapy of cancers.
As one of the most abundant components of the TME, TAMs are derived from circulating monocytes that are recruited to the TME. After entering TME, monocytes need to undergo metabolic adaptations in order to survive in the harsh microenvironment (3). In other immune cells fatty acid oxidation is known to support cellular survival and longevity (47). Consistent with these studies, high levels of fatty acid oxidation and higher proliferation were observed in TAMs compared with control MΦs. However, how the metabolism process initiates and shapes the generation and function of TAMs in TME is completely unknown.
To elucidate the mechanism underlying fatty acid oxidation-mediated differentiation and function of TAMs, we analyzed activated signaling pathways between TAMs and control MΦs with IPA, and found that IL-4, IL-13, IL-10, PPARG, and STAT6 related signaling pathways were significantly activated in TAMs. In MΦs, IL-4 and IL-13 could directly or indirectly activate STAT6 phosphorylation and enhance PPARγ activity via STAT6. Thus, we hypothesized that STAT6 phosphorylation may be a potential target of fatty acid oxidation to initiate and shape the function of TAMs. Indeed, compared with control MΦs, the phosphorylation levels of STAT6 in TAMs were significantly increased. Interestingly, we observed that the activities of JAK1, an important STAT6 activator, and SHP1, an important STAT6 negative regulator, were affected by oxidative stress in TAMs, suggesting that fatty acid oxidation-induced high oxidative stress initiates and shapes the function of TAMs by regulating the JAK1-STAT6 axis in MΦs. However, consistent with a recently published study (48), neither IL-4 nor IL-13 was detected in the TCM used in our study, indicating that STAT6 activation in TAMs may be independent on IL-4/IL-13 signaling but induced by enhanced fatty acid oxidation. Thus, our findings provide a plausible explanation as to why lipid metabolism promotes the generation of M2-like TAM under IL4/IL13-free TME and is crucial for TAM activity.
To summarize, we describe a novel mechanism underlying lipid metabolism-initiated process that promotes the differentiation and function of the protumor TAMs in TME. This finding may not only contribute to a better understanding of the importance of lipid metabolism in the differentiation and function of protumor MΦs but also underscore fatty acid oxidation-dependent signaling pathway as a potential therapeutic target for the treatment of human cancers.
Supplementary Material
Significance:
This study highlights the role of lipid metabolism in the differentiation and function of TAMs and suggests targeting TAM fatty acid oxidation as a potential therapeutic modality for human cancers.
Acknowledgement
Funding
This work was supported in part by NIH/NCI R01s CA200539, CA211073, CA214811, and CA239255 grants and by Cancer Prevention & Research Institute of Texas Recruitment of Established Investigator Award (RR180044).
We thank Research Core services in Lerner research institute, Cleveland Clinic and Houston Methodist Research Institute for their support. We especially thank Tidyomics for their technical support for single-cell sequencing data analysis.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
References:
- 1.Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunological reviews 2014;262:36–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ye H, Zhou Q, Zheng S, Li G, Lin Q, Wei L, et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell death & disease 2018;9:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014;6:1670–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Q, Lu Y, Li R, Jiang Y, Zheng Y, Qian J, et al. Therapeutic effects of CSF1R-blocking antibodies in multiple myeloma. Leukemia 2018;32:176–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang C, He L, He P, Liu Y, Wang W, He Y, et al. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med Oncol 2015;32:352. [DOI] [PubMed] [Google Scholar]
- 6.Zheng Y, Wang Q, Li T, Qian J, Lu Y, Li Y, et al. Role of Myeloma-Derived MIF in Myeloma Cell Adhesion to Bone Marrow and Chemotherapy Response. Journal of the National Cancer Institute 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Ostling J, et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell reports 2016;15:2000–11 [DOI] [PubMed] [Google Scholar]
- 8.Mantovani A, Locati M. Macrophage Metabolism Shapes Angiogenesis in Tumors. Cell metabolism 2016;24:653–4 [DOI] [PubMed] [Google Scholar]
- 9.Osterborg A, Yi Q, Henriksson L, Fagerberg J, Bergenbrant S, Jeddi-Tehrani M, et al. Idiotype immunization combined with granulocyte-macrophage colony-stimulating factor in myeloma patients induced type I, major histocompatibility complex-restricted, CD8- and CD4-specific T-cell responses. Blood 1998;91:2459–66 [PubMed] [Google Scholar]
- 10.Yan H, Dong M, Liu X, Shen Q, He D, Huang X, et al. Multiple myeloma cell-derived IL-32gamma increases the immunosuppressive function of macrophages by promoting indoleamine 2,3-dioxygenase (IDO) expression. Cancer letters 2019;446:38–48 [DOI] [PubMed] [Google Scholar]
- 11.Petty AJ, Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy 2017;9:289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cha HR, Lee JH, Hensel JA, Sawant AB, Davis BH, Lee CM, et al. Prostate cancer-derived cathelicidin-related antimicrobial peptide facilitates macrophage differentiation and polarization of immature myeloid progenitors to protumorigenic macrophages. The Prostate 2016;76:624–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. The Journal of clinical investigation 2016;126:3672–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer research 2014;74:24–30 [DOI] [PubMed] [Google Scholar]
- 15.Osinsky S, Bubnovskaya L, Ganusevich I, Kovelskaya A, Gumenyuk L, Olijnichenko G, et al. Hypoxia, tumour-associated macrophages, microvessel density, VEGF and matrix metalloproteinases in human gastric cancer: interaction and impact on survival. Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico 2011;13:133–8 [DOI] [PubMed] [Google Scholar]
- 16.Vatolin S, Phillips JG, Jha BK, Govindgari S, Hu J, Grabowski D, et al. Novel Protein Disulfide Isomerase Inhibitor with Anticancer Activity in Multiple Myeloma. Cancer research 2016;76:3340–50 [DOI] [PubMed] [Google Scholar]
- 17.Cassol E, Cassetta L, Rizzi C, Alfano M, Poli G. M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J Immunol 2009;182:6237–46 [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Zheng Y, Li T, Wang Q, Qian J, Lu Y, et al. Chemokines CCL2, 3, 14 stimulate macrophage bone marrow homing, proliferation, and polarization in multiple myeloma. Oncotarget 2015;6:24218–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Zhang S, Jiang H, Wang J, Weng L, Mao Y, et al. PA from an H5N1 highly pathogenic avian influenza virus activates viral transcription and replication and induces apoptosis and interferon expression at an early stage of infection. Virology journal 2012;9:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng Y, Yang J, Qian J, Qiu P, Hanabuchi S, Lu Y, et al. PSGL-1/selectin and ICAM-1/CD18 interactions are involved in macrophage-induced drug resistance in myeloma. Leukemia 2013;27:702–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nature reviews Clinical oncology 2017;14:399–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Frontiers in immunology 2014;5:614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahal OM, Wolfe AR, Mandal PK, Larson R, Tin S, Jimenez C, et al. Blocking Interleukin (IL)4- and IL13-Mediated Phosphorylation of STAT6 (Tyr641) Decreases M2 Polarization of Macrophages and Protects Against Macrophage-Mediated Radioresistance of Inflammatory Breast Cancer. International journal of radiation oncology, biology, physics 2018;100:1034–43 [DOI] [PubMed] [Google Scholar]
- 24.Johnson DJ, Pao LI, Dhanji S, Murakami K, Ohashi PS, Neel BG. Shp1 regulates T cell homeostasis by limiting IL-4 signals. The Journal of experimental medicine 2013;210:1419–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nature medicine 2018;24:1277–89 [DOI] [PubMed] [Google Scholar]
- 26.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018;174:1293–308 e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones J, Otu H, Spentzos D, Kolia S, Inan M, Beecken WD, et al. Gene signatures of progression and metastasis in renal cell cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2005;11:5730–9 [DOI] [PubMed] [Google Scholar]
- 28.Beroukhim R, Brunet JP, Di Napoli A, Mertz KD, Seeley A, Pires MM, et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer research 2009;69:4674–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun L, Hui AM, Su Q, Vortmeyer A, Kotliarov Y, Pastorino S, et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer cell 2006;9:287–300 [DOI] [PubMed] [Google Scholar]
- 30.Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, et al. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood 2012;120:376–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bi E, Li R, Bover LC, Li H, Su P, Ma X, et al. E-cadherin expression on multiple myeloma cells activates tumor-promoting properties in plasmacytoid DCs. The Journal of clinical investigation 2018;128:4821–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nature medicine 2010;16:880–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer immunology research 2015;3:1236–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. The Journal of biological chemistry 2014;289:7884–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nature immunology 2014;15:846–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B, et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016;5:e1191731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome biology 2017;18:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Sun Y, Rao E, Yan F, Li Q, Silverstein KA, et al. Fatty acid-binding protein E-FABP restricts tumor growth by promoting IFN-beta responses in tumor-associated macrophages. Cancer research 2014;74:2986–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlager SI, Madden LD, Meltzer MS, Bara S, Mamula MJ. Role of macrophage lipids in regulating tumoricidal activity. Cellular immunology 1983;77:52–68 [DOI] [PubMed] [Google Scholar]
- 40.Zhao J, Zhi Z, Wang C, Xing H, Song G, Yu X, et al. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncology reports 2017;38:2105–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Science translational medicine 2019;11. [DOI] [PubMed] [Google Scholar]
- 42.Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018;37:2285–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Enciu AM, Radu E, Popescu ID, Hinescu ME, Ceafalan LC. Targeting CD36 as Biomarker for Metastasis Prognostic: How Far from Translation into Clinical Practice? BioMed research international 2018;2018:7801202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Scientific reports 2015;5:14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K, et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 2014;32:1746–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017;6:e1344804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nature reviews Cancer 2013;13:227–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell metabolism 2019;29:1376–89 e4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.