

Abstract
Aiming to identify immune molecules with a novel function in cancer pathogenesis, we found the cluster of differentiation 177 (CD177), a known neutrophil antigen, to be positively correlated with relapse-free (RFS), metastasis-free (MFS) or overall survival (OS) in breast cancer. Additionally, CD177 expression is correlated with good prognosis in several other solid cancers including prostate, cervical, and lung. Focusing on breast cancer, we found that CD177 is expressed in normal breast epithelial cells and is significantly reduced in invasive cancers. Loss of CD177 leads to hyperproliferative mammary epithelium and contributes to breast cancer pathogenesis. Mechanistically, we found that CD177-deficiency is associated with an increase in β-Catenin signaling. Here we identified CD177 as a novel regulator of mammary epithelial proliferation and breast cancer pathogenesis likely via the modulation of Wnt/β-Catenin signaling pathway, a key signaling pathway involved in multiple cancer types.
Introduction
CD177 (also referred to as NB1, HNA-2a, or PRV1) is a glycophosphatidylinositol-linked extracellular membrane bound protein that belongs to the Ly-6 family [1, 2]. CD177 is a glycophosphatidylinositol (GPI)-linked extracellular membrane protein and has been shown to be expressed on the surface and in granules of human neutrophils and serves as a marker for myeloproliferative diseases [3, 4]. To study the physiological role of CD177, we have generated a CD177 knockout (Cd177−/−) mouse model [5]. Cd177−/− mice at 8 weeks of age exhibited slightly lower blood neutrophil, but they appear to have a normal function in clearance of bacterial infection and chemotaxis [5]. The roles of CD177 in solid cancers are largely unknown, though there is a correlation between loss of CD177 expression and poor prognosis in colorectal and gastric cancers [6, 7].
β-Catenin is a member of the Catenin family of proteins mainly involved in two distinct and relevant functions, with one being a structural protein involved in adherens junctions and the other as a WNT-induced transcriptional co-activator where it plays an important role in development and tissue homeostasis [8]. β-Catenin transcriptional co-activator activity is regulated by canonical WNT signaling [9]. Cellular β-Catenin largely exists in three pools, i.e. cytoplasmic, nuclear, or membrane-bound. Cytoplasmic β-Catenin is associated with the “destruction complex” which phosphorylates β-Catenin leading to its proteasomal degradation. Upon binding of a canonical WNT ligand to its receptor, the destruction complex becomes associated with the membrane leading to β-Catenin accumulation and nuclear localization. Membrane β-Catenin forms a complex with E-Cadherin at adherens junctions, together with p120-Catenin and α-Catenin that link to cytoskeleton microfilaments. There is strong evidence showing that membrane β-Catenin can be used by WNT signaling when E-Cadherin or adherens junctions are genetically or chemically destroyed [10–13]. Furthermore, expression of E-Cadherin in otherwise negative cells inhibits WNT/β-Catenin-mediated transcription, thus supporting a role of E-Cadherin/adherens junction in sequestrating β-Catenin to prevent its signaling [11, 14–18].
Dysregulation of β-Catenin plays a role in the development of several types of cancer, including breast cancer [19–22]. WNT/β-Catenin signaling is important in all stages of normal mammary gland development through the regulation of mammary stem cells via a paracrine interaction between luminal epithelial cells and basal epithelial cells [19, 23–29]. Hyper-activation of β-catenin in the mammary gland leads to hyperplasia of mammary epithelial cells and often tumorigenesis when this pathway is dysregulated [30–37].
Here we identified CD177 as a predictor for good prognosis in breast cancer and other types of cancer. We further identified an epithelial-cell intrinsic role of CD177 in tumor suppression that is associated with decreased β-Catenin transcriptional activity.
Materials and Methods
Murine models
All animal experiments were in accordance with University of Iowa IACUC guidelines. NOD-scid IL2Rγnull (NSG) (Jackson laboratory, Bar Harbor, Maine) mice were implanted with subcutaneously with estradiol pills (Innovative Research of America, Sarasota, FL). Three days later, 1×106 MCF-7 CD177-myc or GFP cells in 50% Matrigel (Corning, Corning NY)/PBS were orthotopically transplanted. 2×106 MDA-MB-231 overexpressing CD177-myc or GFP cells in 50% Matrigel (Corning, Corning NY)/PBS were orthotopically transplanted. Balb/C female 6-week old mice were injected with 2.5×105 67NR cells or 67NR cells expressing control or CD177 shRNAs (sh1 or sh2) in 50% Matrigel (Corning). All cells were transplanted into the number four mammary fat pad. For the syngeneic orthotopic transplant tumor model, 6-week old Balb/C female mice were obtained from Charles River Laboratories (Burlington, MA). Tumor growth and development was monitored by external palpations and measured using calipers. Tumor volume was calculated as Length × Width2 × 0.502. MMTV-ErbB2 mice were crossed with CD177−/− mice to generate MMTV-ErbB2/WT, MMTV-ErbB2/CD177+/− and MMTV-ErbB2/CD177−/− littermates. All comparisons between WT and CD177−/− as well as those between MMTV-ErbB2/CD177+/− and MMTV-ErbB2/CD177−/− were made between littermates or aged-related mice. Tumor free survival was determined by recording the date the mice first had palpable tumors two weeks in a row. All mice were terminated when their largest tumor reached 2 cm in diameter or when they reached 600 days old. Tumor multiplicity was determined by counting the number of individual tumors at the time when mice were terminated. With a fixed power of 0.8, for transplant models, we expect a variation of 0.5 standard deviation and a 2-fold change, giving 5–6 per group of animals; for spontaneous models, we expect a variation of 1 standard deviation and a 2-fold change, giving 17–18 per group of animals. Randomization and double-blinded measurements were performed for all transplantation animal experiments.
Survival analysis
Kaplan-Meier survival curves were made using GraphPad Prism software (GraphPad Software Inc., San Diego, CA) from publicly available datasets. RFS based on RNA expression of CD molecules were generated using data from a published meta-dataset of 3554 breast cancer specimens and the KMplot online tools (KMplot.com) [38]. MFS curves were generated using data from GEO Dataset GSE2034[39] (n=286) and GSE2603[40] (n=81). Level 3 Illumina RNAseq data was obtained from the TCGA for 30 cancer types from the cBioPortal [41, 42]. Hazard ratios and P values for survival based on CD177 expression were determined using Cox’s proportional hazards model.
Cell lines, transfection, and transduction
SKBR3, MDA-MB-231, MDA-MB-436, HEK293T and MCF7 cells were obtained from ATCC and maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies, Waltham, MA) with 10% fetal bovine serum (FBS). HMLE cells were provided by Dr. Jing Yang (University of California, San Diego) and maintained in F12 media (Life Technologies) supplanted with 10% FBS, 0.1% insulin, 2 μg/ml hydrocortisone and 10 ng/ml epithelial growth factor. H146, obtained from ATCC, and 67NR, 168FARN and 4TO7 cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 media supplemented with 10% FBS. Human colon epithelial cells were obtained from Dr. Jerry Shay (University of Texas Southwestern) and cultured under DMEM with 10% FBS. Human mammary epithelial cell line (AG11132) was obtained from Coriell Institute for Medical Research (Camden, NJ), cultured using MEGM complete medium (Lonza, Basel, Switzerland). MCF7R cells [43] were from Dr. Marc Lippman at the National Cancer Institute using Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies, Waltham, MA) with 10% fetal bovine serum (FBS). For non-adherent 3-D culture of 67NR and H146 cells, plates were coated with 12 mg poly 2-hydroxyethyl methacrylate (polyHEMA; Sigma Aldrich, St Louis MO)/ml of 95% ethanol and allowed to evaporate. 2×105 cells per ml were plated and cultured for 48 hr. ATCC cells were used within 5–6 generations and other cells were tested for mycoplasma using PlasmoTest-Mycoplasma Detection (InvivoGen, San Diego, CA). For CD177 shRNA, Lentivirus containing shRNA sequences were packaged in HEK293T cells and media containing packaged virus was collected. 67NR cells were incubated with media containing the packaged shRNA lentivirus for 24 hr and stable cells lines expressing the CD177 shRNA were generated by selection of transduced cells with 4 μg/ml puromycin (Thermo Fisher Scientific). The mouse CD177 shRNA’s targeting sequences: Sh1 5’-GCCAAGACTTGATAATGCTCC −3’; Sh2 5’-ACCCAGGCGATTGGGACCTTG-3’ were used to silence CD177 in 67NR cells. For soft agar colony assay, 5×104 cells were suspended in 0.4% agarose/media mixture and plated on top of solidified 0.8% agarose/media mixture. Colonies were cultured for two weeks and counted. For monolayer growth curves, 1×105 cells were plated and counted at 24, 72, and 120 h.
Cell lysates, immunoprecipitation and immunoblots
For membrane and cytosolic fractionation, we followed our previously described protocol [44]. For immunoprecipitation, 1 mg of cell lysate was incubated with 1 μg/mL of antibodies at 4 °C overnight. Immunocomplex was precipitated using protein A or G sepharose beads (Thermo Fisher Scientific). Sepharose beads were resuspended in SDS loading buffer and separated by SDS-PAGE and visualized by Western blotting. For in vitro pull-down assay, 1 μg of FC-fusion CD177 (14501-H02H, SinoBiological, Beijing, China) and His-Tag full-length β-Catenin (11279-H20B, SinoBiological), both purified from HEK293T cells, were incubated using RIPA buffer, with or without the presence of 1 mg of cell lysates from MCF-7 or MDA-MB-231 cells. Ni-NTA agarose was used to pull down His-β-Catenin complex, following with SDS-PAGE and Western Blotting.
Mammary gland whole mount
Mammary glands were removed from CD177−/− and WT mice and fixed in Carnoy’s fix (6 parts ethanol, 3 parts chloroform, and 1-part glacial acetic acid) overnight. They were then rehydrated with ethanol washes, stained with carmine alum stain, cleared, and mounted. Whole mount slides of mammary glands were marked an inch above the inguinal lymph node and all branch points within this inch were counted.
Immunohistochemistry
Tissues were processed with standard IHC protocols. High pH 9 (Vector Labs) was used for antigen retrieval and blocked with background punisher (BioCare Medical, Concord CA). Slides were incubated with primary antibody, anti Ki67 antibody (D2H10; Cell signaling), anti-KRT5 antibody (Poly 19055; Biolegend, San Diego, CA), anti-active β-catenin (D13A1; Cell Signaling), anti-ERα (C-311; Santa Cruz Biotechnology, Dallas, Texas), or anti-PR (D8Q2J; Cell Signaling) for 2 h. Next, rabbit or mouse-on-rodent polymers (BioCare Medical) for 30 min, developed with 3,3’-diaminobenzadine (DAB and 0.3% H2O2 in PBS) developing buffer, and counterstained. Positive cells and total cells per mammary duct were counted.
Immunofluorescence staining and confocal microscopy
Frozen tissue samples or plated cells were fixed for 20 min with 35% methanol/65% acetone on ice. The samples were rehydrated with ice-cold PBS, permeabilized, blocked with 5% goat serum and 0.3% Triton X-100, then incubated with primary antibodies, CD177 (Clone 4C4, Sigma Aldrich), β-Catenin (H-102; Santa Cruz Biotechnology), or E-Cadherin (DECMA-1; Santa Cruz Biotechnology) overnight. The slides were then counter stained with ToPro3 and SlowFade diamond antifade with DAPI (Life Technologies). Images were taken using a Zeiss LSM710 or LSM510 confocal microscope.
Luciferase assay
1×105 cells/well were plated in 24 well plates. After 24 hrs. cells were transfected with pRL-RLuc-Myc (Promega, Madison WI) and M50 Super8x TOPflash (Addgene Plasmid # 12456) [45], plasmids at a 1:16 ratio. MDA-MB-436 were co-transfected with 500ng CD177-myc, E-cadherin or empty pcDNA3 plasmids, alone or together. H146 cells were co-transfected with 25 nM CD177 or Non-targeting siRNAs (L-020431-00-0005, D-001810-10-20; Dharmacon, Lafayette, CO). 24 hrs. after transfection, media was collected and 400 ng/mL recombinant Wnt3a (R & D Systems, Minneapolis MN) or vehicle was added and distributed back to the cells. For 67NR cells, following transfection 1×105 cells were plated in polyHEMA coated 24-well plates for 24 hrs. prior to treatment with WNT3a. Luciferase levels were measured using the Dual-Luciferase Reporter Assay System (Promega) 24 hrs. after treatment. Sequence for CD177 siRNA was as follows: ACACGUUGAUGCUCAUUGA, CAGUUCAGCAUGUGUGGAA, GCAACAACCUCGUUAACUC and GGACCACCAUUAUGACACA.
Flow Cytometry
Cells were suspended in PBS with 2% FBS and incubated with CD177-FITC conjugated antibody (MEM-166; Biolegend) for 30 min. Cells were washed and evaluated by flow cytometry using a BD FACSCanto II (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (FlowJo LLC, Ashland, OR).
Real-time PCR
RNA was isolated as described above and cDNA was generated using Superscript III First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). mRNA expression was quantified by real-time PCR using SYBR premix Ex Taq II (Takara, Mountain View, CA) and analyzed using ViiA7 Real-time PCR System (Thermo Fisher Scientific). The following primers were used: Human CD177 F-CCAGGAGGACTTCTGCAACA R-ACTGGGCAC CTCAAGGATCC, Mouse Cd177 F-CCGGGAGAATATGGAGACAC, R-CGCTGCTG CTCATAGACGTA, Mouse Lgr5 F-TCTTCTAGGAAGCAGAGGCG R-CAACCTCAGC GTCTTCACCT, Mouse Axin2 F-TGCATCTCTCTCTGGAGCTG R-ACTGACCGACGATTC CATGT, Mouse TCF4 F-TCTCCATAGTTCCTGGACGG R-GTGGACATTTCAC TGGCTCA, Mouse Fabp5 F-TGTTGTTGCCATCACACGTA R-CGGCTTTGAGGA GTACATGA, Mouse Snai2 F-GATGTGCCCTCAGGTTTGAT R-GGCTGCTTCAA GGACACATT, Mouse Sfrp1 F-GGGTTTCTTCTTCTTGGGGA R-CATCTCTGTGCAAGCGAGTT, Mouse Il2rg F-GAACCC GAAATGTGTACCGT R-ACAGAGATCGAAGCTGGACG, Mouse Ppia F-CAGTGCTCAGA GCTCGAAAGT R-GTGTTCTTCGACATCACGGC, Mouse Ctnnb1 F-ATGGAGCCGGACAGAAAAGC R-CTTGCCACTCAGGGAAGGA.
CRISPR/Cas9
E-Cadherin protein expression was knocked-down using the LentiCRISPRv2 (Addgene plasmid #52961) CRISPR/Cas9 system[46, 47]. The guide sequence, 5’-ATAGGCTGTCCTTTGTCGA-3’, was ligated into the LentiCRISPRv2 plasmid using Roche Rapid Ligation kit (Basel, Switzerland). The lentiviral particles were packaged in HEK293T cells followed by infection of target cells. Puromycin was used for selection, followed by flow cytometry to sort out E-Cadherin negative population.
RNA-sequencing and genomic analysis.
RNA from tumor samples were isolated with RNeasy Plus Mini Kit (Qiagen, Hilden, Germany). Samples with an RNA Integrity Number score > 8.0 as measured by an Agilent BioAnalyzer 2100 were submitted to the University of Chicago Genomics. Paired-end 75 bp on the Illumina HiSeq 2000 was performed. Gene sets related to WNT/β-Catenin signaling enriched in the CD177-low expressing groups with an FDR < 0.25 were identified and graphed using Prism GraphPad software.
Raw data was aligned using the Usegalaxy web platform and TopHat with the mm10 genome [48–50]. The aligned bam files were further processed according the previously established workflow for cufflinks [51] using quartile and bias corrections. Z-scores were calculated using the resulting gene expression file and a heatmap of β-Catenin regulated genes was generated using Graphpad Prism Software (Graphpad Software Inc). Expression versus methylation analysis utilized mean-centralized level 3 Illumina HiSeq 2000 RNAseq expression data and Infinium HumanMethylation450 beta-values. Data was downloaded from the UCSC Cancer Genome Browser with samples being divided into primary tumor and paired normal tissue samples. Copy number analysis was done using cBioportal with the available cancer datasets [41, 42]. Expression data for Breast cancer (GSE2034), LUSC (TCGA) and COAD/READ (TCGA) were separated into groups with high and low expression of CD177 and Gene Set Enrichment analysis was performed using the Molecular Signature Database (MSigDB)[52, 53].
Statistical methods
Log-rank testing was used to determine significance for all survival-curves. One-way ANOVA with multiple comparisons was used to determine significance for dual-luciferase assays. Significance of tumor growth curves was determined with two-way ANOVA. Significance for all other experiments was determined by Student’s t test assuming equal variance. P values of less than or equal to 0.05 were considered significant.
Results
CD177 predicts RFS and MFS in breast cancer patients and other solid tumors
Using a published meta-dataset of 3554 specimens [38], we generated a list of CD molecules whose RNA levels were correlated with recurrence free survival (RFS) in breast cancer (Table S1). CD177 had one of the highest correlations between expression and RFS, higher than many other makers for immune cells with known anti-cancer functions, including those for NK cells (CD314/NKG2d, CD94), toxic T cells (CD8), macrophages (CD163), etc. (Supplementary Table 1, Figure 1A hazard ratio (HR) = 0.62). CD177 is also positively correlated with metastasis-free survival (MFS), in two well-established datasets, GSE2034 [39] (n = 286, Figure 1B, Figure S1A) and GSE2603 [40] (n = 81, Figure S1B), irrelative of bone or lung metastasis (Figure S1A–B). The positive correlation between CD177 and RFS was true among all breast cancer subtypes (Figure 1C and Supplementary Table 2).
Figure 1.
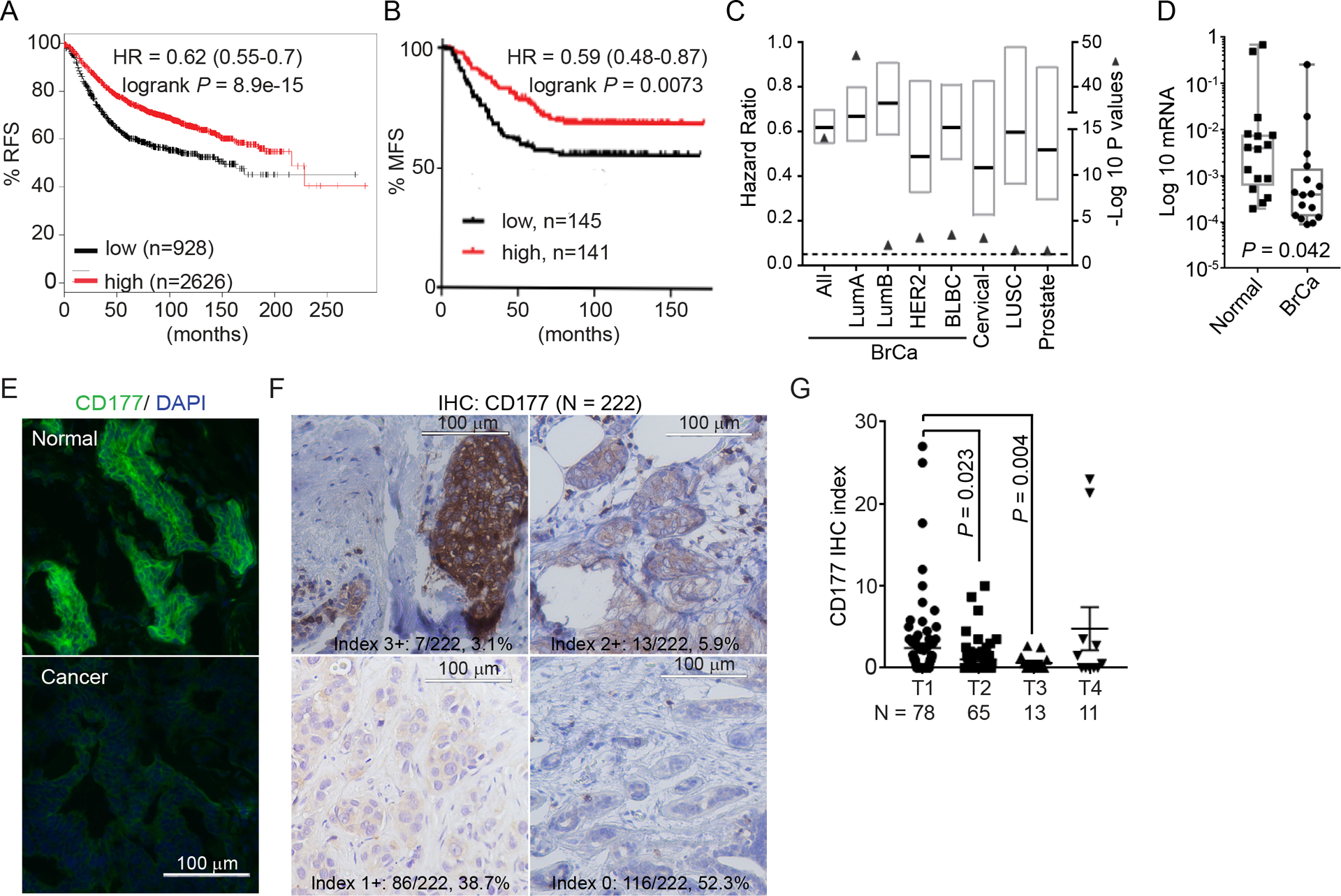
CD177 is commonly lost in invasive cancer and positively correlated with survival.
(A) Correlation between CD177 expression and relapse-free survival (RFS) in all breast cancer samples using an online KMPlot software including a total of 3554 breast cancer specimens.
(B) Correlation between CD177 expression and metastasis-free survival (MFS) in breast cancer among patient populations included in GSE2034 dataset (n=286). Logrank test was done to determine significance.
(C) Correlation between CD177 expression and RFS (breast cancer, BrCa; Lung squamous cell carcinoma, LUSC and prostate cancer), and overall survival (OS, Cervical cancer). BrCa data was generated using KMPlot software with samples separated by intrinsic subtypes (Luminal A, LumA; Luminal B, LumB; HER2+; basal-like breast cancer, BLBC). Cervical, LUSC and prostate data are from TCGA Illumina HiSeq level 3 data. Graph represents the hazard ratio (black bar) and range (box) with the −Log 10 Logrank P value (closed triangle). Dashed line is −Log10(0.05). N numbers for each group is presented in Table S2.
(D) CD177 log 10 mRNA level in breast cancer and paired normal tissue. n = 16, paired t test.
(E) Representative immunofluorescent staining for CD177 in human breast cancer specimen and paired normal tissue (n = 6 pairs, also Figure S2A).
(F) Representative IHC staining for CD177 of a tissue microarray of 222 human breast cancer samples. Samples were scored using index of 0–3+ bases on the intensity of CD177 staining and percentage of positivity. The intensity scores and number/percentages are indicated.
(G) CD177 IHC index for samples from F separated by tumor stage. IHC index was calculated by the following formula: Intensity index × Percentage × 10. One-way ANOVA was used to determine significance with adjusted P values indicated. Only samples with traceable tumor stage information were shown (n = 167).
Also see Supplementary Table S1–S3 and Figure S1–2.
Using Analysis of Illumina HiSeq expression data deposited in The Cancer Genome Atlas (TCGA) for 30 cancer types, we next checked if CD177 expression was correlated with survival in other types as cancer. We found that CD177 expression is positively correlated with survival in 8 carcinomas (HR < 0.7 and Logrank P < 0.05; Figure S1C and Supplementary Table 3), such as overall survival (OS) in cervical squamous cell carcinoma (HR = 0.44), RFS in lung squamous cell carcinoma (LUSC, HR = 0.60), and RFS in prostate cancer (HR = 0.5) (Figure 1C and Supplementary Table 2).
CD177 is expressed in normal mammary epithelial cells but is reduced in breast cancer
Since CD177 is a positive predictor for breast cancer we determined whether CD177 is expressed on cells beyond neutrophils. Immunohistochemistry (IHC) images from Human Protein Atlas [54] reveled that CD177 is expressed on epithelial cells in cervical cancer, prostate cancer and LUSC (Figure S1D–F). In breast cancer, CD177 mRNA expression was reduced compared to normal tissues of 16-paired human breast cancer patients (Figure 1D). Immunofluorescent staining revealed that CD177 was strongly expressed in the normal mammary epithelial cells but reduced in all paired cancer specimens (Figure 1E; Figure S2A). IHC staining for CD177, using a different monoclonal antibody (clone 4c4), of a tissue microarray (TMA) found CD177 staining in cancer cells, with lower levels of CD177 staining correlated with increased tumor stages (Figure 1F–G).
Most breast cancer cell lines had significantly decreased CD177 mRNA levels compared to non-fully transformed HMLE mammary epithelial cells (Figure S2B). These cells had nearly undetectable expression of CD177 by Western blot or surface expression by flow cytometry under monolayer culture. However, when mouse mammary breast cancer 67NR, prostate cancer PC3, lung cancer H146 and a tamoxifen resistant MCF7 (MCF7-R) cell lines were grown in non-adherent conditions, CD177 protein was found on the cell surface by flow cytometry (Figure S2C–E).
To determine if CD177 locus is commonly mutated, deleted or has promoter methylation, we utilized cBioPortal dataset [41, 42] and found no recurrent mutations or chromosomal deletions related to CD177 locus. We also mined TCGA datasets for invasive breast cancer and found no significant correlation between promoter methylation and CD177 expression in either breast cancer samples or normal breast tissues, suggesting that CD177 expression in breast cancer may not be regulated by promoter methylation (Figure S2F).
CD177-deficiency induces hyperproliferation of mammary epithelial cells
As we observed a common loss of CD177 in human breast cancer, we sought to further study the physiological function of CD177 in the mammary gland to better understand the role of CD177 in mammary epithelial cells. We examined the physiological function of CD177 in the mammary gland using Cd177−/− mice [5]. At 7-weeks there was no discernable difference in mammary glands of knockout mice compared to wild-type littermates (Figure 2A). However, mammary glands from 10-months old Cd177−/− mice exhibited significantly increased side branching and ductal structures (Figure 2A–B). Hematoxylin and Eosin (H&E) staining revealed many mammary duct structures with loss of luminal architecture and multiple layers of epithelial cells (Figure S3A, black arrows). This phenotype was not found in the wildtype (WT) littermates (Figure S3A, Figure 2C). The mammary epithelium from the Cd177−/− mice had increased proliferation indicated by Ki-67 IHC (Figure 2D, Figure S3B, upper panels). Cd177−/− mice exhibited similar nuclear progesterone receptor (PR, Figure 2E, Figure S3C, left) and slightly decreased nuclear estrogen receptor (ER, Figure 2F, Figure S3C, right), suggesting the hormone-independent proliferative phenotype within Cd177−/− mammary epithelium. This suggests that CD177-deletion promotes the expansion of mammary epithelial cells, a prerequisite for breast cancer development.
Figure 2.
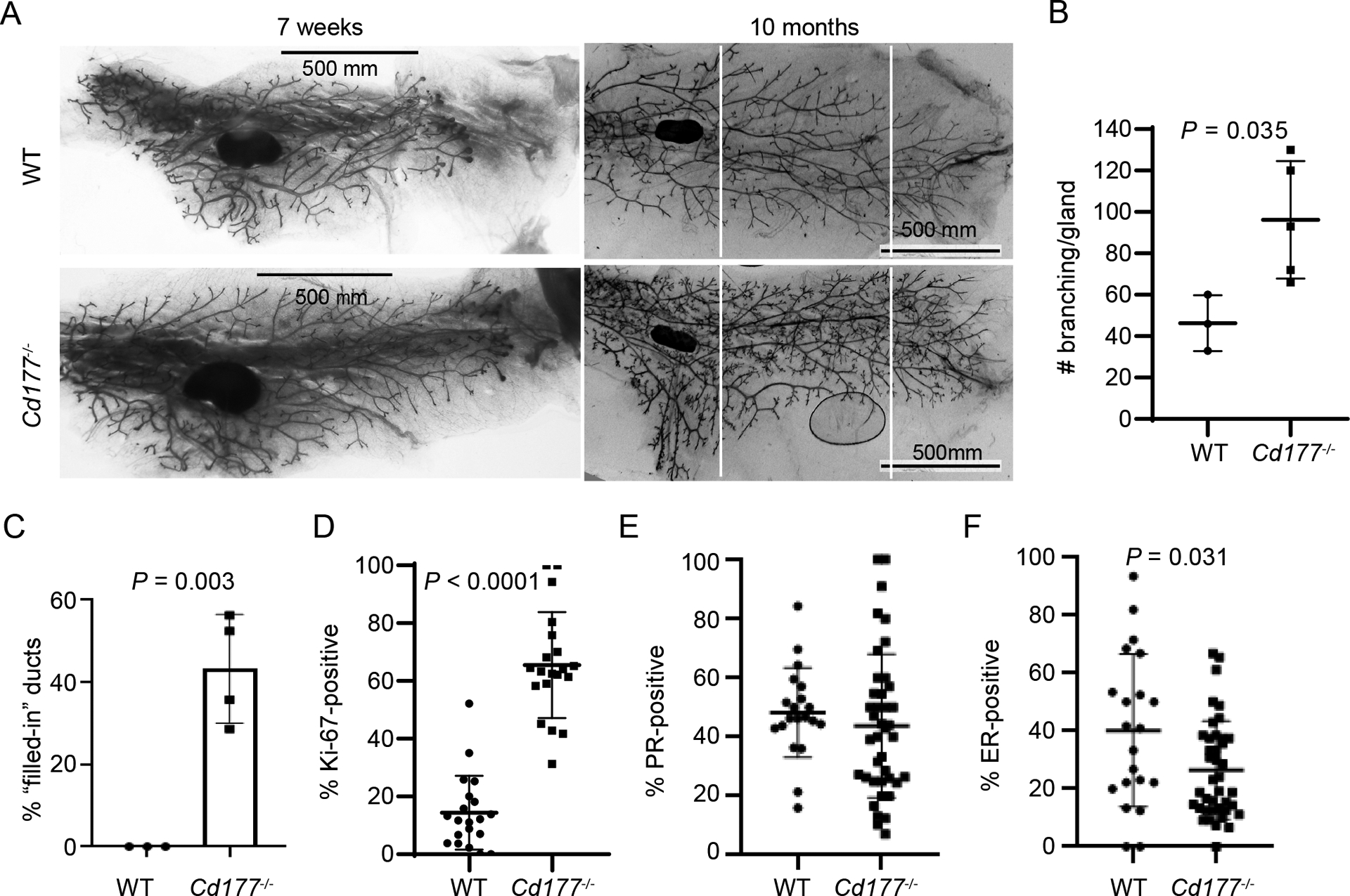
CD177-deficiency leads to proliferative mammary epithelial cells
(A) The No. 4 mammary glands from the indicated mice were collected at 7 weeks (left panels) and 10 months (right panels) of age. The number of branches between the white lines was counted.
(B) Graph depicts the number of branches from (A) from the indicated mice. Significance was determined with by Student’s t test (n=3 WT, n=5 CD177−/−).
(C) Number of filled ducts was counted from age-matched mice at 10 month (n=3 WT, n=5 CD177−/−), based on H&E images shown in Figure S3A.
(D) Percentages of Ki-67 positive cells was calculated from mammary epithelial cells of age-matched mice at 10 month (n=3 WT, n=5 CD177−/−, 5–10 ducts counted per gland), based on IHC shown in Figure S3B.
(E-F) Percentages of PR-(E) or ER-(F) positive cells was calculated from mammary epithelial cells of age-matched mice at 10 month (n=2 WT, n=4 CD177−/−, 10 ducts counted per gland), based on IHC shown in Figure S3C.
Also see Figure S3.
Normal mammary ducts from both Cd177−/− and WT mice contained a single layer of keratin (Krt) 5-positive basal cells and Krt-8-postive luminal cells, whereas the filled mammary ducts from Cd177−/− mice had multiple layers of basal and luminal cells (Figure S3B, lower panels). We observed no difference in the percentage of Krt-5- or Krt-8-positive cells in mammary glands from Cd177−/− mice compared to WT littermates (Figure S3D–E). These data indicate that CD177-deficiency leads to an increase in both luminal and basal epithelial cells while maintaining their ratio.
CD177 is a critical regulator for mammary tumorigenesis
To study the role of CD177 in breast cancer, we stably overexpressed human CD177 in human breast cancer cell lines, including MCF7 (luminal, Figure 3A) and SKBR3 (HER2+, Figure 3B). Overexpression of CD177 in MCF7 and SKBR3 cells reduced monolayer growth (Figure 3A–B) and soft agar colony formation (Figure 3C–D). In concordance, silencing Cd177 in 67NR cells by shRNA (Figure 3E, Figure S4A) resulted in an increase in the number of soft agar colonies, which was reversed when CD177 was reconstituted (Figure S4B, Figure 3F).
Figure 3.
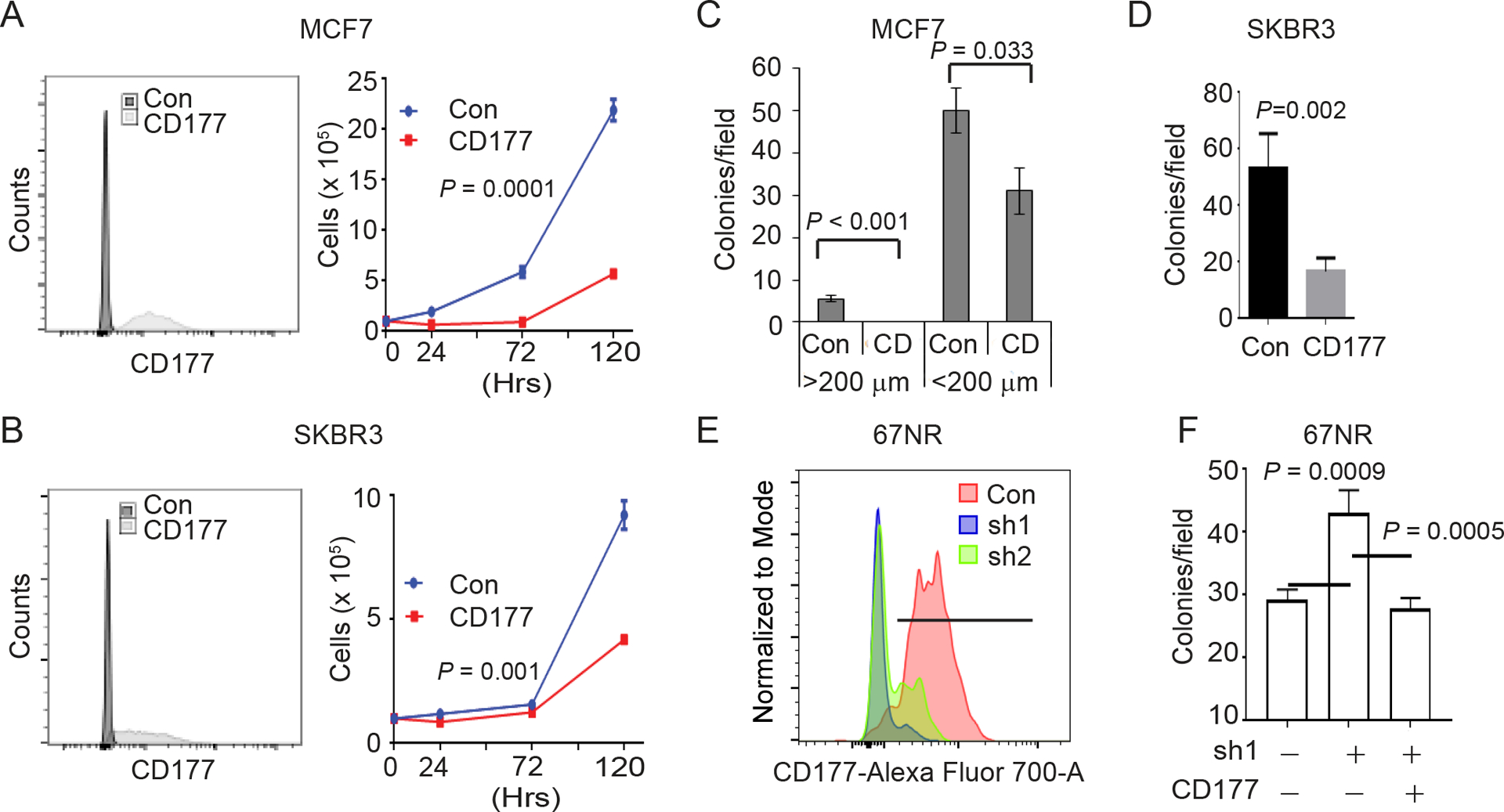
CD177 suppresses breast cancer cell growth and colony formation
(A-B) The indicated cells stably expressing CD177 or GFP (con) vector were purified by surface CD177 staining and flow cytometry. Expression of surface CD177 was determined by flow cytometry (left panels) and monolayer cell growth curves were shown (right panels). Two-way ANOVA was done to determine significance (n=3).
(C-D) 3-dimensional soft agar assay for MCF7 (C) and SKBR3 (D) expressing CD177 (CD) or GFP (Con). Graphs depict the average number of colonies per microscopy field ± s.d. At least 3 fields per sample were counted. Students t test was done to determine significance (n=3). 200 μm in (C) defines the colony size.
(E) 67NR cells were selected for stable expression of CD177 shRNA following lentivirus transduction from two independent shRNAs. Graph depicts the surface CD177 expression determined by flow cytometry.
(F) 3-dimensional soft agar assay for 67NR control of sh1 cells from (E) transfected with human CD177 as indicated. Graphs depict the average number of colonies per microscopy field ± s.d. At least 3 fields per sample were counted. One-way ANOVA was used to determine significance.
Also see Figure S4.
To confirm the role of CD177 as a cancer cell-intrinsic tumor suppressor, we orthotopically injected CD177-overexpressing MCF7 or MDA-MB-231 cells into immunodeficient NOD-scid IL2Rγnull (NSG) mice. Tumor growth was significantly suppressed relative to control tumors for both cell lines (Figure 4A–B). Likewise, silencing Cd177 in 67NR cells (Figure 3E, Figure S4A) resulted in the increased tumor growth compared to 67NR control cells (Figure 4C).
Figure 4.
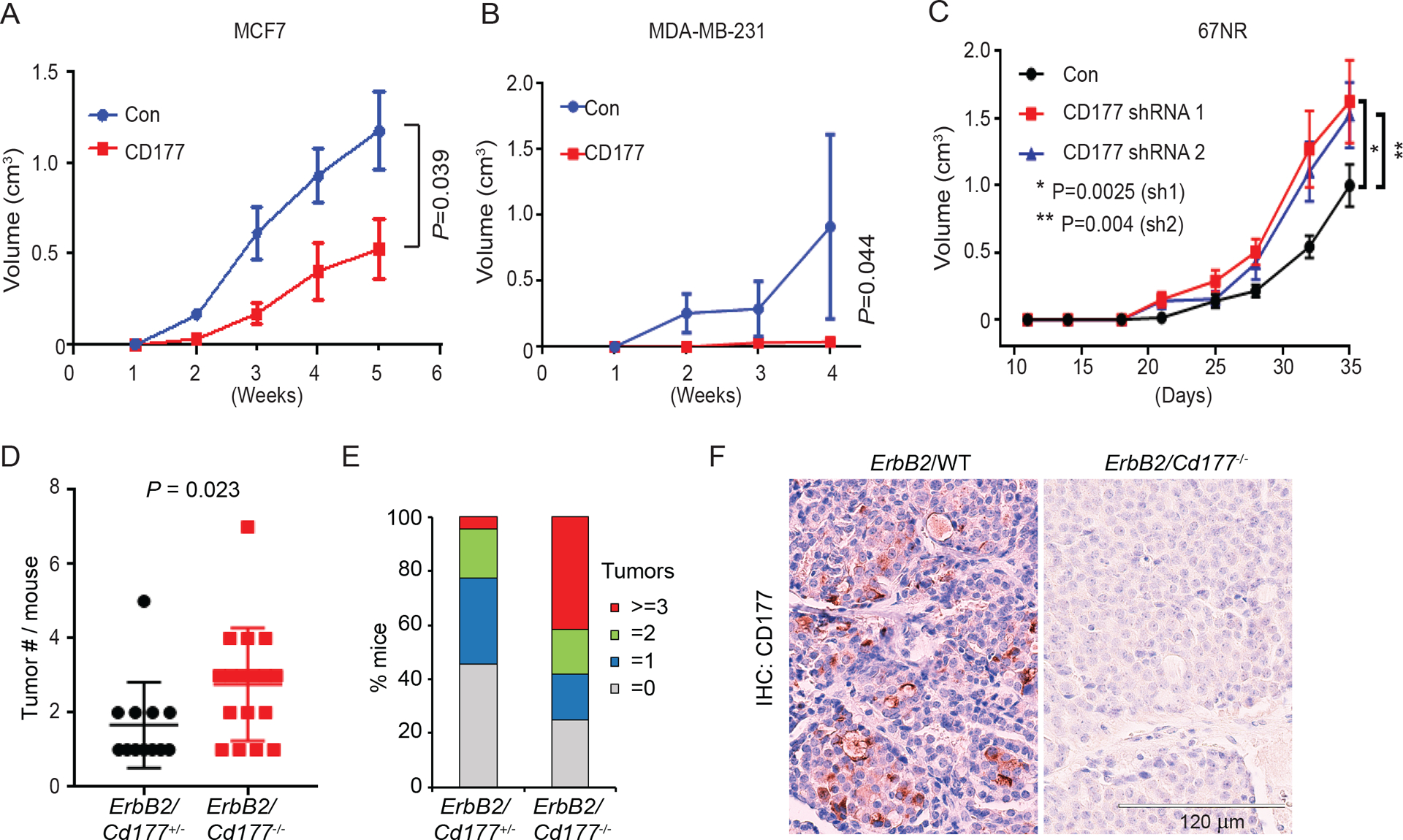
CD177 suppresses tumorigenesis in a cancer cell-intrinsic manner.
(A-B) MCF-7 (A) or MDA-MB-231 (B) cells stably expressing CD177 or control vector (con) were implanted into the number 4 mammary glands of NSG mice and tumor volume was monitored. Graph depicts the average tumor volume ± s.e.m. Two-way ANOVA was used to determine significance (n=4–6).
(C) 67NR cells stably expressing CD177 shRNA1, shRNA2 or control 67NR cells from Figure 3E were implanted into the # 4 mammary fatpad of Balb/c mice and tumor volume was monitored. Graph depicts the average tumor volume ± s.e.m. Two-way ANOVA was used to determine significance.
(D-E) MMTV-ErbB2/Cd177+/− and MMTV-ErbB2/Cd177−/− mice were monitored for tumor development. Mice were euthanized when the largest tumor reached 2cm and the number of tumors per mice was counted. Graph depicts the number of tumors per mouse (D, only tumor-bearing mice were shown) or the percent of mice with the indicated number or tumors (E, all mice were shown) from MMTV-ErbB2/Cd177+/− and MMTV-ErbB2/Cd177−/−.
(F) Representative IHC images for CD177 in tumors from MMTV-ErbB2/WT and MMTV-ErbB2/Cd177−/− mice.
Also see Figure S4.
We also examined the role of CD177 in an oncogene-induced mammary tumor model. We crossed Cd177−/− mice [5] with MMTV-ErbB2-transgenic (tg) mice to produce MMTV-ErbB2/Cd177+/− or MMTV-ErbB2/Cd177−/− littermate progeny. The MMTV-ErbB2/Cd177−/− mice did not shown difference in first tumor onset (Figure S4C), but had increased tumor multiplicity compared to MMTV-ErbB2/Cd177+/− littermates (Figure 4D, P = 0.023); 41% of the MMTV-ErbB2/Cd177−/− mice but only 4.5% of the MMTV-ErbB2/Cd177+/− mice had more than 3 tumors (Figure 4E). Surface expression of CD177 was verified in a small percentage of cancer cells in MMTV-ErbB2/WT mice, indicative of reduced CD177 expression in cancer (Figure S4D, Figure 4F). As expected, no CD177 was observed in tumors from MMTV-ErbB2/Cd177−/− mice (Figure 4F). Unlike the cell line data (Figure 3A–B) or within mammary glands (Figure 2), there was no significant increase in Ki-67-positive cells in these tumors (Figure S4E–F), suggesting that tumors evade CD177-mediated suppression of proliferation at late stages likely by downregulation of surface CD177 (Figure 4F).
CD177 expression is associated with decreased β-Catenin activity
Since nothing is known related to CD177-mediated signaling transduction in cancer, we performed datamining of the TCGA invasive breast cancer (BrCa) dataset. We correlated CD177 expression with known biological pathways using gene set enrichment analysis (GSEA) [52], comparing BrCa samples with low versus high CD177 expression. We found that the low expression of CD177 was correlated with the enrichment of WNT and β-Catenin signaling (Figure 5A). Similar results were found in several other major cancer subtypes including the TCGA lung squamous cell carcinoma (LUSC), colon adenocarcinoma (COAD) and rectal adenocarcinoma (READ) (Figure 5B), where CD177 expression was observed either in corresponding cancer specimens (Figure S1F) or known in literature [6]. Next generation RNA sequencing also revealed that Cd177-deficiency led to decreased expression of WNT/β-Catenin-targeted genes within ErbB2-induced tumors from Figure 4D–F (Supplementary Table 4, Figure 5C) and further validated by real-time PCR (Figure 5D, Figure S5A). We have shown before that human breast cancers rarely have nuclear β-Catenin [55], in agreement with previous publications using clinical approved IHC protocols [37, 56]. Using an antibody that specifically recognizes active β-Catenin, we did not find significant nuclear β-Catenin staining on tissue sections of these ErbB2-induced tumors, either within Cd177+/− or Cd177-KO genotypes. Staining of normal glands from Figure 2A, we found that Cd177-deficiency led to a significant increase in nuclear β-Catenin staining of mammary glands of 10-month old mice, preferably within the basal layer of mammary epithelium (Figure 5E–F), in agreement with the critical role of β-Catenin in basal mammary stem cells.
Figure 5.
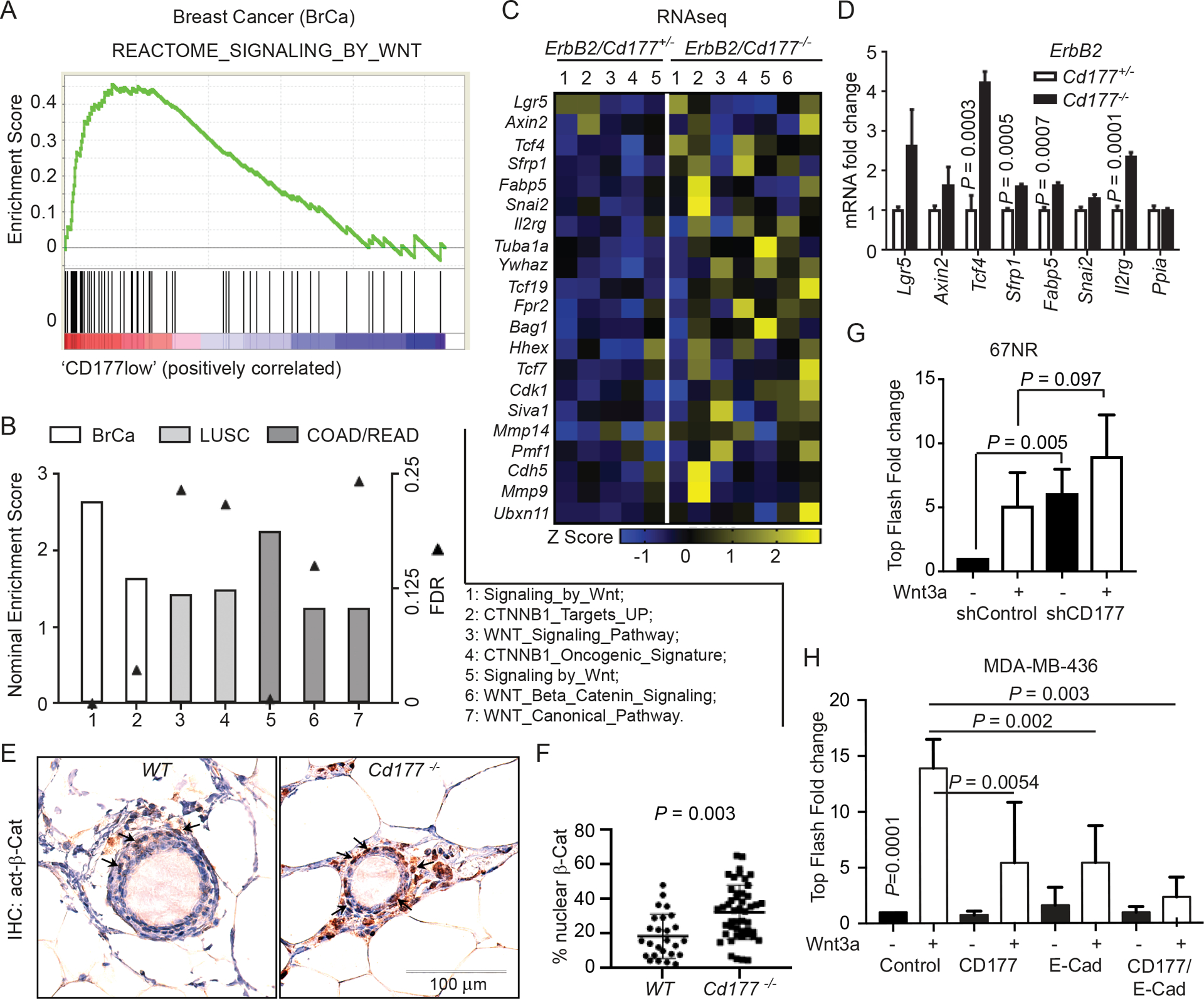
CD177 attenuates the canonical WNT/β-Catenin signaling
(A-B) GSEA showing activation of WNT/β-Catenin pathways in CD177low cohorts of human cancers, including human breast cancer samples (BrCa; GSE2034, 19 CD177low versus 19 CD177high cancer specimens), lung squamous cell carcinoma (LUSC; TCGA), colon adenocarcinoma (COAD; TCGA) and rectal adenocarcinoma (READ; TCGA). Cancer specimens were separated into high versus low CD177 expressing groups (BrCA: low 19 vs high 19; LUSC: low 98 vs high 98; COAD: low 33 vs high 33; READ: low 15 vs high 15). Graph depicts the net enrichment score (bar) and false discovery rate (FDR, solid triangle) for the indicated gene sets in the CD177 low groups.
(C) RNAseq was performed using tumor RNAs collected from MMTV-ErbB2/Cd177+/− and MMTV-ErbB2/Cd177−/− mice used in Figure 4D (5 and 7, respectively, Table S4). Heatmap depicting Z-scores for the expression of β-Catenin regulated genes, defined by GSEA WNT/β-Catenin regulated gene signatures in cancer cells, including a combined genes from the following GSEA genesets: Go_Canonical_Wnt_Signaling_Pathway; Go_Wnt_Signaling_Pathway; Biocarta_Wnt_Pathway.
(D) Real-time PCR verifying the mRNA expression of several genes in tumors from the indicated mice. Graph depicts the average mRNA expression relative to Ppia as a fold change compared to WT group ± s.d. Student’s t test was done to determine significance (n=3 from different tumors).
(E-F) Representative IHC images (E) and percentages of active nuclear β-Catenin (F)-positive cells were calculated from mammary epithelial cells of age-matched mice at 10 month (n=2 WT, n=3 CD177−/−, 12–17 ducts counted per gland).
(G) TOPflash dual-luciferase assay for 67NR cells stably expressing CD177 shRNA (Figure 3E) grown as 3-dimensional colonies in poly-HEMA coated plates. Cells were then treated with or without 400 ng/mL recombinant Wnt3a for 24 hr. Graph depicts the average firefly luciferase intensity as a fold change compared to untreated control ± s.d. One-way ANOVA was used to determine significance. n=6 for all experiments.
(H) TOPflash dual-luciferase assay for β-Catenin activation in MDA-MB-436 cells transfected with pcDNA3.1 empty vector (control), CD177 and pcDNA3.1, E-Cadherin and pcDNA3.1 or both E-cadherin and CD177, treated with or without 400 ng/mL recombinant Wnt3a for 24 hr. Graph depicts the average firefly luciferase intensity as a fold change compared to untreated control ± s.d. One-way ANOVA was used to determine significance (n=6 for all experiments). (G-H), black bars, no treatment; white bars, 400ng/ml of Wnt3A treatment.
Also see Figure S5.
We also determined if CD177 regulates the β-Catenin-mediated transcription using a dual-luciferase TOPflash assay [45]. We found that knockdown of Cd177 in 67NR cells increased basal level of β-Catenin transcriptional activity with slight but not significant increase with Wnt3a treatment (Figure 5G). In H146 cells with intrinsic CD177 expression (Figure S2C, S2E), silencing CD177 by siRNA (Figure S5B) significantly increased both basal and Wnt3a-induced β-Catenin transcriptional activity compared to control cells (Figure S5B). Further supporting the sufficiency of CD177 in attenuation of β-Catenin activity, overexpression of CD177 in the CD177− MDA-MB-436 cells significantly reduced Wnt3a-mediated β-Catenin activity (first two open bars in Figure 5H, S5C). As we have seen silencing CD177 in 67NR cells resulted in the increased in vitro (Figure 3F) and in vivo (Figure 4C) tumor growth, we further silenced β-Catenin in 67NR sh1 cells to determine the causative role of β-catenin downstream of CD177-deficiency. We achieved > 90% reduction of β-catenin protein level in 67NR sh1 cells (Figure S5D, right panels), which led to significantly reduced colony growth from the 67RN sh1 cells (Figure S5D, left panels).
CD177 indirectly interacts with β-Catenin
To gain mechanistic understanding how CD177, a GPI-linked extramembrane protein without a transmembrane domain, regulates intracellular β-Catenin activity, we further characterized the molecular nature of CD177 on cancer cells. CD177 was found to be expressed on the membrane (Figure S2C by surface flow cytometry, Figure S6A by immunofluorescence staining), indicative of physiological membrane localization. Using con-focal microscope, we further found that both CD177 (Figure 6A: green) and β-Catenin (Figure 6B: red) were localized at sites of cell-cell contact (Figure 6A–C) within normal human mammary glands; whereas control IgGs did not yield any fluorescence signal (Figure S6B). The two signals were mostly in parallel with each other (Figure 6D, white arrows), indicating the two are likely at the opposite sides of the plasma membrane. Since extracellular CD177 does not have a transmembrane domain and β-Catenin is an intracellular protein, we reasoned the transmembrane E-Cadherin may mediate the interaction since both E-Cadherin and β-Catenin belong to the protein complex involving adherens junctions. We identified co-localization between CD177 and E-Cadherin along the plasma membrane of normal human mammary ducts (Figure S6C). We further combined FC-fusion CD177 recombinant protein with His-Tag full-length β-Catenin, both purified from HEK293T cells, and did not detect any interaction by pull down assays (Figure 6E and Figure S6D, first two lanes). Co-incubation with cell lysates from MCF-7 (E-Cadherin positive), MDA-MB-231 (E-Cadherin negative) (Figure 6E), or HEK293T (Figure S6D) induced the interaction between CD177 and β-Catenin. We further used an anti-E-cadherin antibody to deplete E-Cadherin protein from HEK293T lysate, using an IgG or non-relevant anti-transmembrane protein DSG2 as negative controls (Figure S6E). Depletion of E-Cadherin did not influence the interaction between CD177 and β-Catenin (Figure S6D). We also depleted E-Cadherin by siRNA with a 90% silencing efficiency in MCF7-R cells (Figure 6F) without altering the levels of endogenous CD177 and β-Catenin (Figure 6F). Same lysates were subject to co-immunoprecipitation (co-IP) where anti-β-Catenin pulled down both β-Catenin and CD177 regardless of E-Cadherin expression (Figure 6F, right panels). The reciprocal co-IP of CD177 showed similar results (Figure 6F). CD177-dependent suppression of β-Catenin activity was independent of E-Cadherin in an E-Cadherin-negative MA-MB-436 cells (Figure 5H). These data indicate that CD177 forms a complex with β-Catenin indirectly along the plasma membrane.
Figure 6.

CD177 indirectly interacts with β-Catenin
(A-D) Normal human breast tissues were stained with anti-CD177 (A: green, clone 4c4) and anti-β-Catenin (B: red). Representative confocal images are shown as individual colors and an overlay (C) with nuclear DAPI staining (blue). (D) Enlarged merged image from (C) with white arrows indicating areas where the two signals are parallel.
(E) 1 μg of FC-fusion CD177 and His-Tag full-length β-Catenin, both purified from HEK293T cells, were incubated using RIPA buffer, with or without the presence of 1 mg of cell lysates from MCF-7 or MDA-MB-231 cells. Ni-NTA agarose was used to pull down His-β-Catenin complex, following with SDS-PAGE and Western Blotting. Also see Figure S6D (n=2).
(F) Immunoprecipitation of β-Catenin or CD177 from MCF7-R cells transfected with E-Cadherin siRNA. IP using non-immunogenic IgG was used as a control. Immunoblotting of whole cell lysates (WCL, left) and IP (right) for E-Cadherin, β-Catenin, CD177, β-Actin, and/or IgG heavy chain (IgH) are shown (n=2).
Also see Figure S6.
Discussion
We identified a novel function of CD177 as a tumor-suppressive protein and biomarker for predicting patient outcomes in breast and other types of cancer. The expression of CD177 is decreased in paired invasive cancers as well as more advanced tumors, which agrees with the general loss of tumor suppressors during cancer development. CD177 expression also predicts a good patient outcome in breast cancer and other cancer types. While the loss of CD177 alone was insufficient to induce tumorigenesis, it did induce hyperproliferation of mammary epithelial cells. Mammary ducts from 10-month Cd177−/− mice exhibited multiple layers of epithelial cells with some filled ducts, a phenotype observed in the early stages of tumorigenesis and puberty. This resembles the activation of β-Catenin in the mammary gland that leads to hyperproliferation of the mammary epithelial cells and adenocarcinoma [30, 31]. Nuclear β-Catenin is also elevated within the mammary epithelial cells from 10-month Cd177−/− mice relative to WT mice.
We also found that CD177 forms a complex with β-Catenin, suppressing WNT-induced β-Catenin transcriptional activity. Though constitutive activation of WNT signaling leads to adenocarcinoma in aged mice, Cd177−/− mice did not develop spontaneous carcinomas. β-Catenin is a known tumor promotor in several other types of cancer where CD177 is a good prognosis marker such as cervical [57–59], prostate [60, 61] and LUSC [62, 63]. CD177 may act as a tumor suppressor in these cancers by attenuating β-Catenin signaling similar to breast cancers.
We have made a long-term commitment to determine a transmembrane protein that potentially mediates the interaction between extracellular CD177 and intracellular β-Catenin. The apparent E-Cadherin fails to fulfill this function. Several attempts using mass spectrometry also failed to identify a membrane protein that bridges CD177 and β-Catenin; whereas the search is ongoing, it is important to add CD177 as a negative regulator of β-Catenin activity. In summary, our results support a model where CD177 indirectly interacts with β-Catenin to the plasma membrane independently of E-Cadherin, preventing β-Catenin activation. This is in agreement with bioinformatics analysis where CD177 is a good prognostic factor in various cancer types.
Supplementary Material
Acknowledgements
WZ was supported by NIH grants CA200673 and CA203834, the V Scholar award, American Cancer Society seed grant, Breast Cancer Research Award and Oberley Award (National Cancer Institute Award P30CA086862) from Holden Comprehensive Cancer Center at the University of Iowa, University of Iowa. N.B. was supported by NIH CA206255. Q.X. was supported by the National Natural Science Foundation of China (No. 81502313). We thank Dr. Foekens for sharing the site-specific metastasis information related to GSE2034. We thank Dr. Shay for sharing the human colon epithelial cells.
Footnotes
Competing interests:
All authors declare no conflict of interest.
References
- 1.Temerinac S, Klippel S, Strunck E, Roder S, Lubbert M, Lange W et al. Cloning of PRV-1, a novel member of the uPAR receptor superfamily, which is overexpressed in polycythemia rubra vera. Blood 2000; 95: 2569–2576. [PubMed] [Google Scholar]
- 2.Kissel K, Santoso S, Hofmann C, Stroncek D, Bux J. Molecular basis of the neutrophil glycoprotein NB1 (CD177) involved in the pathogenesis of immune neutropenias and transfusion reactions. European journal of immunology 2001; 31: 1301–1309. [DOI] [PubMed] [Google Scholar]
- 3.Caruccio L, Bettinotti M, Director-Myska AE, Arthur DC, Stroncek D. The gene overexpressed in polycythemia rubra vera, PRV-1, and the gene encoding a neutrophil alloantigen, NB1, are alleles of a single gene, CD177, in chromosome band 19q13.31. Transfusion 2006; 46: 441–447. [DOI] [PubMed] [Google Scholar]
- 4.Passamonti F, Pietra D, Malabarba L, Rumi E, Della Porta MG, Malcovati L et al. Clinical significance of neutrophil CD177 mRNA expression in Ph-negative chronic myeloproliferative disorders. Br J Haematol 2004; 126: 650–656. [DOI] [PubMed] [Google Scholar]
- 5.Xie Q, Klesney-Tait J, Keck K, Parlet C, Borcherding N, Kolb R et al. Characterization of a novel mouse model with genetic deletion of CD177. Protein Cell 2015; 6: 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol 2011; 29: 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toyoda T, Tsukamoto T, Yamamoto M, Ban H, Saito N, Takasu S et al. Gene expression analysis of a Helicobacter pylori-infected and high-salt diet-treated mouse gastric tumor model: identification of CD177 as a novel prognostic factor in patients with gastric cancer. BMC gastroenterology 2013; 13: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17: 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell 2012; 149: 1192–1205. [DOI] [PubMed] [Google Scholar]
- 10.Eger A, Stockinger A, Schaffhauser B, Beug H, Foisner R. Epithelial mesenchymal transition by c-Fos estrogen receptor activation involves nuclear translocation of beta-catenin and upregulation of beta-catenin/lymphoid enhancer binding factor-1 transcriptional activity. J Cell Biol 2000; 148: 173–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci 1999; 112 (Pt 8): 1237–1245. [DOI] [PubMed] [Google Scholar]
- 12.Kuphal F, Behrens J. E-cadherin modulates Wnt-dependent transcription in colorectal cancer cells but does not alter Wnt-independent gene expression in fibroblasts. Exp Cell Res 2006; 312: 457–467. [DOI] [PubMed] [Google Scholar]
- 13.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 2008; 68: 3645–3654. [DOI] [PubMed] [Google Scholar]
- 14.Stockinger A, Eger A, Wolf J, Beug H, Foisner R. E-cadherin regulates cell growth by modulating proliferation-dependent beta-catenin transcriptional activity. J Cell Biol 2001; 154: 1185–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanson B, White P, Vincent JP. Uncoupling cadherin-based adhesion from wingless signalling in Drosophila. Nature 1996; 383: 627–630. [DOI] [PubMed] [Google Scholar]
- 16.Heasman J, Ginsberg D, Geiger B, Goldstone K, Pratt T, Yoshida-Noro C et al. A functional test for maternally inherited cadherin in Xenopus shows its importance in cell adhesion at the blastula stage. Development 1994; 120: 49–57. [DOI] [PubMed] [Google Scholar]
- 17.Gottardi CJ, Wong E, Gumbiner BM. E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol 2001; 153: 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fagotto F, Funayama N, Gluck U, Gumbiner BM. Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J Cell Biol 1996; 132: 1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Incassati A, Chandramouli A, Eelkema R, Cowin P. Key signaling nodes in mammary gland development and cancer: beta-catenin. Breast Cancer Res 2010; 12: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polakis P The many ways of Wnt in cancer. Curr Opin Genet Dev 2007; 17: 45–51. [DOI] [PubMed] [Google Scholar]
- 21.Koesters R, Ridder R, Kopp-Schneider A, Betts D, Adams V, Niggli F et al. Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms’ tumors. Cancer Res 1999; 59: 3880–3882. [PubMed] [Google Scholar]
- 22.Chiurillo MA. Role of the Wnt/beta-catenin pathway in gastric cancer: An in-depth literature review. World J Exp Med 2015; 5: 84–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badders NM, Goel S, Clark RJ, Klos KS, Kim S, Bafico A et al. The Wnt receptor, Lrp5, is expressed by mouse mammary stem cells and is required to maintain the basal lineage. PLoS One 2009; 4: e6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindvall C, Evans NC, Zylstra CR, Li Y, Alexander CM, Williams BO. The Wnt signaling receptor Lrp5 is required for mammary ductal stem cell activity and Wnt1-induced tumorigenesis. J Biol Chem 2006; 281: 35081–35087. [DOI] [PubMed] [Google Scholar]
- 25.Lindvall C, Zylstra CR, Evans N, West RA, Dykema K, Furge KA et al. The Wnt co-receptor Lrp6 is required for normal mouse mammary gland development. PLoS One 2009; 4: e5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tepera SB, McCrea PD, Rosen JM. A beta-catenin survival signal is required for normal lobular development in the mammary gland. J Cell Sci 2003; 116: 1137–1149. [DOI] [PubMed] [Google Scholar]
- 27.Brisken C, Heineman A, Chavarria T, Elenbaas B, Tan J, Dey SK et al. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev 2000; 14: 650–654. [PMC free article] [PubMed] [Google Scholar]
- 28.Chu EY, Hens J, Andl T, Kairo A, Yamaguchi TP, Brisken C et al. Canonical WNT signaling promotes mammary placode development and is essential for initiation of mammary gland morphogenesis. Development 2004; 131: 4819–4829. [DOI] [PubMed] [Google Scholar]
- 29.Zeng YA, Nusse R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell 2010; 6: 568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teuliere J, Faraldo MM, Deugnier MA, Shtutman M, Ben-Ze’ev A, Thiery JP et al. Targeted activation of beta-catenin signaling in basal mammary epithelial cells affects mammary development and leads to hyperplasia. Development 2005; 132: 267–277. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, Li Y, Liu Q, Lu W, Bu G. Wnt signaling activation and mammary gland hyperplasia in MMTV-LRP6 transgenic mice: implication for breast cancer tumorigenesis. Oncogene 2010; 29: 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imbert A, Eelkema R, Jordan S, Feiner H, Cowin P. Delta N89 beta-catenin induces precocious development, differentiation, and neoplasia in mammary gland. J Cell Biol 2001; 153: 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 1988; 55: 619–625. [DOI] [PubMed] [Google Scholar]
- 34.Hayes MJ, Thomas D, Emmons A, Giordano TJ, Kleer CG. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin Cancer Res 2008; 14: 4038–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozaki S, Ikeda S, Ishizaki Y, Kurihara T, Tokumoto N, Iseki M et al. Alterations and correlations of the components in the Wnt signaling pathway and its target genes in breast cancer. Oncol Rep 2005; 14: 1437–1443. [DOI] [PubMed] [Google Scholar]
- 36.Prasad CP, Gupta SD, Rath G, Ralhan R. Wnt signaling pathway in invasive ductal carcinoma of the breast: relationship between beta-catenin, dishevelled and cyclin D1 expression. Oncology 2007; 73: 112–117. [DOI] [PubMed] [Google Scholar]
- 37.Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol 2010; 176: 2911–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat 2010; 123: 725–731. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005; 365: 671–679. [DOI] [PubMed] [Google Scholar]
- 40.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD et al. Genes that mediate breast cancer metastasis to lung. Nature 2005; 436: 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng Q, Zhang Z, Shea MJ, Creighton CJ, Coarfa C, Hilsenbeck SG et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res 2014; 24: 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuzawa A, Tseng PH, Vallabhapurapu S, Luo JL, Zhang W, Wang H et al. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science 2008; 321: 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol 2003; 13: 680–685. [DOI] [PubMed] [Google Scholar]
- 46.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014; 343: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014; 11: 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goecks J, Nekrutenko A, Taylor J, Galaxy T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 2010; 11: R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M et al. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol 2010; Chapter 19: Unit 19 10 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res 2005; 15: 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011; 27: 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G et al. A pathology atlas of the human cancer transcriptome. Science 2017; 357. [DOI] [PubMed] [Google Scholar]
- 55.Borcherding N, Cole K, Kluz P, Jorgensen M, Kolb R, Bellizzi A et al. Re-Evaluating E-Cadherin and beta-Catenin: A Pan-Cancer Proteomic Approach with an Emphasis on Breast Cancer. Am J Pathol 2018; 188: 1910–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geyer FC, Lacroix-Triki M, Savage K, Arnedos M, Lambros MB, MacKay A et al. beta-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol 2011; 24: 209–231. [DOI] [PubMed] [Google Scholar]
- 57.Bulut G, Fallen S, Beauchamp EM, Drebing LE, Sun J, Berry DL et al. Beta-catenin accelerates human papilloma virus type-16 mediated cervical carcinogenesis in transgenic mice. PLoS One 2011; 6: e27243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Q, Cao HZ, Zheng PS. LGR5 promotes the proliferation and tumor formation of cervical cancer cells through the Wnt/beta-catenin signaling pathway. Oncotarget 2014; 5: 9092–9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang YN, Liu BZ, Zhao QY, Hou T, Huang X. Nuclear localizaiton of beta-catenin is associated with poor survival and chemo-/radioresistance in human cervical squamous cell cancer. Int J Clin Exp Patho 2014; 7: 3908–3917. [PMC free article] [PubMed] [Google Scholar]
- 60.Kypta RM, Waxman J. Wnt/beta-catenin signalling in prostate cancer. Nat Rev Urol 2012; 9: 418–428. [DOI] [PubMed] [Google Scholar]
- 61.Yokoyama NN, Shao S, Hoang BH, Mercola D, Zi X. Wnt signaling in castration-resistant prostate cancer: implications for therapy. Am J Clin Exp Urol 2014; 2: 27–44. [PMC free article] [PubMed] [Google Scholar]
- 62.Nakashima N, Liu D, Huang CL, Ueno M, Zhang X, Yokomise H. Wnt3 gene expression promotes tumor progression in non-small cell lung cancer. Lung Cancer 2012; 76: 228–234. [DOI] [PubMed] [Google Scholar]
- 63.Uematsu K, He B, You L, Xu Z, McCormick F, Jablons DM. Activation of the Wnt pathway in non small cell lung cancer: evidence of dishevelled overexpression. Oncogene 2003; 22: 7218–7221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.