

Abstract
We previously reported that the environmental pollutant and tobacco smoke constituent dibenzo[def,p]chrysene (DBP) induced DNA damage, altered DNA methylation and induced oral squamous cell carcinoma (OSCC) in mice. In the present study, we showed that 5% dietary black raspberry (BRB) significantly reduced (p <0.05) the levels of DBP-DNA adducts in the mouse oral cavity with comparable effect to those of its constitutes. Thus, only BRB was selected to examine if aberrant DNA methylation induced by DBP can be altered by BRB. Using comparative genome-wide DNA methylation analysis, we identified 479 hypermethylated and 481 hypomethylated sites (q < 0.01, methylation difference >25%) between the oral tissues of mice treated with DBP and fed control diet or diet containing BRB. Among the 30 differential methylated sites (DMS) induced by DBP, we found DMS mapped to Fgf3, Qrich2, Rmdn2 and Cbarp were hypermethylated by BRB while hypomethylated by DBP at either the exact position or proximal sites; DMS mapped to Vamp3, Ppp1rB1, Pkm, and Zfp316 were hypomethylated by BRB but hypermethylated by DBP at proximal sites. In addition to Fgf3, 2 DMS mapped to Fgf4 and Fgf13 were hypermethylated by BRB; these fibroblast growth factors are involved in regulation of the epithelial-mesenchymal transition pathway (EMT) as identified by IPA. Moreover, BRB significantly reduced (p<0.05) the tumor incidence from 70% to 46.7%. Taken together, the inhibitory effects of BRB on DNA damage combined with its effects on epigenetic alterations may account for BRB inhibition of oral tumorigenesis induced by DBP.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the 6th most common human cancer worldwide; oral squamous cell carcinoma (OSCC) is the most histologic type of this disease (1–3). Up to 77% of oral cancer cases are diagnosed at an advanced stage (4) and early diagnosis of this disease has not improved over time; the survival rate is stagnant at approximately 50% but in general, it varies with the stage of the disease (4). About one-third of patients treated with surgery, radiotherapy and chemotherapy (5) will experience local or regional recurrence and/or distant metastasis (6).
Exposure to exogenous carcinogens [tobacco smoke, smokeless tobacco, excess alcohol, human papillomavirus (HPV)] can account for 90% of OSCC (1,2). Avoidance of risk factors has only been partially successful in preventing this disease, largely because of the addictive power of tobacco smoking and alcohol consumption. Therefore, novel or improved approaches to prevention and/or early detection are urgently needed for the management and control of this disease.
Prevention remains a desirable approach since treating cancers, including OSCC, at late stages even with improved targeted therapies continues to be a major challenge. Our approach to prevention is based on understanding the molecular mechanisms that account for the induction of OSCC by carcinogens found in the human environment (7) . Preclinical animal models that employ environmental carcinogens, reflect tumor heterogeneity, and accurately reflect the cellular and molecular changes in the multi-step process of oral carcinogenesis in humans could provide a realistic platform. Thus, we introduced a novel OSCC mouse model using the environmental pollutant and tobacco smoke constituent dibenzo[def,p]chrysene, also known as dibenzo[a,l]pyrene (DBP), and its fjord region diol epoxide (DB[a,l]PDE) (8,9).
We found that both DBP and DB[a,l]PDE induced DNA damage in mouse oral tissues (7). The induction of DNA damage by these carcinogens stimulated a follow-up investigation aimed at determining their effects on in vivo mutagenesis in the oral cavity of Big Blue C57BL/6 mice (8,9). Both carcinogens were powerful mutagens and induced mutation profiles in the lacI reporter gene similar to those observed in p53 gene in human HNSCC; specifically, 50% of DBP and DB[a,l]PDE-induced mutations are G:C→T:A and G:C→A:T substitution and about 30% of the mutations at AT base pairs and these percentages are similar to those found in the P53 gene in human HNSCC (7,10). Furthermore, we also showed that upreulation of p53 and COX-2 proteins was observed (8,9). DBP also resulted in a significant upregulation of several inflammatory-related genes in the mouse oral tissue (11). Furthermore, we showed that hypomethylation of Fgf3 is a potential biomarker for early detection of OSCC in mice treated with DBP (12). Fgf3 is among a large fibroblast growth factor superfamily genes which are involved in numerous biological activities, including cell survival and regulation of epithelial-mesenchymal transition (EMT) pathways (13–15). Frequent amplification of Fgf3 gene is observed in HNSCC (16–18).
The development of effective, safe, and easy to administer chemopreventive agents is urgently needed and continues to be an active area in the arena of cancer prevention research. Several sources of phytochemicals have been proposed but one that has shown great promise in cancer prevention in both preclinical and clinical investigations is black raspberry (BRB) (7,19–24). BRB has been shown to inhibit 7,12-dimethylbenz[a]anthracene (DMBA) induced cancer in the hamster cheek pouch (23) and 4-nitroquinoline-N-oxide (4-NQO)-induced tongue cancer in the rat (24). However, both DMBA and 4-NQO have never been detected in the human environment and in contrast to the hamster model, the human oral cavity lacks a cheek pouch. In addition, DMBA is known to induce H-ras mutations which are found in less than 5% of oral cancers in the Western world (7). Taken together, our mouse model offers a realistic platform to mechanistically study cancer prevention in the oral cavity at the genetic and epigenetic levels. Thus, in the present study, we selected BRB powder as a whole food approach and its related constituents to initially determine their effects, using a short-term mouse bioassay, on DBP-induced DNA damage in the oral cavity; the results strongly suggest the protective role of BRB on DBP-induced DNA damage was comparable to other constituents and thus for practical reasons, only BRB was used in follow-up studies as a whole food approach to examine for the first time its effects on epigenetic regulation (DNA methylation) and tumorigenesis-induced by DBP in the mouse oral cavity.
Material and Methods
Chemicals
We prepared DBP according to our published method (8). Structural characterization of this carcinogen was based on NMR and high-resolution mass spectral data, and its purity (≥99%) was determined by HPLC. Protocatechuic acid (PA) and ferulic acid (FA) were purchased from Sigma-Aldrich Chemical Co. BRB powder was provided by Berri Products LLC (OR, USA). BRBE was provided by Dr. Stoner and prepared using a published method (25).
Animals
Species, strain and sex (female B6C3F1 mice 6-8 weeks of age, The Jackson Laboratory) of the animals used in the present study were selected based on our previous report (8). Following one week of quarantine, mice were transferred to the bioassay laboratory; they were kept on a 12-hour light/dark cycle, 50% relative humidity and 21 ± 2°C. Mice were provided with water and food ad libitum. All the bioassays were performed in line with the NIH Guide for the Care and Use of Laboratory Animals; experimental protocols were approved by the Institutional Animal Care and Use Committee.
Effects of dietary BRB, BRB extract (BRBE), FA, and PA on DBP-induced DNA adducts in the mouse oral tissues.
Five groups of mice (6 mice/group) were fed AIN-93 M diet (5% corn oil) as control diet and AIN-93M diet containing BRB (5%); BRBE (1.6%), anthocyanin enriched fraction extracted from BRB, FA (0.05%, a major phenolic compound in raspberries); and PA (0.2%, a major metabolite of anthocyanins) starting 2 weeks prior to the administration of DBP (24 nmol, 3 times per week for 5 weeks) by topical application into the oral cavity of mice (Figure 1A); the various diets were fed until termination of the bioassay.
Figure 1.
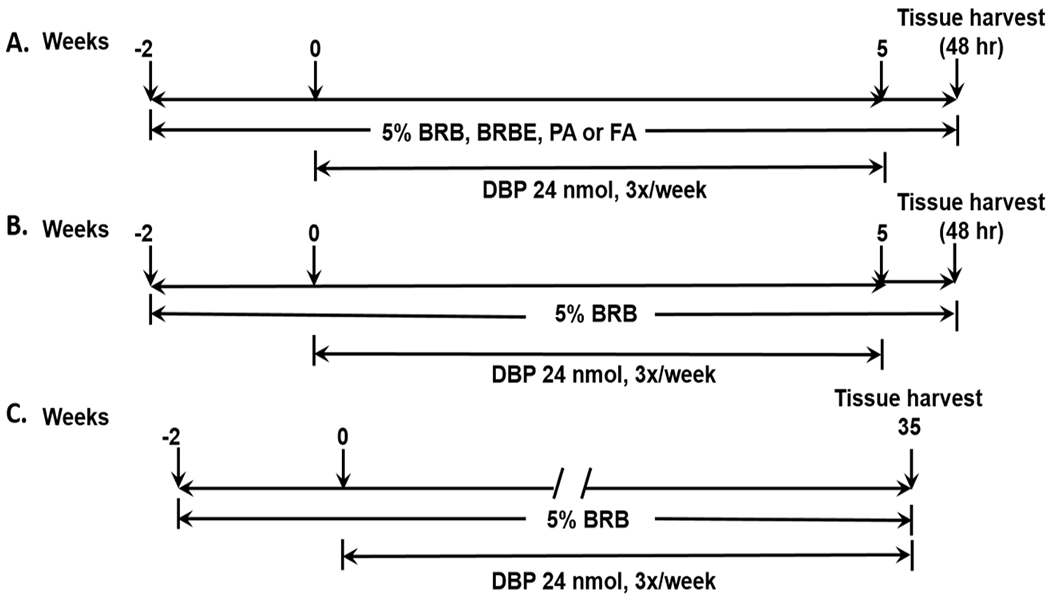
Experimental protocol for the effects of BRB on DBP-induced (A) DNA damage, (B) DNA methylation and (C) tumorigenesis in the mouse oral tissues.
Several previous preclinical studies indicated that 5% BRB was the optimal level for protection against cancer development in animal models [reviewed in (7)]. Levels of the other agents were estimated based on the composition of BRB and PA level was based on a published method (26). The dose of DBP was selected based on our previous studies (8,27). Mice were sacrificed 48h after the last dose of DBP and tissues were harvested for DNA isolation.
The analysis of the major deoxyadenosine (dA) adducts derived from DBP by LC-MS/MS was performed using our previously published method (27,28). Briefly, DNA was isolated from oral tissues using the Qiagen genomic DNA isolation procedure. The level of DNA was quantified by a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). The internal standard [‘15N5]-anti-trans DBPDE-dA adduct (150 pg) was added to about 60 μg DNA prior to enzymatic hydrolysis. Subsequently, in the presence of 10 μL of 1 mol/L MgCl2/mg DNA and DNAse 1 (0.2 mg/mg DNA), DNA was hydrolyzed at 37°C for 1.5 hours, followed by overnight incubation of the mixture with snake venom phosphodiesterase (0.08 U/mg DNA) and alkaline phosphatase (2U/mg DNA). The analysis of dA was performed by HPLC using an aliquot of the DNA hydrolysate. Partial purification of the remaining supernatant was achieved using an Oasis HLB column (1 cm3, 30 mg, Waters Ltd.). The analysis of DBP-dA adduct was performed on an API 3200 LC-MS/MS triple quadrupole mass spectrometer interfaced with an Agilent 1200 series HPLC using an Agilent extend-C18 5μM 4.6x150 mm column. Adducts were monitored in multiple reaction monitoring modes and the MS/MS transition of m/z 604 → m/z 335 and m/z 609 → m/z 335 were monitored for targeted adducts and the internal standard to maximize the sensitivity, respectively.
Effects of dietary BRB on DNA methylation in the oral cavity of mice treated with DBP
We found that the inhibition of DBP-DNA adducts formation by dietary 5% BRB was not significantly different from the other dietary groups (cf Results), and BRB as a whole food approach is easy to obtain at a lower cost. Based on these findings, in the present study, we tested the hypothesis that dietary 5% BRB may alter DNA methylation induced by DBP in the early stage of carcinogenesis prior to any morphological abnormalities but at a stage of maximum DNA damage observed in the mouse oral cavity (12). The goal of this study was to investigate the chemopreventive effects of dietary BRB on oral tumorigenesis induced by DBP and to elucidate the mechanisms that may account for the cancer preventive activity of BRB in this animal model. Thus, using a group of mice treated with DBP as a control to compare with those treated with DBP+BRB allowed us to identify the methylation sites altered by BRB in the presence of DBP. Two groups of mice (n=3 per group), after one week of quarantine, were fed either control AIN-93M diet or AIN-93M diet containing 5% BRB for two weeks prior to topical application of DBP (24 nmol in DMSO) 3 times a week for five weeks (Figure 1B). Mice were sacrificed 48h after the last dose of DBP. Oral tissues were isolated from the same anatomic sites of mice and pooled together for DNA extraction.
Genomic DNA from oral tissues of mice treated with DBP and fed control diet or a diet containing 5% BRB as described above were extracted and purified according to the DNeasy Blood and Tissue kit (Qiagen). The DNA was then subjected to Enhanced Reduced Representation Bisulfite Sequencing (ERRBS) analysis as described previously by our group (12). Briefly, DNA was digested by MspI followed by end repair, adenylation and adapter ligation with a modification of bead size selection to capture MspI fragments of 70-320 bp size. The resulting libraries were bisulfite-converted followed by PCR amplification and read by 1×50 bp on HiSeq and the resulting CASAVA-demultiplexed.fastq files were subjected to downstream analyses. Base calls of bisulfite-treated sequencing reads were mapped to the mm9 mouse assembly and methylation calls were performed using Bismark v0.10.1 (Babraham Bioinformatics, UK). The differential methylation between DBP-BRB vs DBP was calculated using methylKit v0.9.2 R package with “normalizeCoverage” function to avoid bias introduced by systematically more sequenced samples. Differentially methylated bases with q-value < 0.01 and percent methylation difference > 25% were extracted. The methylKit only analyzes bases covered in all samples and takes into account samples size to calculate both p- and q-values. In particular, methylKit accepts either a single sample per group in which case it uses the Fisher’s exact test to calculate p-values or multiple samples per group in which case it uses logistic regression. These differentially methylated sites (DMS) were annotated with genic parts information from the UCSC Table Browser (mm9 refGene table). Ingenuity Pathways version 2014-10-25 was then performed on the genes with altered methylation patterns to obtain significant canonical pathways associated with dietary BRB.
The effect of BRB on DBP-induced tumorigenesis in the mouse oral tissues
The design of this protocol consisted of two groups of mice (30/group) at the age of 8 weeks. Mice were treated (by topical application into the oral cavity) with DBP (24 nmol) that had been dissolved in DMSO 3 times per week for 35 weeks (Figure 1C). In group 1, mice were fed AIN-93M control diet and those in group 2 were fed AIN-93M diet containing 5% BRB. Feeding started two weeks before DBP administration and continued until termination of the bioassay. In our previous study (29), no histological abnormalities were observed in the oral cavity of mice treated with the vehicle (DMSO) by topical application and fed AIN-93M diet containing 5% BRB, and thus such a group of mice was not included in the present study. During the progress of the bioassay, mice were weighed weekly in the first month and then every two weeks until termination. Mice were culled from each group and sacrificed by CO2 asphyxiation if a sudden weight loss (>20%) or tumors size over 0.5 cm in diameter were observed. At sacrifice, we collected soft tissues of the oral cavity which included tongue, pharynx, and other oral tissues (hard palate, buccal mucosa and floor of the mouth). Tissues were fixed in 10% neutral-buffered formalin and then processed in an automated Tissue-Tek VIP processor and paraffin-embedded with a Tissue-Tek TEC embedding station. Staining with hematoxylin and eosin (H&E) were applied to sections that were cut at 6 μm. A board-certified veterinary pathologist (HA) blinded to treatment protocol examined all tissues and lesions received and provided diagnoses based on established International Harmonization of Nomenclature and Diagnostic Criteria (INHAND) for Lesions in Rats and Mice (30,31).
Results
Effects of BRB powder and related agents on DBP-induced DNA adducts in oral tissues of mice
The goal of this short-term animal bioassay was to rank the potency of BRB and related agents to inhibit DNA damage induced by DBP, a prerequisite step in the multi-step carcinogenesis process. Here we showed that BRB (5%), BRBE (1.6%), PA (0.2%) or FA (0.05%) in the diet, starting 2 weeks before carcinogen treatment and continued until termination of the bioassay significantly (p<0.05) reduced the levels of (-)-anti-trans-DBPDE-dA in mouse oral tissues as shown in Figure 2; these treatments significantly resulted in 19.2%, 29.6%, 23.0% and 25.3% reduction of the level of (-)-anti-trans-DBPDE-dA in murine oral tissues, respectively. The percent of inhibition was comparable following the various interventions; therefore we selected, for practical purposes, only 5% BRB as the agent in the subsequent bioassays.
Figure 2.
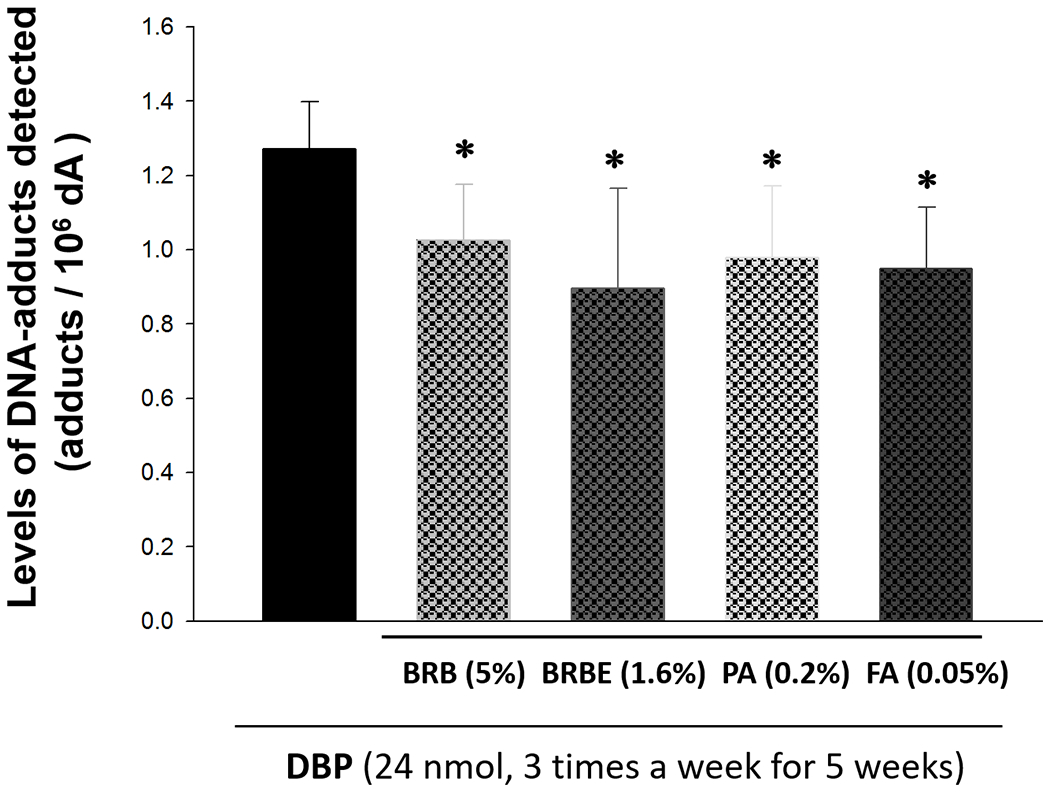
The effects of BRB (5%), BRBE (1.6%), PA (0.2%) or FA (0.05%) in the diet on the levels of (-)-anti-trans-DBPDE-dA in mouse oral tissues. *p < 0.05
Effect of BRB on DBP-induced DNA methylation in the mouse oral tissues
To examine if dietary BRB may alter the DNA methylation induced by DBP, we isolated DNA from histologically normal oral tissues of mice treated with multiple doses of DBP and fed with a control diet or diet containing 5% BRB powder and performed ERRBS analysis coupled with next generation sequencing. This approach provides a quantitative, single nucleotide resolution on the status of DNA methylation both within and outside CpG islands. Our data showed that an average of 8.8 million Illumina sequencing reads was generated per sample and the percentages of methylated C in CpG context were 35.2% and 32.6% for DBP and DBP+BRB respectively. Approximately 72% of the sequencing reads were mapped to either strand of the mouse genome (mm9). These sequencing data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (Accession No:GSE8991).
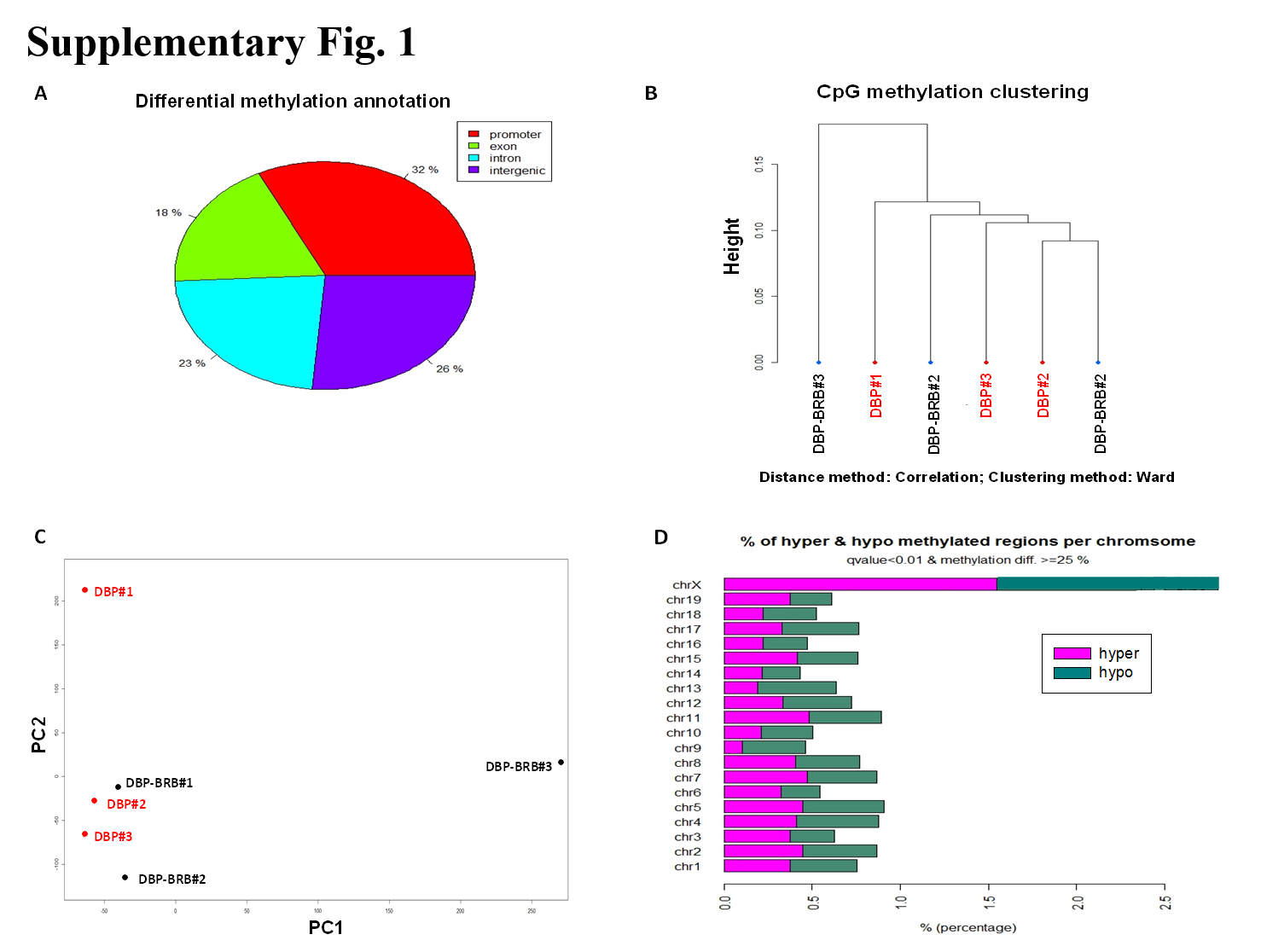
Comparative methylation analysis was conducted between DBP+BRB vs DBP groups to identify differentially DNA methylated sites (DMS) using methylKit (version 0.9.2) R package (32). Our results showed that about 32% DMS are located in promoters, 18% in exons, 23% in introns, and 26% in intergenic regions [Supplementary Fig S1 (A)]; both unsupervised analyses of hierarchical clustering (1-Pearson correlation distance + Ward clustering method) and principal component analysis (PCA) were performed to examine the correlations among all of these samples as shown in Supplementary Fig S1 (B) and (C), respectively. Due to limited sample size, no distinct clusterings were observed; however, large variations were noted from DBP+BRB treated samples in PC1; in PC2, DBP-treated samples showed larger variations as compared to DBP+BRB-treated. The percentage of hyper and hypomethylated differential methylation sites (DMS) out of all covered CpGs for each chromosome (Chr) is shown in Supplementary Fig S1 (D).
A total of 960 differential methylation sites (DMS) were identified between DBP vs. DBP+BRB with q-value <0.01 and percent methylation difference >25%; among them 479 were hypermethylated and 481 were hypomethylated as listed in Supplementary Table S1 (A) and (B), respectively. Among 960 DMS, we found 4 DMS mapped to genes Fgf3, Qrich2, Rmdn2, and Cbarp were hypermethylated by dietary BRB but these loci were hypomethylated by DBP either at the exact position or at proximal site (12). On the other hand, DMS mapped to genes Vamp3, Ppp1r13l, Pkm, and Zfp316 were hypomethylated by BRB but they were hypermethylated by DBP at proximal sites. Although DBP induced two hypermethylated sites mapped to Disc1, BRB induced both hyper- and hypomethylation at proximal sites (Table 1).
Table 1.
Comparisons of differentially methylated sites identified in both ERRBS of DBP-BRB vs. DBP and DBP vs. DMSO
| DBP-BRB vs DBP | DBP vs. DMSO | ||||
|---|---|---|---|---|---|
| Chr | Gene | methylation differences | position | methylation differences | position |
| chr7 | Fgf3 | 34.18234 | 152014445 | −37.6374 | 152014445 |
| chr11 | Qrich2 | 28.62254 | 116317982 | −36.7897 | 116317982 |
| chr17 | Rmdn2 | 35.05828 | 80062330 | −28.5125 | 80062330 |
| chr10 | Cbarp | 34.17839 | 79594923 | −27.812 | 79594761 |
| chr4 | Vamp3 | −26.8916 | 150809404 | 38.833 | 150503856 |
| chr7 | Ppp1r13l | −32.7257 | 19955765 | 43.2683 | 19955390 |
| chr9 | Pkm | −29.0348 | 59503922 | 43.3803 | 59504569 |
| chr5 | Zfp316 | −29.0043 | 144014872 | 25.2916 | 144049680 |
| −28.8456 | 144014905 | ||||
| chr8 | Disc1 | −28.7854 | 127736702 | 36.25 | 127736689 |
| 27.96859 | 127615449 | 35.3156 | 127736699 | ||
Ingenuity Pathway Analysis® (IPA) for the annotated genes identified the top canonical pathways (−log (p) ≥ 1.3) as Glutamate receptor signaling, IGF-1 signaling, Relaxin signaling, Molybdenum cofactor biosynthesis, regulation of epithelial-mesenchymal transition (EMT) pathway, and glycolysis I (Supplementary Table S2). It is noted that in addition to Fgf3, 2 DMS mapped to genes Fgf4 and Fgf13 were also hypermethylated by BRB treatment; these fibroblast growth factors are involved in the EMT pathway.
Effect of BRB on DBP-induced oral tumorigenesis
The inhibitory effects of BRB on DNA damage induced by DBP combined with its effects on epigenetic alterations induced by this carcinogen in the mouse oral cavity provided a strong rationale to further examine the chemopreventive efficacy of BRB against DBP induced oral cancer in the same mouse model as described below.
Body Weights (mean ± SE) of mice treated with DBP and fed control diet containing 5% BRB, and those treated with DBP and fed control diet are provided in Figure 3A. The body weights of mice in both groups were increased gradually in a similar pattern. The cumulative mortality of mice during the progress of the bioassay is shown in Figure 3B. The probability of survival is displayed using a Kaplan-Meier plot with death as an endpoint. A log-rank test was used to evaluate the difference between these groups and no significant difference was observed in survival analysis.
Figure 3.
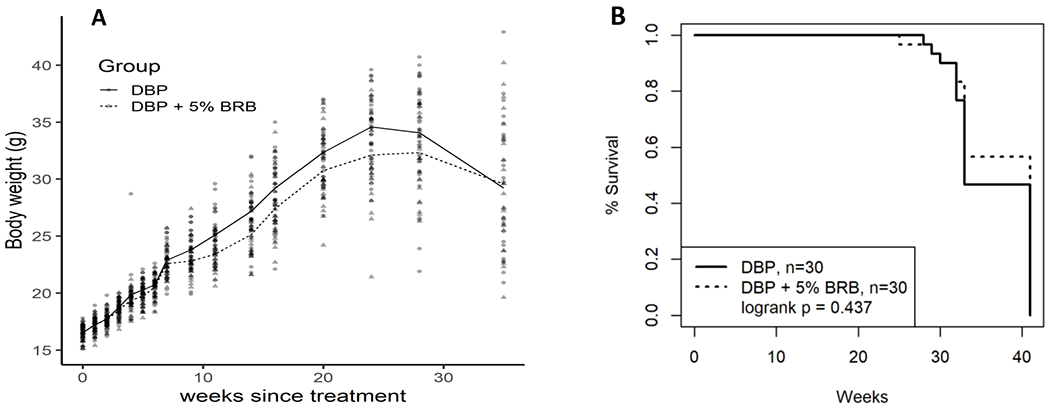
Body weights (A) and percentage survival (B) of B6C3F1 mice treated by topical application of DBP (24 nmol, 3 times a week) and DBP + 5% BRB during the progress of the bioassay.
The tumor incidence (including papilloma, carcinoma in situ (CIS) and SCC) in the oral cavity of mice treated with DBP and fed diet containing 5% BRB was significantly (p<0.05) reduced from 70% to 46.7% (Table 2 and Figure 4), and the incidence of papilloma was also significantly (P<0.05) reduced from 33.3% to 13.3%. SCC in the oral mucosa was reduced from 40% to 33.3%, and CIS was reduced from 13.3% to 3.3%. The overall histological findings in mice treated with DBP and DBP+BRB in the lip, oral cavity and tongue were summarized in Table S3 and showed in Fig S2; BRB had no protective effect in tumors developed in the lip; only one animal treated either with DBP or DBP+BRB developed SCC.
Table 2.
Inhibition of Tumorigenesis by BRB in the oral cavity of DBP-treated mice.
| Treatments | ||
|---|---|---|
| DBP | DBP + BRB | |
| Number of mice | 30 | 30 |
| CIS | 4 (13.3)1 | 1 (3.3) |
| Papilloma | 10 (33.3)1 | 4 (13.3)* |
| SCC | 12 (40.0)1 | 10 (33.3) |
| Total Tumors | 21 (70.0)2 | 14 (46.7)* |
Number in parentheses, percentage of mice developed tumors.
Tumors induced by DBP in the oral cavity consist of papilloma, CIS and SCC.
Tumor incidence was significantly reduced compared to DBP treatment, p < 0.05.
Figure 4.
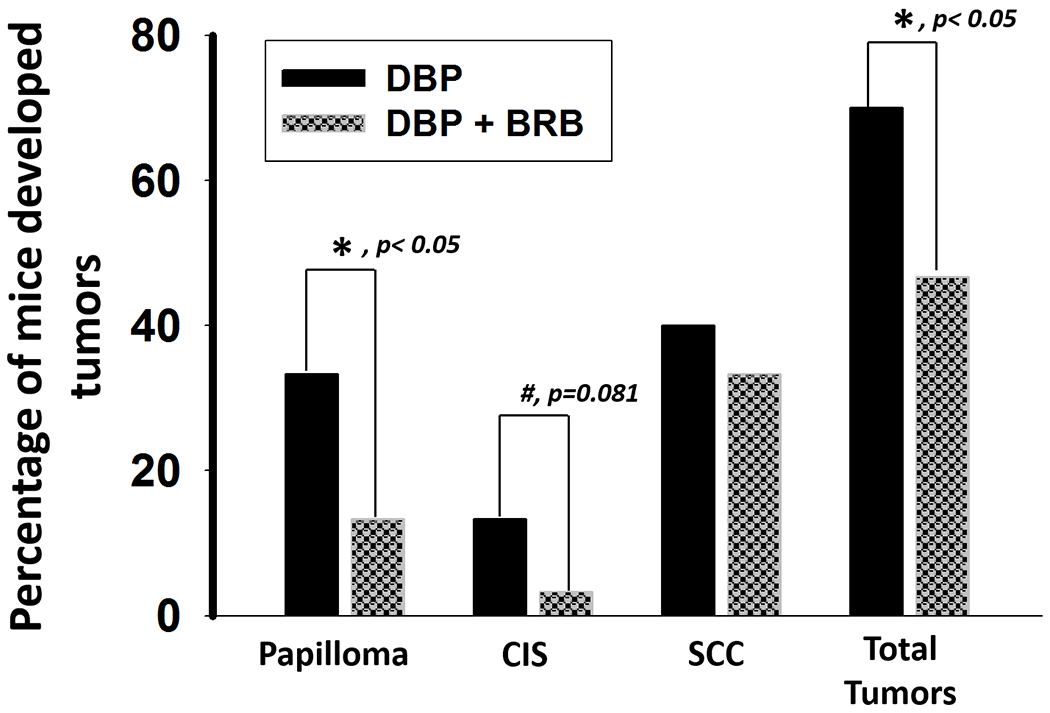
5% BRB inhibits DBP-induced tumorigenesis in the oral cavity of mice. *p < 0.05
Discussion
Genotoxicity (DNA damage, genetic mutations, chromosomal abnormalities) induced by chemicals found in the human environment has been shown to play an essential step in the multi-step carcinogenesis process (33–35). Our first goal of this study was to rank in a short-term in vivo study the efficacy of BRB, some of its constituents and a major metabolite (PA) derived from a major and a biologically active component of BRB (anthocyanins) on DBP-induced DNA damage in the mouse oral cavity. PA accounts for about 70% of the metabolites of anthocyanins in humans (36) and is an inhibitor of chemically-induced cancers in rodents (37). FA, a phenolic acid, is also present in raspberries and can account, in part, for their chemopreventive activities (38). We demonstrated for the first time using LC-MS/MS method that BRB and related agents have comparable but significantly inhibited the levels of (-)-anti-trans-DB[a,l]PDE-dA in the oral tissues of mice treated with DBP. These in vivo findings are consistent with our previous in vitro studies (28,39), demonstrating that anthocyanin components of BRB and its metabolite, PA, are in part responsible for the inhibitory effects of BRB on the DBP-induced DNA adducts formation. Our results also support that dietary consumption of BRB and its related components can inhibit the metabolic activation of DBP or enhance the detoxification of DB[a,l]PDE and the DNA repair efficacy in mouse oral cavity; collectively, these effects of BRB may account for the inhibition of the subsequent mutagenesis and carcinogenesis resulting from exposure to DBP. Although anthocyanins are the most abundant compounds in BRB and can account for much of their antioxidant activity (40,41), they are expensive for routine feeding studies. Considering the cost and easy access, 5% BRB in the diet was used for the remaining mouse bioassays performed in this study.
In addition to genotoxicity, emerging evidence strongly indicates that epigenetic alterations can also play a critical role in the initiation and progression of environmental carcinogenesis (42,43). Many types of DNA damage (covalent DNA adducts, oxidative lesion, abasic sites, photodimers, etc.) have been shown to alter DNA methylation via various mechanisms associated with the formation of DNA lesions and/or by alteration of DNA methyltransferases (12,44). Previously we reported that DBP was able to alter DNA methylation profile in the mouse oral cavity at a stage prior to any morphological abnormalities; we also identified that Fgf3, a gene frequently amplified in HNSCC, was hypomethylated by DBP and may serve as a potential biomarker for early detection of oral carcinogenesis (12). Given the reversibility, although not all of the epigenetic changes, we examined for the first time if BRB may modulate DNA methylation in a manner consistent with prevention of oral carcinogenesis induced by DBP. The effect of RBR on DNA methyltransferase 1 (DNMT1) has been reported by others (45), indicating the BRB can decrease the protein expressions of DNMT1. In our previous study (12), we reported that changes in mRNA expressions of DNMT1, DNMT3a and DNMT3b induced by DBP were observed, but not significant; however, we did not determine the efect of DBP on the activities of DNA methyltransferases. Consistent with the effect of BRB on DNMT1(45), we noted that dietary BRB reduced the percentages of methylated C in CpG context from 35.2% to 32.6 %. Induction of genomic hypomethylation has also been shown to preferentially inhibit the development of squamous cell carcinoma in the tongue and esophagus induced by the synthetic 4-nitroquinoline-1-oxide in a mouse model (46).
In light of the effects of BRB on aberrant DNA methylation induced by DBP, in this study we conducted a genome-wide DNA methylation and identified 960 differentially methylated sites as well as the top canonical pathways including EMT that may account for the epigenetic effect of BRB. DNA methylation is one of the epigenetic mechanisms that may regulate gene expression. Many aberrant DNA methylation occurred in the early stage of carcinogenesis without changes in gene/protein expression; thus, methylation changes can be a powerful biomarker for early detection (12) as well as for monitoring the effects of chemopreventive agents. Several DMS that were induced by DBP treatment were found to be altered by dietary BRB; these DMS were mapped to genes including Fgf3, Qrich2, Rmdn2, Cbarp, Vamp3, Ppp1r13l, and Pkm. It is noted that BRB induced hypermethylation of Fgf3 at the same position that was hypomethylated by DBP. In addition to Fgf3, we also found 2 hypermethylated sites mapped to genes Fgf4 and Fgf13 at promoter region. Although BRB induced hypermethylation of Fgf4 and Fgf13 located in the promoter region, hypermethylation of Fgf3 was located at 10 kb upstream of the Fgf3 transcription start site and is not within a CpG island. All these three fibroblast growth factors were invovled in the regulation of EMT pathway, suggesting that BRB may prevent DBP-induced oral carcinogenesis through epigenetic modulation of genes involved in the EMT pathway. In fact, the reversal of the EMT pathway can account for the inhibition of cancer invasion in a preclinical lung cancer models (47). Encouraged by the results of the effects of BRB on genetic (DNA damage) and epigenetic modulations (DNA methylation), the third goal of this study was to examine the effect of BRB on DBP-induced oral tumorigenesis in a long-term bioassay in mice.
Our results demonstrated that mice tolerated 5% BRB in the diet based on survival and body weights. Furthermore, our current finding is in line with our previous report demonstrating the protective effect of BRB against the ultimate and direct-acting carcinogenic metabolite (DB[a,l]PDE) of DBP. The modulating effects of BRB on Phase I and Phase II drug metabolism enzymes and DNA repair enzymes can account for the inhibition of DNA damage induced by DBP and its metabolite DB[a,l]PDE (7). Similar to our previous finding using DB[a,l]PDE, a significant protective effect of BRB was observed in benign tumors, but the inhibition of malignant formation (CIS, SCC) did not reach significance. We believe that significant inhibition of malignancy can also be reached by extending the duration of the bioassay in order to provide an ample opportunity for benign tumors to transition to malignant tumors; unfortunately, the bioassay was not extended because the majority of the tumors’ size were >0.5cm that hinder eating and drinking and thus the animals had to be sacrificed. In contrast to DB[a,l]PDE, which induced both oral and tongue tumors, DBP induced primarily oral tumor and thus the inhibitory effect of BRB on tongue tumors cannot be assessed (only one animal developed SCC in the tongue induced by DBP or DBP + BRB). Literature data demonstrate that preferential inhibition of tumorigenesis in target organs which come in contact with BRB (7). Exposure of the mouse lip to BRB may be extremely short as compared to the oral cavity within which BRB may retain longer and such differential contact of the two sites may, in part, explain the lack of protection on DBP-induced tumors in the lip.
As discussed above, we showed that dietary 5% BRB inhibits DBP-induced oral tumorigenesis through mechanisms including inhibition of DNA adduct formation and alteration of DNA methylation induced by DBP. In our long-term efficacy study, since BRB was fed before carcinogen treatment and continued until termination, we could not distinguish the effect of BRB on both phases of carcinogenesis (initiation vs promotion/progression). However, the inhibitory effects of BRB on DNA damage observed in this study support its protective role during the initiation phase of carcinogenesis. It is also important to acknowledge additional limitations in the present study. Only female mice were used, and thus our results cannot provide insights on sex differences regarding the higher incidence of oral cancer in men than in women (48). Concerning DNA adducts, we focused only on the detection and quantification of the major dA adducts derived from DBP. However, as reported by us recently, the minor dG adducts could be critical in the induction of mutagenesis induced by DBP in vivo (49). Considering its safe usage and ease of administration, combined with its effects on modulating markers critical in carcinogenesis in both preclinical and clinical studies, future studies should be aimed at determining the effect of BRB on the initiation stage of carcinogenesis by assessing levels of DNA adducts derived from select tobacco carcinogens in smokers which are at high risk of developing oral cancer.
Supplementary Material
{kind=link}
{kind=link}
Significance.
We provided mechanistic insights that can account for the inhibition of oral tumors by BRB, which could serve as the framework for future chemopreventive trials for addicted smokers and former smokers.
Footnotes
Conflict of Interest
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Tanaka T, Ishigamori R. Understanding carcinogenesis for fighting oral cancer. Journal of oncology 2011;2011:603740 doi 10.1155/2011/603740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians 2015;65(2):87–108 doi 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians 2017;67(1):7–30 doi 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68(1):7–30 doi 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 5.Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet 2008;371(9625):1695–709 doi 10.1016/s0140-6736(08)60728-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.da Silva SD, Hier M, Mlynarek A, Kowalski LP, Alaoui-Jamali MA. Recurrent oral cancer: current and emerging therapeutic approaches. Front Pharmacol 2012;3:149 doi 10.3389/fphar.2012.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-Bayoumy K, Chen KM, Zhang SM, Sun YW, Amin S, Stoner G, et al. Carcinogenesis of the Oral Cavity: Environmental Causes and Potential Prevention by Black Raspberry. Chem Res Toxicol 2017;30(1):126–44 doi 10.1021/acs.chemrestox.6b00306. [DOI] [PubMed] [Google Scholar]
- 8.Guttenplan JB, Kosinska W, Zhao ZL, Chen KM, Aliaga C, DelTondo J, et al. Mutagenesis and carcinogenesis induced by dibenzo[a,l]pyrene in the mouse oral cavity: a potential new model for oral cancer. Int J Cancer 2012;130(12):2783–90 doi 10.1002/ijc.26344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen KM, Guttenplan JB, Zhang SM, Aliaga C, Cooper TK, Sun YW, et al. Mechanisms of oral carcinogenesis induced by dibenzo[a,l]pyrene: an environmental pollutant and a tobacco smoke constituent. Int J Cancer 2013;133(6):1300–9 doi 10.1002/ijc.28152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med 2007;357(25):2552–61 doi 357/25/2552 [pii] 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen KM, Schell TD, Richie JP Jr., Sun YW, Zhang SM, Calcagnotto A, et al. Effects of chronic alcohol consumption on DNA damage and immune regulation induced by the environmental pollutant dibenzo[a,l]pyrene in oral tissues of mice. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2017;35(4):213–22 doi 10.1080/10590501.2017.1391514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun YW, Chen KM, Imamura Kawasawa Y, Salzberg AC, Cooper TK, Caruso C, et al. Hypomethylated Fgf3 is a potential biomarker for early detection of oral cancer in mice treated with the tobacco carcinogen dibenzo[def,p]chrysene. PLoS One 2017;12(10):e0186873 doi 10.1371/journal.pone.0186873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizukami T, Togashi Y, Naruki S, Banno E, Terashima M, de Velasco MA, et al. Significance of FGF9 gene in resistance to anti-EGFR therapies targeting colorectal cancer: A subset of colorectal cancer patients with FGF9 upregulation may be resistant to anti-EGFR therapies. Molecular carcinogenesis 2017;56(1):106–17 doi 10.1002/mc.22476. [DOI] [PubMed] [Google Scholar]
- 14.Katoh M, Katoh M. WNT signaling pathway and stem cell signaling network. Clinical cancer research : an official journal of the American Association for Cancer Research 2007;13(14):4042–5 doi 10.1158/1078-0432.CCR-06-2316. [DOI] [PubMed] [Google Scholar]
- 15.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology 2014;15(3):178–96 doi 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Somers KD, Cartwright SL, Schechter GL. Amplification of the int-2 gene in human head and neck squamous cell carcinomas. Oncogene 1990;5(6):915–20. [PubMed] [Google Scholar]
- 17.Lese CM, Rossie KM, Appel BN, Reddy JK, Johnson JT, Myers EN, et al. Visualization of INT2 and HST1 amplification in oral squamous cell carcinomas. Genes, chromosomes & cancer 1995;12(4):288–95. [DOI] [PubMed] [Google Scholar]
- 18.Worsham MJ, Lu M, Chen KM, Stephen JK, Havard S, Schweitzer VP. Malignant and nonmalignant gene signatures in squamous head and neck cancer. Journal of oncology 2012;2012:752860 doi 10.1155/2012/752860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen KM, Guttenplan JB, Sun YW, Cooper T, Shalaby NA, Kosinska W, et al. Effects of Black Raspberry on Dibenzo[a,l]Pyrene Diol Epoxide Induced DNA Adducts, Mutagenesis and Tumorigenesis in the Mouse Oral Cavity. Cancer Prev Res (Phila) 2017. doi 10.1158/1940-6207.capr-17-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoner GD. Foodstuffs for preventing cancer: the preclinical and clinical development of berries. Cancer Prev Res (Phila) 2009;2(3):187–94 doi 10.1158/1940-6207.capr-08-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mallery SR, Tong M, Shumway BS, Curran AE, Larsen PE, Ness GM, et al. Topical application of a mucoadhesive freeze-dried black raspberry gel induces clinical and histologic regression and reduces loss of heterozygosity events in premalignant oral intraepithelial lesions: results from a multicentered, placebo-controlled clinical trial. Clin Cancer Res 2014;20(7):1910–24 doi 10.1158/1078-0432.ccr-13-3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knobloch TJ, Uhrig LK, Pearl DK, Casto BC, Warner BM, Clinton SK, et al. Suppression of Proinflammatory and Prosurvival Biomarkers in Oral Cancer Patients Consuming a Black Raspberry Phytochemical-Rich Troche. Cancer prevention research (Philadelphia, Pa 2016;9(2):159–71 doi 10.1158/1940-6207.CAPR-15-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casto BC, Kresty LA, Kraly CL, Pearl DK, Knobloch TJ, Schut HA, et al. Chemoprevention of oral cancer by black raspberries. Anticancer Res 2002;22(6c):4005–15. [PubMed] [Google Scholar]
- 24.Oghumu S, Casto BC, Ahn-Jarvis J, Weghorst LC, Maloney J, Geuy P, et al. Inhibition of Pro-inflammatory and Anti-apoptotic Biomarkers during Experimental Oral Cancer Chemoprevention by Dietary Black Raspberries. Front Immunol 2017;8:1325 doi 10.3389/fimmu.2017.01325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peiffer DS, Zimmerman NP, Wang LS, Ransom BW, Carmella SG, Kuo CT, et al. Chemoprevention of esophageal cancer with black raspberries, their component anthocyanins, and a major anthocyanin metabolite, protocatechuic acid. Cancer prevention research (Philadelphia, Pa 2014;7(6):574–84 doi 10.1158/1940-6207.CAPR-14-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka T, Kawamori T, Ohnishi M, Okamoto K, Mori H, Hara A. Chemoprevention of 4-nitroquinoline 1-oxide-induced oral carcinogenesis by dietary protocatechuic acid during initiation and postinitiation phases. Cancer Res 1994;54(9):2359–65. [PubMed] [Google Scholar]
- 27.Zhang SM, Chen KM, Sun YW, Aliaga C, Lin JM, Sharma AK, et al. Simultaneous detection of deoxyadenosine and deoxyguanosine adducts in the tongue and other oral tissues of mice treated with Dibenzo[a,l]pyrene. Chemical research in toxicology 2014;27(7):1199–206 doi 10.1021/tx5001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guttenplan JB, Chen KM, Sun YW, Kosinska W, Zhou Y, Kim SA, et al. Effects of Black Raspberry Extract and Protocatechuic Acid on Carcinogen-DNA Adducts and Mutagenesis, and Oxidative Stress in Rat and Human Oral Cells. Cancer prevention research (Philadelphia, Pa 2016;9(8):704–12 doi 10.1158/1940-6207.CAPR-16-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen KM, Guttenplan JB, Sun YW, Cooper T, Shalaby NAE, Kosinska W, et al. Effects of Black Raspberry on Dibenzo[a,l]Pyrene Diol Epoxide Induced DNA Adducts, Mutagenesis, and Tumorigenesis in the Mouse Oral Cavity. Cancer prevention research (Philadelphia, Pa 2018;11(3):157–64 doi 10.1158/1940-6207.CAPR-17-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nolte T, Brander-Weber P, Dangler C, Deschl U, Elwell MR, Greaves P, et al. Nonproliferative and Proliferative Lesions of the Gastrointestinal Tract, Pancreas and Salivary Glands of the Rat and Mouse. J Toxicol Pathol 2016;29(1 Suppl):1s–125s doi 10.1293/tox.29.1S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mecklenburg L, Kusewitt D, Kolly C, Treumann S, Adams ET, Diegel K, et al. Proliferative and non-proliferative lesions of the rat and mouse integument. J Toxicol Pathol 2013;26(3 Suppl):27s–57s doi 10.1293/tox.26.27S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome biology 2012;13(10):R87 doi 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nature Luch A. and nurture - lessons from chemical carcinogenesis. Nat Rev Cancer 2005;5(2):113–25 doi 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- 34.Loeb LA, Harris CC. Advances in chemical carcinogenesis: a historical review and prospective. Cancer Res 2008;68(17):6863–72 doi 10.1158/0008-5472.can-08-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA. Environmental and chemical carcinogenesis. Semin Cancer Biol 2004;14(6):473–86 doi 10.1016/j.semcancer.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 36.Vitaglione P, Donnarumma G, Napolitano A, Galvano F, Gallo A, Scalfi L, et al. Protocatechuic acid is the major human metabolite of cyanidin-glucosides. The Journal of nutrition 2007;137(9):2043–8. [DOI] [PubMed] [Google Scholar]
- 37.Mori H, Tanaka T, Sugie S, Yoshimi N, Kawamori T, Hirose Y, et al. Chemoprevention by naturally occurring and synthetic agents in oral, liver, and large bowel carcinogenesis. Journal of cellular biochemistry 1997;27:35–41. [PubMed] [Google Scholar]
- 38.Calderon-Montano JM, Burgos-Moron E, Perez-Guerrero C, Lopez-Lazaro M. A review on the dietary flavonoid kaempferol. Mini reviews in medicinal chemistry 2011;11(4):298–344. [DOI] [PubMed] [Google Scholar]
- 39.Guttenplan JB, Chen KM, Sun YW, Lajara B, Shalaby NAE, Kosinska W, et al. Effects of Black Raspberry Extract and Berry Compounds on Repair of DNA Damage and Mutagenesis Induced by Chemical and Physical Agents in Human Oral Leukoplakia and Rat Oral Fibroblasts. Chem Res Toxicol 2017;30(12):2159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen T, Hwang H, Rose ME, Nines RG, Stoner GD. Chemopreventive properties of black raspberries in N-nitrosomethylbenzylamine-induced rat esophageal tumorigenesis: down-regulation of cyclooxygenase-2, inducible nitric oxide synthase, and c-Jun. Cancer Res 2006;66(5):2853–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoner GD, Wang LS, Casto BC. Laboratory and clinical studies of cancer chemoprevention by antioxidants in berries. Carcinogenesis 2008;29(9):1665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012;13(7):484–92 doi 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 43.Ghantous Y, Schussel JL, Brait M. Tobacco and alcohol-induced epigenetic changes in oral carcinoma. Curr Opin Oncol 2018;30(3):152–8 doi 10.1097/cco.0000000000000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teneng I, Montoya-Durango DE, Quertermous JL, Lacy ME, Ramos KS. Reactivation of L1 retrotransposon by benzo(a)pyrene involves complex genetic and epigenetic regulation. Epigenetics 2011;6(3):355–67 doi 10.4161/epi.6.3.14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang LS, Arnold M, Huang YW, Sardo C, Seguin C, Martin E, et al. Modulation of genetic and epigenetic biomarkers of colorectal cancer in humans by black raspberries: a phase I pilot study. Clinical cancer research : an official journal of the American Association for Cancer Research 2011;17(3):598–610 doi 10.1158/1078-0432.CCR-10-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baba S, Yamada Y, Hatano Y, Miyazaki Y, Mori H, Shibata T, et al. Global DNA hypomethylation suppresses squamous carcinogenesis in the tongue and esophagus. Cancer science 2009;100(7):1186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsieh YS, Chu SC, Hsu LS, Chen KS, Lai MT, Yeh CH, et al. Rubus idaeus L. reverses epithelial-to-mesenchymal transition and suppresses cell invasion and protease activities by targeting ERK1/2 and FAK pathways in human lung cancer cells. Food Chem Toxicol 2013;62:908–18 doi 10.1016/j.fct.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 48.Tota JE, Anderson WF, Coffey C, Califano J, Cozen W, Ferris RL, et al. Rising incidence of oral tongue cancer among white men and women in the United States, 1973-2012. Oral Oncol 2017;67:146–52 doi 10.1016/j.oraloncology.2017.02.019. [DOI] [PubMed] [Google Scholar]
- 49.Guttenplan JB, Chen KM, Sun YW, Shalaby NAE, Kosinska W, Desai D, et al. Effects of the Tobacco Carcinogens N’-Nitrosonornicotine and Dibenzo[a,l]pyrene Individually and in Combination on DNA Damage in Human Oral Leukoplakia and on Mutagenicity and Mutation Profiles in lacI Mouse Tongue. Chem Res Toxicol 2019;32(9):1893–9 doi 10.1021/acs.chemrestox.9b00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.