

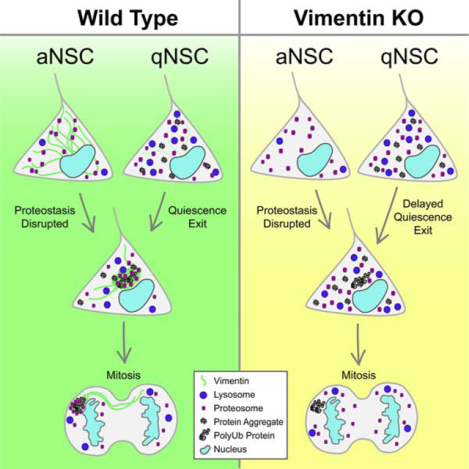
Summary
Maintaining a healthy proteome throughout life is critical for proper somatic stem cell function, yet the complexities of the stem cell response to increases in damaged or aggregated proteins remain unclear. Here we demonstrate that adult neural stem cells (NSCs) utilize aggresomes to recover from disrupted proteostasis, and describe a novel function for the intermediate filament vimentin in proteostasis as a spatial coordinator of proteasomes to the aggresome. In the absence of vimentin, NSCs have a reduced capacity to exit quiescence, a time when NSCs are required to clear a wave of aggregated proteins, and demonstrate an early age-dependent decline in proliferation and neurogenesis. Taken together, these data reveal a significant role for vimentin and aggresomes in the regulation of proteostasis during quiescent NSC activation.
eTOC Blurb
Recent evidence suggests neural stem cell (NSC) quiescence exit is a significant barrier to adult neurogenesis. Morrow et al reveal that NSCs rely on vimentin-mediated proteasome localization to the aggresome to clear proteins during quiescence exit, and uncover a critical role for vimentin as a regulator of cellular proteostasis.
Graphical Abstract
Introduction
Proper somatic stem cell function for lifelong tissue maintenance relies on the cell’s ability to maintain proteostasis, a balance between the synthesis and degradation of proteins which is controlled by a network of protein pathways (Vilchez et al., 2014). Failure to maintain proteostasis leads to an accumulation of misfolded, damaged or aggregated proteins, which is a hallmark of aging and is correlated with stem cell dysfunction (Lopez-Otin et al., 2013, Audesse et al., 2019, Rodriguez-Fernandez et al., 2019).
One way that stem cells regulate this imbalance is through mitosis, where they can asymmetrically segregate damaged or ubiquitinated proteins to one daughter cell (Moore et al., 2015, Moore and Jessberger, 2017, Bufalino et al., 2013, Rujano et al., 2006). Neural stem cells (NSCs) co-segregate ubiquitinated proteins with the intermediate filament (IF) vimentin during mitosis, suggesting vimentin as a player in protein clearance (Moore et al., 2015). While a number of studies have investigated the role of IFs in NSCs, revealing an essential role for the IF nestin during development and a role for phosphorylated vimentin in NSC differentiation, vimentin’s role in protein clearance in NSCs remains unknown (Chen et al., 2018, Park et al., 2010).
Previously it has been shown in cell lines that following impaired proteostasis, accrued misfolded or damaged proteins can be trafficked to the centrosome and surrounded by a vimentin cage in a structure called the aggresome (Ogrodnik et al., 2014, Johnston et al., 1998, Rujano et al., 2006). While much is known about how aggresomes form, their endogenous role, and how they specifically enhance recovery from disrupted proteostasis has remained elusive (Kawaguchi et al., 2003, Ouyang et al., 2012, Ogrodnik et al., 2014). Here we asked if NSCs utilize aggresomes to maintain NSC proteostasis, and what function vimentin plays in these structures.
Results
Tagging of endogenous vimentin in primary mouse NSCs to monitor aggresome formation
To unravel vimentin’s role in maintaining NSC proteostasis, we used CRISPR/Cas9 to add an mNeon fluorophore to genomic vimentin at its 3’ end in mouse primary hippocampal NSCs to dynamically visualize vimentin without overexpression (Fig. 1A). We performed fluorescence activated cell sorting (FACS) to purify mNeon-expressing NSCs, and immunostained against vimentin protein. Vimentin antibody signal strongly colocalized with mNeon (Fig. 1A), suggesting the tag accurately reported vimentin filament formation (Fig. S1A). Vimentin tagging also did not affect NSC morphology, proliferation, differentiation, or motility (Fig. 1A, S1B–E).
Figure 1. In response to a loss of proteostasis, NSCs form aggresomes surrounded by vimentin cages that are asymmetrically inherited during mitosis.
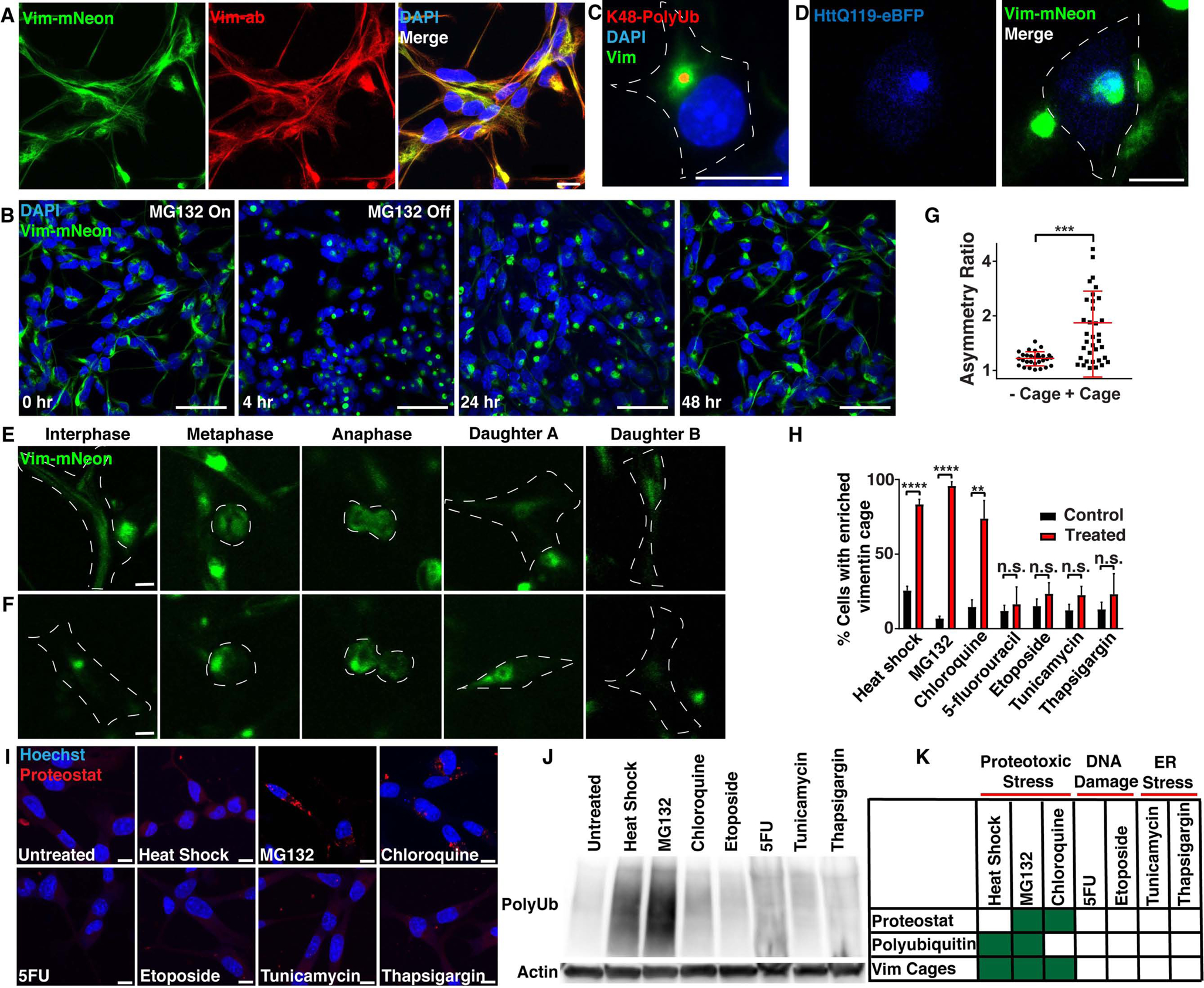
A) Vimentin-mNeon (green) NSCs were immunostained for vimentin (red). B) Vimentin-mNeon NSCs (green) were treated with 5 μM MG132 for 4 hours to transiently disrupt proteostasis, and allowed to recover for up to 48 hours. C) Vimentin-mNeon NSCs were treated with 2 μM MG132 or 0.02% DMSO for 18 hours and then immunostained for K48pUb (red). D) Vimentin-mNeon NSCs (green) were live-imaged 24 hours after electroporation with a construct that expresses a mutant Huntingtin protein (HttQ119; blue). E-G) Representative frames and quantitation of asymmetry from timelapse imaging of vimentin-mNeon NSCs in either untreated conditions or following a 20 minute heat shock at 45°C, that proceeded through mitosis either with or without a vimentin cage formed in interphase (n≥29; Mann-Whitney test; mean ± SD). H) Quantification of vimentin cage formation in response to a heat shock of 20 minutes at 45°C, 5 μM MG132 or 10 μM chloroquine for 6 hours, 50 nM 5-fluorourucil or 100 nM etoposide for 24 hours, and 1 μg/mL tunicamycin or 1 μM thapsigargin for 6 hours (red bars) next to their appropriate DMSO or untreated controls (black bars) (N=3; Two-way ANOVA with post-hoc Tukey’s test; mean ± SD). I) NSCs treated as indicated in Fig. 1H were stained with Proteostat (aggregated proteins; red). (N=3; Two-way ANOVA with post-hoc Tukey’s test; mean ± SD). J) Western blot against polyubiquitin and β-actin in total fraction (soluble and insoluble) protein lysates generated from NSCs stressed as indicated in Fig. 1H alongside an untreated control. K) Chart depicting cellular stress conditions that induced changes in aggregated protein levels (Proteostat), polyubiquitin levels, or vimentin cages. Scale bars, 10 μm (A, C-F, I) 50 μm (B). White line denotes edge of the cell. Nuclei were labeled with DAPI or Hoechst (blue). **p<0.01, ***p<0.001, ****p<0.0001. See also Figure S1.
To identify if vimentin-mNeon NSCs could form vimentin cages that surround aggresomes in response to a loss of proteostasis, we treated WT and vimentin-mNeon NSCs with the reversible proteasome inhibitor MG132 or DMSO for 4 hours, and then immunostained against vimentin throughout the treatment and during recovery (Fig. 1B, S1F–H). We defined a vimentin cage as a cage-like structure at the centrosome with >50% of the vimentin signal within each cell (Fig. S1F). We found that NSCs form a vimentin cage with similar dynamics, as visualized with both mNeon signal and vimentin antibody staining (Fig. S1G–H). Importantly, we confirmed that vimentin cages indeed surrounded an aggresome, as vimentin-mNeon surrounded K48-linked polyubiquitin-rich (K48pUb) aggregates, Proteostat-labeled aggresomes, and overexpressed aggregation-prone proteins at the centrosome (Fig 1C–D, S1I–M).
Aggresomes form during interphase in response to increased levels of ubiquitinated or aggregated proteins
We previously found that over-expressed vimentin is asymmetrically segregated between two daughter cells during NSC mitosis (Moore et al., 2015). To better understand vimentin dynamics and aggresome formation throughout the cell cycle in endogenously tagged vimentin-mNeon NSCs, we performed timelapse imaging. Similar to reports in cell lines (Ogrodnik et al., 2014, Rujano et al., 2006), we found that vimentin asymmetry during mitosis in NSCs was tightly linked to aggresome formation in interphase (Fig. 1E–G, S1N). Thus, we next asked what drives vimentin cage formation in NSCs during interphase.
Vimentin has been shown to react to cellular stress by becoming upregulated and collapsing to form a cage around the aggresome (Rujano et al., 2006, Johnston et al., 1998, Ogrodnik et al., 2014, Vilaboa et al., 1997). To determine if vimentin responds to all cellular stresses with cage formation, we measured the percentage of NSCs which formed a vimentin cage in response to different challenges. We found that heat shock, inhibition of proteasomes (MG132), and inhibition of autophagy (chloroquine) increased the percentage of NSCs forming a vimentin cage, whereas drugs that caused DNA damage (5-fluorouracil, etoposide), or ER stress (tunicamycin, thapsigargin) did not affect vimentin cage formation (Fig. 1H), even if taken to a concentration that kills the cells (Fig. S1O–P). As the aggresome has been shown to consist of aggregated and ubiquitinated proteins (Ogrodnik et al., 2014, Johnston et al., 1998), in these treatments we also measured the levels of aggregated proteins using the dye Proteostat, and the total (soluble and insoluble) amount of polyubiquitinated proteins in western blots. We found that Proteostat signal was increased only with MG132 or chloroquine treatment, whereas total polyubiquitinated protein levels were increased with heat shock and MG132 (Fig. 1I–J). These experiments support the hypothesis that NSCs form aggresomes specifically in response to a loss of proteostasis (Fig. 1K).
Vimentin is not necessary for aggresome formation in NSCs
Despite reports of vimentin at the aggresome, its role has been unclear (Johnston et al., 1998). To determine if vimentin is necessary for aggresome formation, we first used CRISPR/Cas9 to create three vimentin KO lines in primary mouse hippocampal NSCs (KO), as well as a control NSC line treated with a non-targeting guide RNA (WT; Fig. 2A, S2A). Vimentin KO NSCs had decreased expression of the IFs glial fibrillary acidic protein (GFAP) and nestin (Fig. S2B, Table S1), however displayed no differences in NSC proliferation, differentiation, or motility (Fig. S2C–F). To determine if vimentin is required for aggresome formation, we treated WT and vimentin KO NSCs with MG132 to induce an increase in misfolded or damaged proteins, and found that both WT and vimentin KO NSCs made K48pUb aggresomes in the nuclear bay (Fig. 2B). These data reveal that vimentin is not necessary for aggresome formation, or basic NSC function.
Figure 2. Vimentin KO NSCs fail to localize proteasomes to the aggresome and demonstrate a reduced capacity to recover proteostasis.
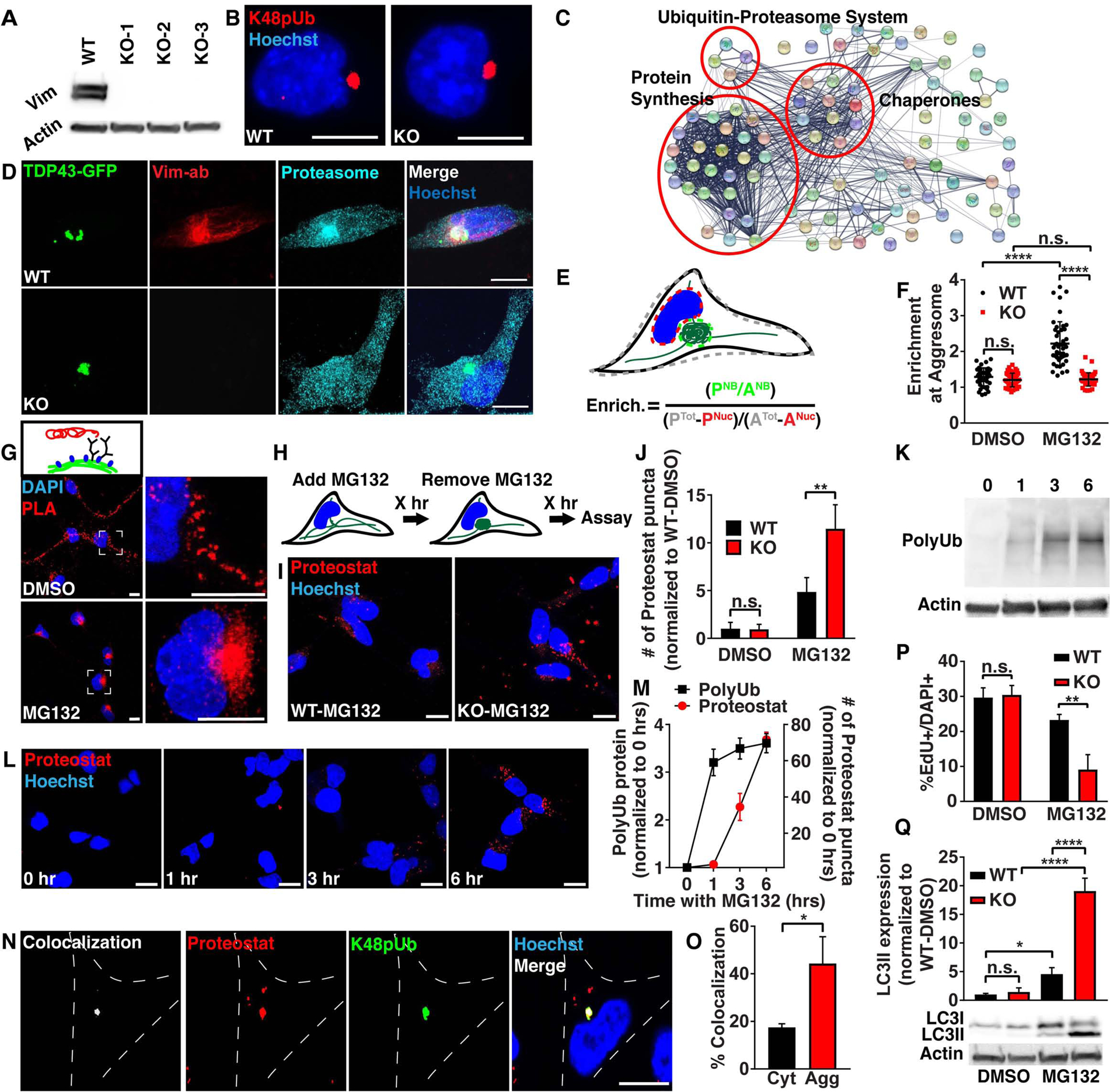
A) WT and 3 vimentin KO clones were probed by western blot for vimentin and β-actin protein levels. B) WT and vimentin KO NSCs were treated with 2 μM MG132 for 16 hours and then immunostained for K48pUb (red). C) Functional protein association network generated from LC-MS/MS on proteins co-immunoprecipitated with vimentin-mNeon following 5 μM MG132 for 4 hours, and visualized using String. D) WT and vimentin KO NSCs 24 hours after electroporation with a construct expressing the C-terminal fragment of TDP-43-GFP were fixed and immunostained for vimentin (red) and the α5 subunit of the proteasome (cyan). E-F) WT (black dots) and vimentin KO (red dots) NSCs were stressed with 0.05% DMSO or 5 μM MG132 for 4 hours and then fixed and immunostained for the α5 subunit of the proteasome and quantitated for proteasome enrichment at the aggresome by measuring Proteasome Intensity (P) and Area (A) of the nuclear bay (NB), nucleus (Nuc) or entire cell (Tot). (n≥45 cells; Mann-Whitney test; mean ± SD). G) WT NSCs were treated with 0.05% DMSO or 5 μM MG132 for 4 hours and then fixed and probed with a proximity ligation assay (red) against vimentin and the α5 subunit of the proteasome. H) Graphic outlining treatment and recovery scheme for experiments in Fig. 2I, P and Q. I-J) WT (black bar) and vimentin KO (red bar) NSCs were treated with 0.05% DMSO or 5 μM MG132 for 4 hours and then allowed to recover for 2 hours before fixing and staining aggregated proteins with the dye Proteostat (red). (N=3; Two-way ANOVA with post-hoc Tukey’s test; mean ± SD). K-M) WT NSCs were treated with 5 μM MG132 for 0, 1, 3 and 6 hours and then stained with Proteostat (red) or analyzed for total fraction (soluble and insoluble) polyubiquitin levels on a western blot. (N=3; Two-way ANOVA with post-hoc Tukey’s test; mean ± SD). N-O) WT NSCs were treated with 5 μM MG132 for 4 hours and then fixed and immunostained for K48pUb (green) and stained for Proteostat (red). Colocalization of Proteostat signal overlapping with K48pUb either in the cytosol (Cyt) or within the vimentin cage (Agg) was quantified using Imaris. (n=30; Student’s t-test; mean ± SD) P) WT (black bar) and vimentin KO (red bar) NSCs were treated with 0.05% DMSO or 5 μM MG132 for 4 hours and then allowed to recover for 2 hours. During the last hour of treatment, cells were pulsed with EdU for 1 hour and then fixed and analyzed for percent EdU+ NSCs. (N=3; Two-way ANOVA with post-hoc Tukey’s test; mean ± SD). Q) WT (black bar) and vimentin KO (red bar) NSCs were treated with 0.02% DMSO or 2 μM MG132 for 24 hours and then allowed to recover for 1 hour before analyzing LC3I and LC3II protein on a western blot. (N=3; Two-way ANOVA with post-hoc Tukey’s test; mean ± SD). Scale bars, 10 μm. Nuclei were labeled with Hoechst (blue). *p<0.05, **p<0.01, ****p<0.0001. See also Figure S2 and Tables S1–3.
Vimentin interacts with proteins important in the maintenance of NSC proteostasis
To identify what proteins interact with vimentin at the aggresome, we performed a co-immunoprecipitation against mNeon in vimentin-mNeon NSCs in both control and MG132-stressed conditions, followed by liquid chromatography-mass spectrometry (LC-MS/MS) to identify proteins that interact either directly or indirectly with vimentin (Fig. 2C). Interestingly, in comparison to control untagged NSCs transfected with cytosolic mNeon, we found the presence of nodes important for protein homeostasis, such as heat shock proteins and chaperones for protein re-folding, ribosomal proteins and elongation factors for protein translation, and proteasomes for protein degradation (Fig. 2C, Table S2). These results suggest that vimentin is acting as an organizer of proteins that regulate the balance of protein synthesis, refolding, and degradation.
Vimentin spatially localizes proteasomes to aggresomes in response to cellular stress
Proteasomes, multi-subunit protein complexes which primarily process polyubiquitinated proteins for degradation, previously have been identified at aggresomes and inclusion bodies, and also were present in our vimentin interactome (Table S2) (Schipper-Krom et al., 2014, Hao et al., 2013, Wigley et al., 1999, Ogrodnik et al., 2014). To visualize this interaction, we electroporated the aggregation prone C-terminal domain of TDP43-GFP into NSCs to induce aggresome formation and then performed immunostaining against the α5 subunit in the catalytic 20S portion of the proteasome (Fig. 2D). WT NSCs made a vimentin cage surrounding TDP43-GFP at the aggresome, and as our mass spectrometry data suggested, the vimentin cage was strongly enriched with proteasomes (Fig. 2D–F). Using a proximity ligation assay (PLA) which results in labeling if proteins are within 40nm of each other, we applied antibodies against vimentin and the same α5 proteasome subunit and detected a strong signal at the aggresome in WT NSCs, supporting an interaction between vimentin and proteasomes (Fig. 2G, S2G). Interestingly, interactions between proteasomes and vimentin were also present along vimentin filaments in unstressed conditions (Fig. 2G), suggesting these interactions are maintained following stress as vimentin filaments collapse to the nuclear bay, bringing proteasomes to the aggresome. Strikingly, while vimentin KO NSCs could still form an aggresome, there was no enrichment of proteasomes at the aggresome, revealing that vimentin is required for proteasome localization to the aggresome (Fig. 2D–F). Thus, our data suggest that proteasome-mediated protein turnover at the aggresome may be impaired in vimentin KO NSCs and may impact their ability to recover from disrupted proteostasis.
Vimentin KO NSCs demonstrate a reduced ability to recover from a loss of proteostasis
Initial papers characterizing vimentin KO mice reported limited phenotypes in these animals (Colucci-Guyon et al., 1994); however, further studies showed that in conditions of stress, vimentin KO mice had poor outcomes (Lundkvist et al., 2004, Terzi et al., 1997). We thus asked how vimentin KO NSCs would respond when challenged with a loss of proteostasis if they are unable to deliver proteasomes to the aggresome. We created a stress/recovery paradigm where we treated WT or vimentin KO NSCs with MG132 or DMSO for 4 hours to disrupt proteostasis, followed by a 2 hour recovery prior to analysis (Fig. 2H). Using the dye Proteostat to measure the levels of aggregated proteins, we found that whereas basally there was no difference in the amount of aggregated proteins in WT and vimentin KO NSCs, following disrupted proteostasis, vimentin KO NSCs had significantly increased levels of aggregated proteins during recovery (Fig. 2H–J). This finding suggests that vimentin KO NSCs are less able to degrade proteins that accumulate as a result of proteasome inhibition with MG132. To better understand the temporal dynamics of aggregated protein formation, we measured total (soluble and insoluble) polyubiquitinated protein levels and Proteostat labeling, indicative of aggregated proteins, at different timepoints during MG132 treatment in WT NSCs. We found that after treatment with MG132, ubiquitin levels increased and were maintained, followed by a delayed increase of Proteostat signal, suggesting that when ubiquitinated proteins are unable to be degraded, they aggregate (Fig. 2K–M). Further, we observed a high degree of co-localization between K48pUb proteins and Proteostat labeled aggregated proteins at the aggresome (Fig. 2N–O). These data propose a model whereby the lack of proteasome localization in vimentin KO NSCs to the aggresome may result in inefficient degradation of misfolded or polyubiquitinated proteins, leading to an increase in aggregated proteins.
We next asked if the increased amount of aggregated protein following a loss of proteostasis in vimentin KO NSCs affects NSC behavior. Using a one hour pulse of EdU to label NSCs in S phase following the stress/recovery paradigm, we found that whereas the percentage of EdU+ NSCs under basal conditions was not different between WT and vimentin KO cells with DMSO treatment, disruption of proteostasis with MG132 treatment resulted in decreased proliferation in vimentin KO NSCs during a 2 or 8 day recovery period (Fig. 2P, S2H–I). Thus, although basally vimentin KO NSCs have no appreciable phenotype, during a recovery following disruptions in proteostasis they have significantly increased levels of aggregated proteins, and decreased proliferation that failed to improve even after an extended recovery time.
Proteasome expression and activity is not changed in vimentin KO NSCs
To confirm that these effects were not due to differential expression of proteasome components in vimentin KO NSCs, we performed RNA sequencing of WT and vimentin KO NSCs, and found no difference in the amount of mRNA of proteasome components (Fig. S2J, Table S3). Further, to determine if proteasome function is altered in vimentin KO NSCs, we lysed WT and KO NSCs, losing all spatial information, and subjected them to a proteasome activity assay. All measured substrates testing chymotrypsin, trypsin, and caspase activities showed similar proteasome activity levels in WT and vimentin KO NSCs during recovery after a loss of proteostasis (Fig. S2K–M), indicating that proteasomes in vimentin KO NSCs have normal function. These experiments demonstrate that there is no change in proteasome expression or activity in vimentin KO NSCs, suggesting the lack of spatial localization of proteasomes to the aggresome due to vimentin KO may be the main driver of increased protein aggregation and decreased proliferation measured in vimentin KO NSCs.
Vimentin KO NSCs upregulate autophagy in response to increased levels of aggregated proteins
Another arm of cellular protein degradation is autophagy, where autophagosomes engulf large aggregates which fuse with acidic lysosomes to degrade proteins (Yu et al., 2018). To determine if vimentin KO NSCs compensate for defects in proteasome-mediated protein degradation, we measured LC3II protein levels in WT and vimentin KO NSCs treated with DMSO or MG132 followed by recovery (Fig. 2Q). In line with our previous observations, we observed no differences between WT and vimentin KO NSCs in the DMSO condition, but during recovery from MG132 treatment, vimentin KO NSCs had increased autophagic flux indicated by increased levels of LC3II, suggesting that vimentin KO NSCs are more dependent on autophagy to recover proteostasis (Fig. 2Q). This was further confirmed using lentiviral overexpression of mCherry-GFP-LC3 in WT and vimentin KO NSCs. This peptide localizes to autophagosomes (GFP+ and mCherry+). When autophagosomes fuse with an acidified lysosome (mCherry+), GFP is denatured, giving a readout for autophagic flux (Fig. S2N). Similarly, following MG132 treatment to disrupt proteostasis, vimentin KO NSCs expressing mCherry-GFP-LC3 demonstrated increased autophagic flux (Fig. S2O), as well as increased mCherry signal demonstrating increased utilization of lysosomes (Fig. S2P). This increase in lysosomes following MG132 treatment and recovery in vimentin KO NSCs was also visualized using Lysotracker (Fig. S2Q). These data support a model where, following an inability to spatially localize proteasomes to the aggresome, polyubiquitinated proteins begin to aggregate, and vimentin KO NSCs increase autophagy to increase aggregate clearance.
Vimentin protein is downregulated in qNSCs, and forms cages only during quiescence exit
Recent studies have shown that quiescent NSCs (qNSCs) of the subventricular zone (SVZ) have increased levels of protein aggregates (Proteostat), which are removed through autophagy during activation into the cell cycle (Leeman et al., 2018). Using a well-established protocol to induce quiescence in NSCs in vitro (Mira et al., 2010, Martynoga et al., 2013, Knobloch et al., 2017) (Fig. 3A, S3A), we found that quiescent primary mouse hippocampal NSCs also had increased levels of protein aggregates, as measured by Proteostat, which were cleared during quiescence exit (Fig. 3A–B). We next asked if vimentin would respond to increased protein aggregates in qNSCs with aggresome formation. We imaged vimentin-mNeon NSCs prior to quiescence induction, after full quiescence was reached, and during qNSC activation (Fig. 3A). We found that qNSCs downregulate vimentin protein, and surprisingly, do not form vimentin cages in the presence of increased protein aggregates (Fig. 3B–E). Using timelapse microscopy of vimentin-mNeon NSCs during quiescence exit, we found that prior to the first cell division following BMP4 removal, vimentin is upregulated and forms a cage in interphase, resulting in asymmetric inheritance of the vimentin cage to one of the daughter cells (Fig. 3F–H). Interestingly, vimentin mRNA is upregulated in qNSCs (Fig. 3I), suggesting the NSCs are primed to respond with upregulation and cage formation in the transition to activation.
Figure 3. NSCs form aggresomes during quiescence exit, and vimentin KO NSCs have a reduced capacity to exit quiescence in vitro.
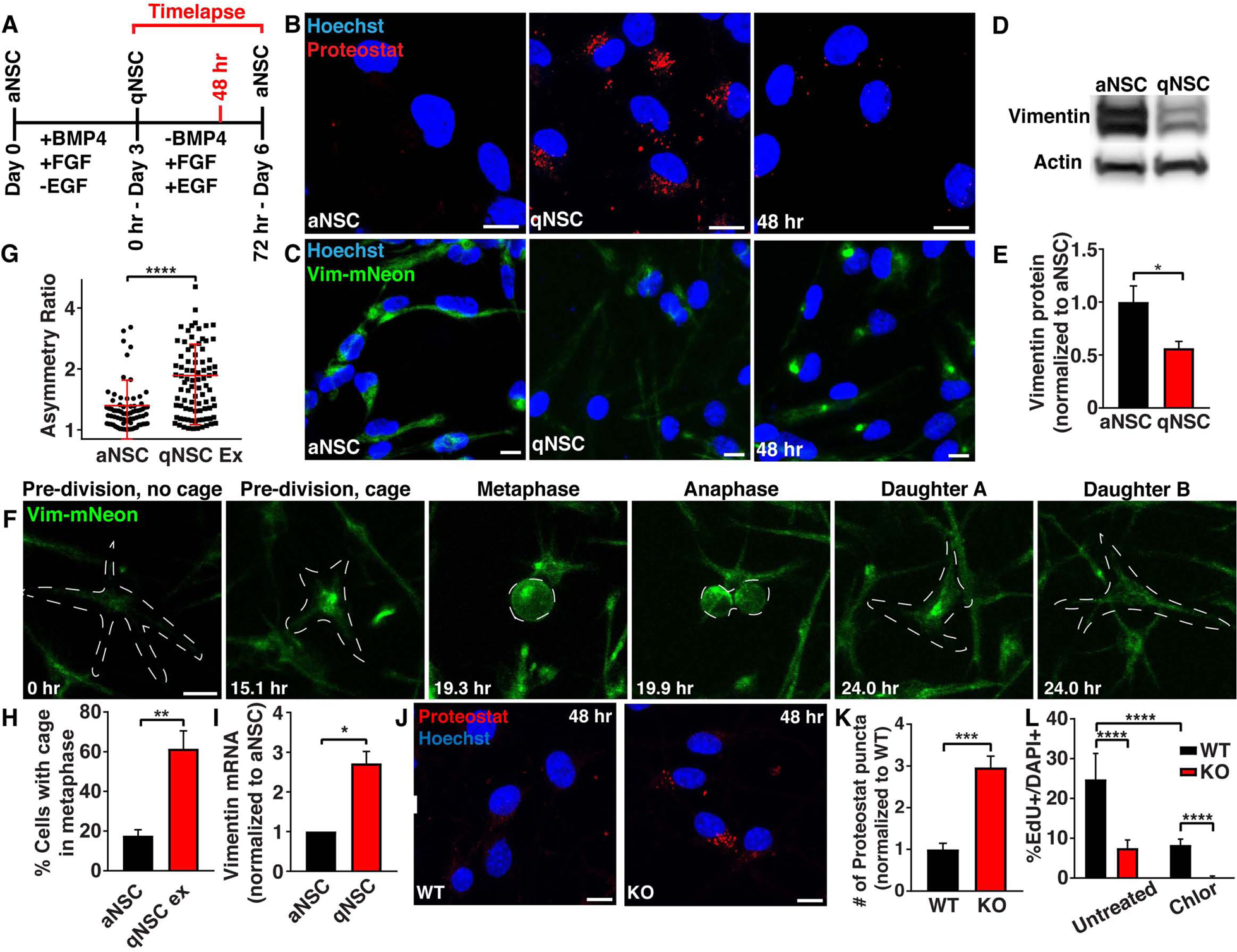
A) Schematic outlining critical experimental time points examined during an in vitro quiescence paradigm. B) aNSCs, qNSCs or qNSCs exiting quiescence for 48 hours were fixed and stained with Proteostat (aggregated proteins; red). C) Images of vimentin-mNeon (green) aNSCs, qNSCs or qNSCs exiting quiescence for 48 hours. D-E) aNSCs and qNSCs were probed by western blot for vimentin and β-actin protein levels. (N=3; Student’s t-test; mean ± SD). F) Representative division of a vimentin-mNeon NSC during quiescence exit. G-H) Quantification of vimentin asymmetry during mitosis (G) and presence of vimentin cages in metaphase prior to mitosis (H) in vimentin-mNeon aNSCs or qNSCs during quiescence exit. (n≥71; Mann-Whitney test; mean ± SD). I) aNSCs and qNSCs were analyzed by qRT-PCR for vimentin mRNA expression levels. (N=3; Student’s t-test; mean ± SD). J-K) WT or vimentin KO NSCs exiting quiescence for 48 hours were stained with Proteostat (aggregated proteins; red). (N=3; Student’s t-test; mean ± SD). L) WT or vimentin KO NSCs exiting quiescence for 48 hours in either an untreated condition or in the presence of 1 μM chloroquine were pulsed for 1 hour with EdU before analysis. (N≥3; Two-way ANOVA with post-hoc Tukey’s test; mean ± SD). Scale bars, 10 μm. Nuclei were labeled with Hoechst (blue). White line denotes edge of the cell. *p<0.05, **p<0.001 ***p<0.001, ****p<0.0001. See also Figure S3.
Vimentin KO NSCs demonstrate a reduced capacity to exit quiescence in vitro
To test if vimentin was necessary for quiescence exit, we induced full quiescence in WT and vimentin KO NSCs, and then began the transition to activation. Forty-eight hours into this transition (Fig. 3A), we stained for Proteostat and found that vimentin KO NSCs have increased levels of aggregated proteins (Fig. 3J–K) and lysosomes (Fig. S3B–C), mimicking our results disrupting proteostasis using MG132, and suggesting an impairment in protein degradation during activation. Further, we found that vimentin KO NSCs had decreased proliferation as measured by EdU (S phase), Ki67 (cell cycle), and phosphohistone H3 (PH3; late G2 and M) (Fig. 3L, S3D–E), suggesting a reduced ability to exit quiescence. Proliferation rates during quiescence exit were approximately half in vimentin KO NSCs (Fig. 3L), suggesting this is not the only mechanism driving quiescence exit. As autophagy was previously shown to contribute to quiescence exit (Leeman et al., 2018), we induced quiescence in WT and vimentin KO NSCs, this time adding chloroquine to inhibit autophagy during quiescence exit. We found that autophagy inhibition during quiescence activation in vimentin KO NSCs rendered NSCs fully unable to exit quiescence (Fig. 3L), suggesting that vimentin’s caging and proteasome localization, as well as autophagy are both playing roles in quiescence exit in NSCs.
During quiescence exit, vimentin KO NSCs fail to asymmetrically segregate proteasomes during mitosis
Our finding that NSCs asymmetrically segregate vimentin during quiescence exit suggested that vimentin may be involved in coordinating the asymmetric segregation of different cellular cargoes such as polyubiquitin and proteasomes during mitosis. To address this question, we imaged vimentin-mNeon and vimentin KO NSCs during quiescence exit and quantified asymmetry of vimentin, K48pUb, and proteasomes in these divisions (Fig. S3F–K). In WT NSCs, K48pUb and proteasomes were primarily asymmetrically inherited during mitosis by the daughter cell that inherited more vimentin (Fig. S3F, H; (Moore et al., 2015)). Vimentin KO NSCs retained the ability to asymmetrically segregate K48pUb proteins during mitosis (Fig. S3F–G), a finding unsurprising in light of our observation that vimentin is not involved in aggresome formation (Fig. 2B). While vimentin was not required for asymmetric segregation of K48pUb proteins, vimentin was critical for the asymmetric segregation of proteasomes during mitosis (Fig. S3I–J). These data suggest that vimentin KO NSCs asymmetrically segregate polyubiquitin to daughters without providing them with an enrichment of proteasomes to clear this burden.
NSCs asymmetrically segregate the aggresome in vivo
To determine if vimentin cage formation and asymmetric segregation of vimentin during mitosis is conserved in a physiologic setting in vivo, we performed immunofluorescence to visualize endogenous vimentin in the dentate gyrus of 7 week-old Nestin-GFP mice. While NSC divisions in brain slices are rare, we observed the asymmetric segregation of vimentin in a dividing Nestin+ NSC, supporting our in vitro data, and previously published overexpression studies in embryonic NSCs (Fig. 4A, S4B) (Moore et al., 2015). Additionally, we also observed enrichment of vimentin in the nuclear bay of a small number of NSCs in interphase (Fig. 4B). These data collectively suggest that in vivo, NSCs can utilize aggresomes during quiescence exit.
Figure 4. NSCs in vimentin KO mice demonstrate a reduced capacity to exit quiescence following a stimulus and during early aging.
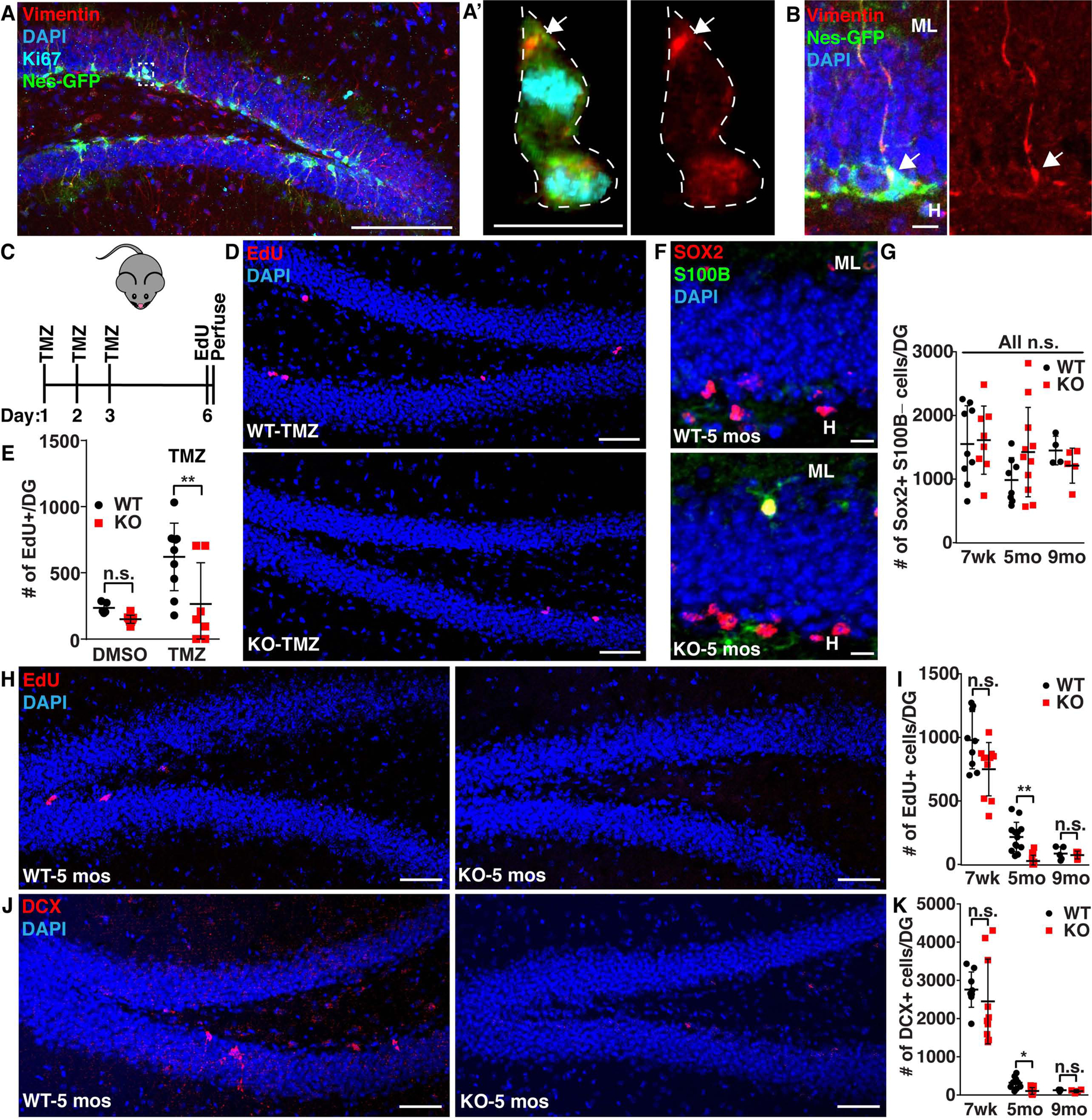
A-B) 7 week old Nestin-GFP mouse brain sections were immunostained for GFP (NSCs; green), vimentin (red) and Ki67 (cyan). A’ was created by making a 3-D mask of the cytoplasmic GFP signal around the dividing cell (Imaris), revealing only intracellular staining. White line denotes edge of cells. C-E) 7 week old WT or vimentin KO mice were treated with 25 mg/kg TMZ or DMSO once per day for 3 days and then allowed to recover for 3 days before being injected with 50 mg/kg EdU 3 times every 3 hours, perfused 3 hours after the last EdU injection and analyzed for proliferative (EdU+) NSCs in the SGZ. (n≥4; Two-way ANOVA with post-hoc Tukey’s test; mean ± SD). F-K) 7 weeks (wk), 5 months- (mo) old, or 9 months-old WT and vimentin KO mice were injected with 50 mg/kg EdU once per day for 4 days, sacrificed 1 day following the last EdU injection and then analyzed for proliferative (EdU+) NSCs in the SGZ, total newborn neuron (doublecortin (DCX; red)) number and total NSC number (Sox2+ (red) S100B- (green)). The hilus (H) and the molecular layer (L) are denoted in F. (n≥4; Kruskal-Wallis Test with post-hoc Dunn’s test; mean ± SD). Scale bars, 100 μm (A), 50 μm (D, H, J), 10 μm (A’, B, F). Nuclei were labeled with Hoechst or DAPI (blue). *p<0.05, **p<0.001. See also Figure S4.
NSCs in vimentin KO mice have a decreased ability to exit quiescence in vivo
As vimentin KO leads to a decreased ability to exit from quiescence in vitro, and we observed vimentin cages to be formed in vivo, we asked if vimentin KO animals would demonstrate a reduced ability of NSCs to exit quiescence in vivo. Previously it was shown that following temozolamide (TMZ) treatment, dividing NSCs are depleted, leading to qNSC activation to replenish the population (Knobloch et al., 2017, Garthe et al., 2009) (Fig. S4A). We performed 3 days of TMZ injections in WT and vimentin KO mice followed by a 3 day recovery. Prior to perfusion, mice received 3 EdU injections (Fig. 4C). We found that vimentin KO NSCs had decreased levels of EdU+ cells in the subgranular zone (SGZ) following TMZ treatment as compared to WT mice, suggesting that vimentin is necessary for efficient quiescent exit in hippocampal NSCs in vivo (Fig. 4D–E).
Recently, it has been proposed that a major factor driving reduced neurogenesis during aging is an increased time spent in quiescence, suggesting that vimentin KO animals may sustain a faster decline in proliferation and neurogenesis during aging (Kalamakis et al., 2019). To measure the differences in adult neurogenesis at different ages, we performed 4 EdU injections once per day for four days in 7 weeks-old, 5 month-old, or 9 month-old WT and vimentin KO littermates prior to perfusion. We found that vimentin KO mice exhibit a faster age-dependent decline in NSC proliferation (EdU+) and neurogenesis (DCX+), but display similar numbers of NSCs (Sox2+/S100B-) (Fig. 4F–K), suggesting that vimentin KO NSCs have difficulty exiting quiescence, and further supporting a role for vimentin in NSC quiescence exit.
Discussion
qNSC activation has become increasingly recognized as one of the critical barriers to neurogenesis (Kalamakis et al., 2019). Recently, Leeman et al showed that one key component driving qNSC activation is a shift in proteostasis, where a wave of aggregated proteins (Proteostat) are cleared (Leeman et al., 2018). Induction of autophagy enhanced clearance of these aggregated proteins and increased the rate of qNSC activation, suggesting a direct connection between a qNSC’s ability to activate and the ability to clear aggregated proteins (Leeman et al., 2018). Our study further validates the findings that qNSCs carry an increased load of Proteostat+ aggregated proteins, here in hippocampal NSCs (Fig. 3B), and provides a mechanism of aggresome formation as a means for qNSCs to turn up protein turnover during qNSC activation (Fig. 3C).
During aging, NSCs have a decreased ability to exit quiescence (Kalamakis et al., 2019, Ziebell et al., 2018, Leeman et al., 2018). While other regions of the body, including other stem cell niches, experience changes with aging in rodents at more advanced ages such as 18–24 months, hippocampal NSCs show decreases in proliferation as early as 2 months, which saturate by 9 months (Ben Abdallah et al., 2010, Cho et al., 2008, Lukjanenko et al., 2019). We find that NSC proliferation and neurogenesis decline more rapidly in vimentin KO animals over time (Colucci-Guyon et al., 1994), further supporting a role for vimentin in regulating quiescence exit in vivo (Fig. 4C–K).
Our study provides insight into the two-decade old question of vimentin’s function at the aggresome, demonstrating its role as an organizer of proteostasis-related proteins to the aggresome, and a critical regulator of proteostasis in NSCs. Further, we provide a function for aggresomes within a biological system, as a program that NSCs use to clear protein during qNSC activation. Our findings pave the way for studies aimed at understanding the mechanisms driving vimentin cage formation, the significance of aggresome asymmetry during mitosis as a mechanism of stem cell regulation, and the application of our findings to other cell types and diseases.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Darcie L. Moore (darcie.moore@wisc.edu). Plasmids generated in this study have been deposited to Addgene under catalog numbers listed in the “Key Resources Table.” All other resources used for this study are available through the Lead Contact, Darcie L. Moore (darcie.moore@wisc.edu) at reasonable request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Vimentin - Immunofluorescence | Sigma | Cat#ab5733 |
| Vimentin - Western Blot | Cell Signaling Technology | Cat#5741S |
| Vimentin - Used for proximity ligation assay | Abcam | Cat#ab22651 |
| Polyubiquitin (FK1) | Enzo | Cat#BML-PW8805-0500 |
| K48-linked Polyubiquitin | Millipore Sigma | Cat#05-1307-AF488 |
| α5-Proteasome | Abcam | Cat#ab189855 |
| Sox2 | Santa Cruz Bio | Cat#Sc-17320 |
| S100B | Abcam | Cat#ab52642 |
| Nestin | Thermo | Cat#MA1110 |
| GFAP | Dako | Cat#Z033401-2 |
| Ki67 | Abcam | Cat#ab15580 |
| Phospho-histone-3 (PH3) | Abcam | Cat#ab14955 |
| Pericentrin | Abcam | Cat#ab4448 |
| LC3B | Abcam | Cat#ab48394 |
| GFP | Abcam | Cat#ab5450 |
| Doublecortin | Cell Signaling Technology | Cat#4604S |
| Bacterial and Virus Strains | ||
| DH5α | Invitrogen | Cat#18265-017 |
| MDS 42 Low Mut Delta recA | Scarab Genomics | Cat#C-6262-10k |
| Biological Samples | ||
| N/A | N/A | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FGF-2 | PeproTech | Cat#100-18B |
| EGF | PeproTech | Cat#AF-100-15 |
| Heparin | Sigma | Cat#H3149 |
| BMP-4 | Fisher Scientific | Cat#5020BP010 |
| Laminin | Sigma | Cat#L2020 |
| MG 132 | Sigma | Cat#M7449 |
| Chloroquine | Sigma | Cat#C6628 |
| Etoposide | UW Madison Oncology | N/A |
| 5-Fluorouracil | UW Madison Oncology | N/A |
| Tunicamycin | Sigma | Cat#SML1287 |
| Thapsigargin | Sigma | Cat#T9033 |
| Temozolomide | Sigma | Cat#T2577 |
| Z-Leu-Leu-Glu-AMC | Enzo | Cat#BML-ZW9345 |
| Suc-Leu-Leu-Val-Tyr-AMC | Enzo | Cat#BML-P802 |
| Boc-Leu-Arg-Arg-AMC | Enzo | Cat#BML-BW8515 |
| DMSO | Sigma | D2650 |
| Critical Commercial Assays | ||
| MACS Neural Tissue Papain Dissociation Kit | Miltenyi Biotec | Cat#130-092-628 |
| Mouse Neural Stem Cell Nucleofector™ Kit | Lonza | Cat#VPG-1004 |
| Proteostat Aggresome Detection Kit | Enzo | Cat#51035-K100 |
| LysotrackerRED | Thermo | Cat#L7528 |
| Living Cell Assay with Calcein | Invitrogen | Cat#L3224 |
| EdU Pulse Kit | Invitrogen | Cat#C10337 |
| DuoLink Proximity Ligation Assay | Millipore Sigma | Cat#DUO92101 −1KT |
| iSCRIPT cDNA Synthesis Kit | Bio Rad | Cat#1708890 |
| SYBR Green Premix Ex Taq (Tli RNase H Plus) | Thermo | Cat#4385610 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| mNeonGreen Trap-A Linked Agarose Beads for IP | ChromoTek | Cat#nta-20 |
| Deposited Data | ||
| Vimentin KO RNA Sequencing Data | GEO | GSE136553 |
| Experimental Models: Cell Lines | ||
| WT NSCs | This paper | N/A |
| Vimentin KO NSC Clones (1, 2, 3 and control strain) | This paper | N/A |
| Vimentin-mNeon NSCs | This paper | N/A |
| HEK293FT | Gift from Dr. Subhojit Roy | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BI/6J Mice | The Jackson Laboratory | N/A |
| 129S-Vimtm1Cba/MesDmarkJ Mice | The Jackson Laboratory | N/A |
| Nestin-GFP Mice | Yamaguchi et al 2000 | N/A |
| Oligonucleotides | ||
| F Psma5: AGTAGCATTGTTGTTTGGAG | This paper | N/A |
| R Psma5: CTTCAGAGTCATAGACTTATGG | This paper | N/A |
| F 18S: GAACTGAGGCCATGATTAAGAG | This paper | N/A |
| R 18S: CATTCTTGGCAAATGCTTTC | This paper | N/A |
| F Vim: CGGCTGCGAGAGAAATTGC | This paper | N/A |
| R Vim: CCACTTTCCGTTCAAGGTCAAG | This paper | N/A |
| Nontargeting gRNA: GCGAGGTATTCGGCTCCGCG | This paper | N/A |
| Vimentin 1 gRNA: CGGGTCACATAGGCGCCACC | This paper | N/A |
| Vimentin 2 gRNA: GTGGCTCCGGCACATCGAGC | This paper | N/A |
| Vimentin 3 gRNA: GTGGCTCCGGCACATCGAGC | This paper | N/A |
| Recombinant DNA | ||
| Vimentin-mNeon Homology Directed Repair Template | This paper | N/A |
| Vimentin-mNeon gRNA | This paper | N/A |
| Vimentin KO gRNA 1 | This paper | N/A |
| Vimentin KO gRNA 2 | This paper | N/A |
| HttQ119-eBFP overexpression | This paper, HttQ119 insert from Dr. Harm Kampinqa | N/A |
| Tdp43-gfp overexpression | Addgene | Cat#28197 |
| mNeon overexpression | Gift from Dr. Erik Dent, mNeon cDNA from Allele Biotechnology & Pharmaceuticals, Inc | N/A |
| VHL-BFP overexpression | This paper, VHL insert from Addgene #21053 | N/A |
| GFP-Ubiquitin | Addgene | Cat#11928 |
| mCherry-GFP-LC3 | Gift from Dr. Ashley Webb | N/A |
| psPAX2 | Addgene | Cat#12260 |
| pCMV-VSV-G | Addgene | Cat#8454 |
| Software and Algorithms | ||
| Microsoft Excel | Microsoft | N/A |
| Prism | GraphPad | N/A |
| Adobe Illustrator | Adobe.com | N/A |
| Imaris | Oxford Instruments | N/A |
| Other | ||
| N/A | N/A | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
In this study, male and female mice between the ages of 6 weeks and 9 months were used. Nestin-GFP mice were obtained from Xinyu Zhao (UW-Madison) which were developed by Kensaku Mori’s laboratory (Yamaguchi et al., 2000). 129S-Vimtm1Cba/MesDmarkJ and C57BL/6J mice were obtained from The Jackson Laboratory. All techniques associated with animals are described in further detail in the “Method Details” section of this document. All facilities and protocols for animal work were approved by the Research Animal Resources and Compliance (RARC) at UW-Madison. Animals had ad libitum access to food and water and were kept under a 12hr light-12hr dark cycle. All procedures were performed in strict accordance with all federal and institutional policies.
Cell Lines
All cell lines (aside from HEKs discussed in the viral particle generation part of the methods) in this study were NSCs derived from ~6 week old male C57BL/6 mouse hippocampi. Cultures were grown at 5% CO2 at 37°C. Cultures were periodically tested for mycoplasma to ensure no contamination was present. More detail on specific experiments and techniques used to maintain these cell lines is listed below in the “Method Details” section.
METHOD DETAILS
NSC Dissection and Culture
NSCs were obtained by pooling cells extracted from the hippocampi of approximately 3–5 mice around 6 weeks old, similar to a previously described protocol (Moore et al., 2015). Hippocampi were dissected in cold HBSS and dissociated using the GentleMACS Dissociator (Miltenyi Biotec) and MACS Neural Tissue Papain Dissociation Kit (Miltenyi Biotec 130-092-628) using the manufacturer’s protocol with added myelin removal. NSCs were cultured as described previously (Moore et al., 2015). Activated NSCs (aNSCs) were cultured with 37°C/5% CO2 in serum-free media: DMEM/F12 GlutaMax (Invitrogen 10565018) with penicillin-streptomycin-fungizone (1:100, Invitrogen 15140122), B27 (1:50, Invitrogen 17504044), and 20 ng/mL FGF-2 and EGF (PeproTech 100–18B and AF-100–15). Cells grown as monolayers were additionally given 5ug/mL Heparin (Sigma H3149).
For imaging experiments, aNSCs were plated onto poly-L-ornithine (PLO; 10 μg/mL plastic, 50 μg/mL glass, Sigma P3655) and laminin (5 μg/mL, Sigma L2020) coated cuvettes (Fisher Scientific 12-565-337 or Ibidi 80826-G500). Cell culture dishes reaching confluency were trypsinized and split into lower densities using previously described methods (Moore et al., 2015). Briefly, single cells were spun down at 120xg for 4 minutes, resuspended in 0.05% trypsin (Invitrogen 25300–054) in Versene (Thermo Fisher 15040066) and placed at 37°C for 5 minutes. Twice the volume of trypsin inhibitor (Sigma T6522) was added for 2 minutes, cells were mechanically triturated, and then cells were spun again at 120xg for 4 minutes before being resuspended in media and plated onto new dishes.
Quiescent NSCs (qNSCs) were cultured using a previously established protocol (Mira et al., 2010, Martynoga et al., 2013, Knobloch et al., 2017) similarly to aNSCs with the exception of the removal of EGF and the addition of 50 ng/mL BMP-4 (Fisher Scientific 5020BP010). All qNSC experiments were cultured in qNSC medium on PLO- and laminin-coated wells for at least 3 days before performing quiescence experiments. For experiments observing quiescence exit, after at least 3 days of BMP-4 treatment the media was changed back to proliferation media (-BMP-4, +FGF, +EGF).
Transfections
Electroporations were performed on 4 million trypsinized NSCs resuspended in Mouse Nucleofector solution (Lonza VPG-1004) with 7 μg of Endotoxin-free DNA using the Mouse Neural Stem cell program in the Nucleofector II machine (Lonza). Electroporations were plated immediately onto glassware coated with PLO and laminin with a media change 24 hours later to remove dead cells. In overexpression experiments of varying aggregation-prone proteins, cells were imaged 24–48 hours after electroporation.
Molecular Cloning and Constructs
For design and delivery of guide RNAs (gRNAs) we used the tool at crispr.mit.edu to choose gRNA sequences, and cloned identified sequences (see KRT) into pSpCas9(BB)-2A-Puro (PX459) V2.0, (Addgene #62988) or pSpCas9(BB)-2A-GFP (PX458) (Addgene #48138) using a previously established cloning protocol for this vector (Ran et al., 2013).
The vimentin-mNeon homology directed repair template was constructed by performing a four piece Gibson assembly (NEB E5510S) to ligate homology arms corresponding to vimentin’s carboxyl terminus with a linker sequence (GGTGGTGGCGGTTCAGGCGGAGGTGGCTCTGGCGGTGGCGGATCG) and mNeon fluorophore sequence (Allele Biotechnology) into pENTR4-HaloTag w876–1 (Addgene #29644). To construct CMV-HttQ119-eBFP, eBFP was PCR amplified from CMV-eBFP-2A-H2B-eGFP (gift from Dr. Murray Blackmore) and ligated into CMV-HttQ119-eYFP (gift from Dr. Harm Kampinga). To construct CAG-BFP-VHL, a four piece Gibson assembly was performed ligating eBFP from CMV-eBFP-2A-H2B-eGFP, VHL-WPRE from pESC-LEU-GFP-VHL (Addgene #21053) and a linker between VHL and BFP into a CAG-GFP-PRE vector (Kaganovich et al., 2008). Tdp43-eGFP (Addgene #28197) and GFP-Ubiquitin (Addgene #11928) were obtained from Addgene (Yang et al., 2010, Dantuma et al., 2006). The CAG-mNeon vector was a gift from Dr. Erik Dent.
Generation of CRISPR Mutant Model Cell Lines
CRISPR/Cas9-based mutant NSCs harboring vimentin-mNeon were generated by electroporating 4 million NSCs with the construct pSpCas9(GAATAAAAATTGCACACACT)-2A-Puro (PX459) V2.0 harboring an expression cassette for a gRNA targeting vimentin’s carboxyl terminus and Cas9 (3.5 μg), as well as pENTR-vimentin-mNeon harboring a template for repairing vimentin’s endogenous locus with the insertion of a linker peptide sequence and an mNeon fluorophore (3.5 μg) as described in the transfection section of the methods. Seven to ten days after electroporation, a pool of vimentin-mNeon NSCs were isolated through fluorescence-activated cell sorting (FACS) using a BD FACSAria high speed cell sorter. CRISPR/Cas9-based vimentin KO NSCs were generated by electroporating NSCs with two gRNAs (CGGGTCACATAGGCGCCACC and GTGGCTCCGGCACATCGAGC) targeting vimentin’s amino terminus in the backbone. NSCs were single cell sorted using a BD FACSAria high speed cell sorter into a plastic flat-bottomed 96 well plate coated with PLO and laminin, and allowed to expand for approximately one month. Approximately 20.83% of these clones survived, and were initially screened for vimentin protein expression by immunostaining for vimentin on a duplicated plate. Clones with a lack of vimentin filaments by immunostaining were further validated for vimentin KO by a western blot. Alongside vimentin KO NSCs, a control strain was generated by treating NSCs with a non-targeting (NT) gRNA (GCGAGGTATTCGGCTCCGCG) that were similarly sorted and expanded. All experiments with vimentin KO and NT NSCs are examining a pooling of 3 full vimentin KO or NT clones.
Microscopy/Live Cell Imaging
All images displayed in this study were taken using either a Nikon C2 confocal microscope or a Zeiss widefield epifluorescent microscope. Live cell time-lapses were performed on the Nikon C2 confocal with humidity, CO2 and temperature control. Typically, imaging of NSCs involved 1 μm step sizes with total stack size ranging from 10–20 μm. Live cell time-lapses typically had images collected every 8 minutes to ensure no divisions were missed unless otherwise noted.
Differentiation Assays
For assessing the ability of NSCs to differentiate in different conditions, aNSCs were plated at a seeding density of 50,000 per one well on a 12 well cuvette (Fisher Scientific 12-565-337). 24 hours later FGF, EGF and heparin were removed from the media and cells were fed with this differentiation media every other day for two weeks. After two weeks, cells were fixed and stained for MAP2ab (neurons) and GFAP (astrocytes). A blinded observer counted the total number of MAP2ab or GFAP positive cells in 10 images of three technical replicates per condition. The experiment was repeated three times on three different days.
Motility Assays
To determine if there were any differences in the migratory behaviors of NSCs in any conditions listed in the text, we performed live cell imaging of NSCs in different conditions acquiring a single image every 5 minutes and used the “manual track” plug-in in FIJI to assess average velocity and total distance traveled over the course of the timelapse. At least 30 cells were traced for each condition for as long as possible before the timelapse ended or the cells were lost. The experiment was repeated three times on three different days.
Proteasome Activity Assays
To measure proteasome activity in protein lysates of NSCs in different conditions we adapted a protocol from Vilchez et al (Vilchez et al., 2012). Cells were cultured as described in the text and then pelleted. Immediately cells were resuspended in proteasome activity buffer (50mM Tris-HCl, pH 7.5, 250mM sucrose, 5mM MgCl2, 0.5mM EDTA and 1mM dithiothreitol) and lysed by passing through a 27-gauge needle ten times. Lysates were clarified by centrifugation at 15,000xg for 10 minutes at 4°C. Protein concentration was quantified using the DC protein assay (Bio-Rad 5000112). 15–25 μg of protein were loaded into wells in a flat bottom 96 well plate for the assay. Each well received the same amount of protein, 2 mM adenosine triphosphate (Fisher ICN15026605), 0.37 mM proteasome substrate (caspase – Z-Leu-Leu-Glu-AMC (Enzo BML-ZW9345), chymotrypsin – Suc-Leu-Leu-Val-Tyr-AMC (Enzo BML-P802), trypsin – Boc-Leu-Arg-Arg-AMC (Enzo BML-BW8515)), and then diluted to a final volume of 100 μL per well with proteasome activity buffer. Plates were immediately placed in a plate reader set to excite at 380 nm and collect 460 nm, taking a datapoint every 5 minutes for 1 hour at 37°C. Three 96-well reactions were averaged for each technical replicate, repeated on three separate days. For each substrate that was loaded in the plate a reaction was set up with no protein and subtracted from each experimental well.
Detecting vimentin cage formation in response to treatment with different cellular stressors
To determine doses of drugs used to stress NSCs, a dose response curve was utilized to determine lethal doses and timings with each drug. Cells were treated with MG132 (Sigma M7449), chloroquine (Sigma C6628), etoposide (from UW-Madison oncology department), thapsigargin (Sigma), 5-fluorouracil (from UW-Madison oncology department), and tunicamycin (Sigma) as indicated in the text. Cells were heat shocked at 45°C for 20 minutes. All experiments involving stressed cells were performed on monolayers plated on PLO and laminin at a starting density of 50,000 cells per well of a 4 well cuvette (Fisher Scientific 12-565-337). NSCs in each condition were imaged in their entirety with a 1 μm step size – 10 images per condition with three technical replicates per condition. For quantitating the presence of vimentin cages in response to cellular stress we defined a cell as having a vimentin cage if >50% of its vimentin was localized to the nuclear bay in a tight cage-like conformation. Cages were counted manually by a blinded observer that was instructed on this definition analyzing data from 3 experiments completed on three different days.
Quantitating asymmetry in vimentin-mNeon NSCs based on prior cage formation in interphase
To assess asymmetry of vimentin during mitosis in the presence or absence of a vimentin cage being formed in interphase, we performed live cell imaging of vimentin-mNeon NSCs either in untreated conditions, or 12 hours after a heat shock, taking an image every 8 minutes with 1 μm step size. Divisions were categorized into two categories based on if the NSCs (1) had a cage or (2) did not have a cage in interphase before mitosis occurred. For each of these two bins, asymmetry was quantified by drawing a region of interest (ROI) around each daughter cell in late anaphase and placing the inheritance of the daughter with more vimentin normalized to area over the sum of the total vimentin in the division normalized to area to create an asymmetry ratio.
Proteostat Staining
For assays involving labelling with the dye Proteostat (Enzo 51035-K100), NSCs were fixed with 4% paraformaldehyde for 15 minutes at room temperature. NSCs were permeabilized with 0.5% Triton and 3 mM EDTA in PBS for 30 minutes at room temperature, and then stained with Proteostat 1:10,000 in PBS for 30 minutes at room temperature. Proteostat was washed twice with PBS for 10 minutes at room temperature. To quantify changes in Proteostat labeling across different treatments, we instructed a blinded observer to count the number of puncta present in each individual cell. For each condition, 10 images of at least three technical replicates were quantified.
Lysotracker Staining
For labeling NSC lysosomes with the dye Lysotracker, we incubated NSCs in medium with 1:1000 LysotrackerRED (Thermo L7528) for 20 minutes with 2 μM Hoechst at 37°C. Cells were washed twice with warm media before imaging immediately. For analysis of Lysotracker staining in different conditions, raw integrated density was measured in a region of interest (ROI) drawn by a blinded observer around each cell using Fiji. Either 10 images of at least 3 replicates were fully counted or at least 300 cells per replicate were analyzed. Each Lysotracker experiment was repeated at least 3 times.
Calcein Live Cell Staining
Calcein AM (Invitrogen L3224) was diluted 1:1,000 with 2 μM Hoechst in NSC media and applied to living cells for 15 minutes at 37°C. Cells were immediately imaged after 15 minutes. For quantification of calcein, 10 images were taken randomly throughout each well of each of 3 technical replicates. For every image in each condition, calcein positive cells were counted by a blinded observer. The experiment was repeated 3 times. Graphs depict 3 technical replicates from one repeat that are representative of the observations of the other replicates completed on different days.
mNeon co-immunoprecipitation/Mass spectrometry
20 million vimentin-mNeonGreen NSCs that were (1) treated with 0.05% DMSO for 4 hours, (2) treated with 5 μM MG132 for 4 hours, or (3) WT NSCs overexpressing mNeon for 72 hours and treated with 0.05% DMSO for 4 hours were pelleted, and resuspended in lysis buffer (10 mM TrisHCl, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40) for 30 minutes. Lysates were incubated with mNeonGreen TRAP-A Linked Agarose Beads (Chromotek, nta-20) for 1 hour. Beads were subsequently pelleted and washed with dilution/wash buffer (10 mM TrisHCl, 150 mM NaCl, 0.5 mM EDTA). Bound proteins were eluted by incubation with 0.2M glycine pH 2.5 followed by neutralization with 1M Tris pH 10.4. Protein samples were reduced with 10 mM DTT for 30 minutes at 37°C, alkylated in the dark with 55 mM indole-3-acetic acid for 45 minutes at 37°C, and digested overnight at 37°C with two 0.5 μg Sequencing Grade Trypsin (Promega PRV5111) additions throughout the incubation. The digest reaction was stopped by acidification.
For LC/MS/MS, digested samples were subsequently dried on a speed vac, and processed with ZipTip (Millipore ZTC18S096) according to the manufacturer’s protocol. Desalted peptides were dried under nitrogen, and resuspended in Buffer A (0.1% formic acid/water) and Buffer B (0.1% formic acid in acetonitrile). Samples were transferred to a Total Recovery Vial (Waters 186005663CV) and run on a 120 minute increasing AcN gradient, using top 10 MS/MS Q Exactive methods with 5 second dynamic exclusion enabled. A blank injection was run in-between samples to minimize carryover. Protein identification, quantification, and analysis were done with Proteome Discoverer 1.4/2.2, SEQUEST HT, and MASCOT using Uniprot mus_musculus protein database. A decoy database was added to each search to establish control variability and false discovery rates.
For analysis of the LC/MS/MS data, genes appearing at least once in 3 technical replicates were pooled together to make one list of genes for each experimental condition. Genes appearing in any of the three control IP conditions (mNeon overexpression) were removed from the list of genes that were pulled down by vimentin in experimental conditions. All genes appearing in any of each replicate of vimentin-mNeon MG132 treated cells were put into String and used to generate gene ontology maps (Szklarczyk et al., 2015).
Immunostaining
Immunostaining for vimentin (1:1000, Sigma Aldrich AB5733), α5 subunit of the proteasome (1:500, Abcam ab189855), phosphohistone H3 (1:500, Abcam 14955), K48pUb (1:500, Millipore Sigma 05–1307-AF488) and Ki67 (1:500, Abcam ab15580) was performed using the following protocol. Cells were fixed in 4% PFA for 15 minutes at room temperature and then permeabilized in 0.25% triton in PBS for 30 minutes at room temperature. Cells were blocked with 20% donkey serum (Millipore Sigma S30) in antibody buffer (150mM sodium chloride, 50mM tris base, 1% bovine serum albumin, 100mM L-lysine, 0.04% sodium azide, pH 7.4) for 1 hour at room temperature (Moore et al., 2009). Primary antibodies were incubated in antibody buffer overnight at 4°C. The following day cells were washed with PBS three times, 10 minutes per wash, at room temperature. Secondary antibodies were diluted 1:500 in antibody buffer and incubated on cells for 1.5 hours at room temperature. Cells were washed with PBS three times, 10 minutes per wash, at room temperature. To stain nuclei, either Hoechst (2 μM, Thermo 62249) or DAPI (2 μM, Invitrogen D1306) diluted in TBS, were added during one of the wash steps following secondary antibody incubation for 10 minutes.
Immunostaining for MAP2ab (1:500, Sigma M2320), GFAP (1:500, Dako Z0334), nestin (1:500, Thermo MA1110), and pericentrin (Abcam ab4448) were performed using the following protocol. Cells were fixed in 4% PFA for 15 minutes at room temperature and then permeabilized and blocked in 0.25% triton and 3% donkey serum (Millipore Sigma S30) in TBS (TBS++) for 30 minutes at room temperature. Cells were incubated in TBS++ with primary antibody diluted 1:100 overnight at 4°C. The following day cells were washed in TBS three times, 5 minutes per wash, at room temperature, followed by TBS++ for 15 minutes at room temperature. Secondary antibodies were diluted 1:500 in TBS++ and incubated with cells for 1 hour at room temperature. Cells were washed with TBS three times, 10 minutes per wash, at room temperature.
For immunostaining free-floating brain slices with Doublecortin (1:250, Cell Signaling Technology 4604S), SOX2 (1:250, Santa Cruz sc-17320) and S100B (1:250, Abcam ab52642), slices were washed 3 times for 5 minutes with TBS in net wells at room temperature, and blocked for 30 minutes at room temperature in TBS++ while shaking. Sections were then incubated overnight in primary antibodies diluted in TBS++ while shaking without nets. The next day, sections were washed 2x for 10 minutes with TBS in net wells while shaking, and incubated for 1 hour 30 min in secondary antibodies (1:250) diluted in TBS++ shaking without nets. After secondary incubation, sections were washed 2x for 5 minutes, incubated with DAPI (1:5000 in TBS) for 5 minutes shaking at room temperature, washed 2x for 5 minutes, and mounted on electrostatically treated slides.
For immunostaining brain slices with GFP (1:250, Abcam ab5450), vimentin (1:250, Sigma-Aldrich AB5733) and Ki67 (1:250, Abcam ab15580), 40 μm cryosections from perfused male and female 7 week old Nestin GFP mice were prepared. Sections were fixed for 15 minutes at room temperature in 4% PFA and then blocked in TBS++ for 30 minutes at room temperature. Sections were then incubated overnight in primary antibodies diluted in TBS++. The next day, sections were washed 2x for 10 minutes with TBS, and incubated for 1 hour 30 min in secondary antibodies (1:250) diluted in TBS++. After secondary incubation, sections were washed 2x for 5 minutes, incubated with DAPI (1:5000 in TBS) for 5 minutes.
In vitro EdU pulses
Cells in S-phase were labeled by a 1 hour incubation with 10 μm EdU (Invitrogen C10337) at 37°C. NSCs were fixed in 4% PFA for 15 minutes at room temperature. Visualization of EdU was achieved using a Click-it kit (Invitrogen C10337) for performing click chemistry as described in the manufacturer’s protocol. For quantification of all EdU pulses, either 10 images of at least 3 replicates were fully counted or at least 300 cells per replicate were counted by a blinded observer. Each EdU experiment was repeated at least 3 times.
Proximity Ligation Assay
To perform the proximity ligation assay we used Sigma’s DuoLink Proximity Ligation assay kit (Millipore Sigma DUO92101–1KT). Cells were fixed and stained as described in the immunostaining section of the methods for vimentin and proteasomes through the primary incubation step, this time using a mouse vimentin antibody (Abcam ab20346; 1:1,000). After overnight incubation with primary antibodies, NSCs were washed with buffer A from the kit twice for 5 minutes at room temperature. PLUS and MINUS probes were diluted in antibody buffer (see immunostaining section of methods) 1:5 and then applied to NSCs for 1 hour at 37°C. NSCs were washed with buffer A from the kit twice for 5 minutes at room temperature. 1xDuoLink Ligation Buffer (diluted in water) and ligase diluted 1:40 were applied to NSCs for 30 minutes at 37°C. NSCs were washed with buffer A from the kit twice for 5 minutes at room temperature. 1x DuoLink Amplification Buffer (diluted in water) with polymerase diluted 1:80 was applied to NSCs for 100 minutes at 37°C. NSCs were washed with buffer B from the kit twice for 10 minutes at room temperature and stained with Hoechst as described in the immunostaining section of the methods.
Western blot
Soluble protein fraction preparations: Frozen cell pellets were thawed on ice and resuspended in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1mM ethylenediaminetetraacetic acid (EDTA), 1% NP-40, 50 mM Tris pH 7.6). The resuspended samples were spun in a tube rotator (VWR) at 18 rpm for 20 minutes at 4°C. Spun samples were sonicated for 1 minute at room temperature using an ultrasonic bath (Branson) set at 5 degas/minute. Centrifugation was performed at 15,000xg (Beckman Coulter) for 15 minutes at 4°C. Following centrifugation, supernatants were collected and protein concentrations of the cell lysates were measured using the DC Protein Assay (Bio-Rad 5000112) and a BioMate 3 Spectrophotometer (Thermo Fisher Scientific) at 750 nm.
Total (insoluble and soluble – for polyubiquitin blots) protein fraction preparations: To prepare total protein fractionation lysates we adapted the protocol used by Leeman et al (21). NSC pellets were resuspended in 150 mM sodium chloride (Sigma S9625), 4% SDS, 50 mM N-ethylmaleimide (Invitrogen 23030), 25mM TCEP (Invitrogen 77720) (Total fraction buffer –TFB) and lysed by passing through a 27 gauge needle ten times. Cells were sonicated at 40 kHz for 5 minutes in a water bath sonicator (Branson 2510 Ultrasonic Cleaner). Resulting lysate was spun down at 15,000xg for 10 minutes at 4°C. Protein concentrations were determined using the DC protein assay (Bio-Rad 5000112). 20–30 μg of sample was loaded into each well for polyacrylamide gel electrophoresis.
Lysates were heated for 10 minutes at 90°C, loaded at 30 μg and run on 4–20% SDS-PAGE gels (Bio-Rad) for 45mins at 150V. Proteins were transferred onto PVDF membranes (Bio-Rad) at 100V for 90 minutes at 4°C. 5% milk in 0.25% triton and 3% donkey serum (Millipore Sigma S30) (TBS-T) was used to block the membranes unless otherwise noted. To detect the presence of specific proteins, the membranes were incubated overnight (rotating at 4°C) in the appropriate primary antibodies diluted per manufacturer recommendations in either 5% Milk in TBST unless otherwise noted – see antibodies below. The membranes were washed 3x (10 minutes each) in 1x TBS-T at room temperature. Blots were then incubated at room temperature in block solution with secondary antibodies listed below at room temperature for 90 minutes. Membranes were again washed three times for 10 minutes with TBS-T before probing the membrane with a SuperSignal femto (Thermo 34095) or pico (Thermo 34577) kit. Primary antibodies were used as follows: β-actin (Bio-rad VMA00048 – 1:1,000), vimentin (Cell signaling 5741S – 1:1,000), polyubiquitin/FK1 (Enzo BML-PW8805 – 1:1,000 in BSA), LC3B (Abcam ab48394 – 1:1,000). Polyubiquitin western blots required a 5% BSA block instead of milk in all steps. Secondary antibodies conjugated to horseradish peroxidase (HRP) used were α-chicken (Promega G1351 – 1:1,000), α-rabbit (Bio-Rad 1706515 – 1:3,000), α-mouse (Bio-Rad 1706516 – 1:3,000). Images were taken using a UVP Imaging system (UVP) to detect protein bands at exposures set so that bands were below saturation.
To quantify western blot protein bands, a region of interest was drawn around each correctly sized band in Fiji to measure raw integrated density. Each integrated density was normalized to each sample’s β-actin control and then had background subtracted before comparing across conditions. Graphs display the average of three representative technical replicates. All experiments were repeated at least three times on three different days.
mCherry-GFP-LC3 Experiments
HEK293FT cells were cultured in DMEM, high glucose, GlutaMAX Supplement, pyruvate (Thermo 10569044) supplemented with 10% FBS (Invitrogen 16000044), 1X MEM Non-Essential Amino Acids (Thermo 11140050) and 600 μM L-glutamine (Thermo 25030081) at 5% CO2 and 37°C. For viral particle generation, HEK293FT cells were transfected with pCMV-VSV-G (Addgene #8454), psPAX2 (Addgene #12260) and FUW mCherry-GFP-LC3 (Addgene #110060) using polyethylenimine (PEI; Polysciences, Inc. 23966–1) (Joung et al., 2017). Viral particles were harvested 48 hours post transfection and purified by ultracentrifugation as previously described (Moore et al., 2015).
WT and vimentin KO NSCs were transduced with LC3-GFP-mCherry at a similar multiplicity of infection (MOI), and allowed to express virus for at least 7 days before performing any experiments. For experiments, 100,000 WT or vimentin KO NSCs in triplicates were treated with either 0.02% DMSO or 2 μM MG132 for 24 hours and then had drug removed for 1 hour before trypsinizing and analyzing GFP and mCherry fluorescence using a BD LSRII flow cytometer. The experiment was repeated 3 times and the data in this manuscript depicts one representative experiment.
Quantitative real time polymerase chain reaction (qRT-PCR)
Total RNA for all conditions was collected using an RNeasy Mini Kit (Qiagen). cDNA was synthesized from isolated RNA with iScriptTM cDNA Synthesis Kit (BioRad). Real time PCR primers sequences from Sigma’s KiCqStart® SYBR® Green Predesigned Primers were obtained from Sigma against Psma5, vimentin, and 18S (see KRT). qRT-PCR and data analysis were performed on the DNA Engine Opticon 2 System (BioRad). The 20 μl amplification mixture consisted of 2X SYBR Green Premix Ex Taq (Tli RNase H Plus) (Takara Bio), 0.2μM forward and reverse primers, and 4μl of cDNA generated from 1 μg of RNA. Thermal cycling conditions were as follows: 95°C for 1 min; 40 cycles of 94°C for 10s, 60°C for 30s, varying plate read temperatures for 1s; 68°C for 5 min; melting curve from 65°C to 95°C every 0.2°C for 1 sec each; 72°C for 5 min. Plate read temperatures were: 79°C for vimentin, and 81°C for 18S. qRT-PCR products were assessed via melt curve and gel electrophoresis analysis. All reactions were run in triplicate. All data was normalized to 18S RNA levels. Fold changes were calculated using the ΔΔCt method (Livak and Schmittgen, 2001).
Quiescent exit timelapse quantifications
To observe vimentin morphology, expression, presence of vimentin cages during mitosis and asymmetry during quiescence and quiescence activation, we plated vimentin-mNeon NSCs at a seeding density of 40,000 NSCs per well of a 4-well cuvette (Fisher Scientific 12-565-337) in aNSC media. 24 hours later quiescence induction began with addition of qNSC media. After 3 days in quiescence media, NSCs were allowed to reactivate by switching back to aNSC media. Quantifications of asymmetry, kinetics of vimentin cage formation and degradation and prevalence of vimentin cage formation were performed by analysis of at least 4 movies of vimentin-mNeon NSCs in either aNSC media or during quiescence exit. To image aNSC cultures to compare to NSCs exiting quiescence, aNSCs were plated in aNSC medium at densities of 40,000 NSCs per well of a 4-well cuvette (Fisher Scientific 12-565-337) and allowed to sit on to laminin for 24 hours before imaging began. Data displayed were pooled from 4 timelapse experiments.
Quiescent exit proteostasis assays
To observe the efficacy of NSC quiescence exit in a variety of conditions, we seeded aNSCs at 10,000 NSCs per well of a 4-well cuvette (Fisher Scientific 12-565-337). Twenty-four hours later qNSC media was added. Three days after qNSC media was first added, cells were switched back to aNSC media. Forty-eight hours into reactivation after the reintroduction of aNSC media, cells were analyzed by the assays indicated in the main text. Each condition/experiment was analyzed by averaging at least 3 technical replicates and repeating the experiment at least 3 times.
Quiescent exit quantification of vimentin, proteasome and K48pUb asymmetry
50,000 vimentin-mNeon NSCs or vimentin KO NSCs were induced into a state of quiescence in an 8-well cuvette (Ibidi 80826) using BMP-4 and then allowed to exit quiescence. During quiescence exit, when vimentin-mNeon expression came up and asymmetry was observed, cells were fixed and immunostained for K48pUb or proteasomes and then asymmetry was quantitated by measuring the raw integrated density (RID) and area of each daughter cells and then normalizing RID/area and comparing each daughter’s inheritance of each cargo respectively. Cell divisions collected represent data from three experiments completed on three different days.
Quantification of K48pUb and Proteostat overlap
WT NSCs were treated with 5 μM MG132 for 4 hours and then were fixed and immunostained for K48pUb and stained for Proteostat as described in the immunostaining and Proteostat staining sections of the methods. Cells were imaged and then loaded into Imaris, where a colocalization channel was built, thresholded to only analyze punctate signal from Proteostat or K48pUb. For each cell, the percentage of Proteostat signal that colocalized with K48pUb was quantified for Proteostat puncta that were either 1) in the aggresome within the vimentin cage or 2) in the cytosol outside the vimentin cage.
Quantification of vimentin cage and Proteostat overlap
WT NSCs were treated with 5 μM MG132 for 4 hours and then were fixed and immunostained for vimentin and stained for Proteostat as described in the immunostaining and Proteostat staining sections of the methods. Cells were imaged and then loaded into Imaris, where a mask was constructed around the vimentin cage and around the cell body. Proteostat puncta for each cell were then binned into two categories: 1) Proteostat puncta that were touching or within the vimentin cage mask, or 2) Proteostat puncta that were within the cytosol but not touching or within the vimentin cage mask. A percentage was calculated and reported for the number of cells including at least one Proteostat puncta either at or within the vimentin cage. The intensity histogram was generated using Elements.
RNA sequencing
For total RNA sequencing of vimentin KO or WT NSCs, three pellets of each cell type were spun down at the same time and sent to LC Sciences for total RNA sequencing. Cutadapt (Martin, 2011) and perl scripts in house were used to remove the reads that contained adaptor contamination, low quality bases and undetermined bases. Then sequence quality was verified using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). We used Bowtie2 (Langmead and Salzberg, 2012) and Tophat2 (Kim et al., 2013) to map reads to the genome of Mus musculus (Version: v88). The mapped reads of each sample were assembled using StringTie (Pertea et al., 2015). Then, all transcriptomes from 6 samples were merged to reconstruct a comprehensive transcriptome using perl scripts and gffcompare (https://github.com/gpertea/gffcompare/). After the final transcriptome was generated, StringTie (Pertea et al., 2015) and Ballgown (Frazee et al., 2015) was used to estimate the expression levels of all transcripts.
StringTie (Pertea et al., 2015) was used to perform expression level for mRNAs and lncRNAs by calculating FPKM (FPKM=[total_exon_fragments/mapped_reads(millions)×exon_length(kB)]). The differentially expressed mRNAs and lncRNAs were selected with log2 (fold change) >1 or log2 (fold change) <−1 and with parametric F-test comparing nested linear models (p value < 0.05) by R package Ballgown (Frazee et al., 2015).
Animals
Male C57BL/6J mice were purchased from The Jackson Laboratory and used for isolating NSCs in in vitro experiments and generating in vitro vimentin KO NSCs. 7 week-old, 5 month-old, and 9 month-old 129S-Vimtm1Cba/MesDmarkJ (vimentin KO) mice from The Jackson Laboratory were used for in vivo vimentin KO experiments. In short, heterozygote mice carrying the mutated allele were intercrossed to produce experimental KO progeny and WT littermate controls. All intercrosses were maintained within the same familial generation. These mice were then genotyped according the strain specific master protocol provided by The Jackson Laboratory (Strain number 025692) through digesting tissue from ear punches, extracting genomic DNA and running a polymerase chain reaction (PCR). Nestin-GFP mice were obtained from Dr. Xinyu Zhao’s laboratory (Yamaguchi et al., 2000).
Mice used in in vivo experiments were transcardially perfused with 0.9% saline solution followed by 4% paraformaldehyde (PFA)/0.2M phosphate buffer. Brains were removed, postfixed overnight in 4% PFA, and subsequently stored in 30% sucrose/PBS solution. These brains were then either sectioned into 40 μm thick sections using a sliding microtome and stored in CPS (25% ethylene glycol, 25% glycerol, 0.05M phosphate buffer) or sectioned on a cryostat 40 μm, immediately mounted and then frozen at −80°C. Free-floating sections were immunostained as described above. For analysis, we immunostained, imaged and quantified 2 of 12 total prepared series of slices (10 slices) per staining pair and then calculated the number of cells per dentate gyrus. Due to vimentin’s disruption of the GFAP and nestin network, we used S100B as a marker for astrocytes in the subgranular zone of the dentate gyrus.
In vivo early aging study paradigm
In the C57BL/6J mouse strain, neurogenesis is observed to decrease to minimal levels by 7 months (Ben Abdallah et al., 2010). Due to even lower levels of neurogenesis in the Vimtm1Cba/MesDmarkJ (vimentin KO) strain (base strain 129S2/SvPas), we used 5 months and 9 months as our early aging timepoints (Colucci-Guyon et al., 1994). EdU (Life Technologies A10044) was dissolved in 0.9% saline solution at 5 mg/mL. For the aging study, animals received four intraperitoneal (i.p.) injections of EdU (50 mg/kg) once per day for 4 days. On the fifth day (one day after the last EdU injection), the mice were perfused. All injections were administered at the same time each day.
In vivo temozolomide study paradigm
Temozolomide (TMZ) experiments were adapted from previously established protocols (Knobloch et al., 2014, Garthe et al., 2009). Stock solutions of TMZ (Sigma T2577) were dissolved fresh in DMSO at 25 mg/mL, and further diluted to 5 mg/mL in PBS the day of injections.
No recovery paradigm:
Animals received 3 i.p. injections of TMZ (25 mg/kg) or were control injected with DMSO/PBS once per day for 3 days. Two hours following the last injection on the third day, mice received a single injection of EdU (50 mg/kg). Three hours after the last EdU injection, mice were sacrificed and perfused.
Three day treatment, three day recovery paradigm:
Animals received 3 i.p. injections of TMZ (25 mg/kg) or were control injected with DMSO/PBS once per day for 3 days, and then left to recover for 3 days. On the third and final day of recovery, animals were injected with a single dose of EdU (50 mg/kg) every 3 hours, 3 times. Three hours after the last EdU injection, mice were sacrificed and perfused.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed either in Microsoft Excel or in GraphPad Prism using tests as indicated in the figure legends. First, all data were tested for normal distribution using the Shapiro-Wilk normality test. Data sets with a normal distribution underwent parametric tests for significance, whereas data sets without a normal distribution underwent nonparametric tests for significance. When comparing two groups with normally distributed data to each other, an unpaired Student’s t-test was utilized. When comparing two groups with non-normally distributed data to each other, a Mann-Whitney test was utilized. When comparing more than two groups with normally distributed data, a two-way ANOVA was used followed by a post hoc Tukey’s test for group comparisons. When comparing more than two groups with non-normally distributed data, a Kruskal-Wallis test was used followed by a post hoc Dunn’s test for group comparisons. Results were found significant if p < 0.05. For cell culture experiments, “n” is equal to the number of cells analyzed. For cell culture experiments, “N” represents the number of times the experiment was repeated on different days with at least three technical replicates. For animal experiments, “n” refers to the number of animals used.
DATA AND CODE AVAILABILITY
RNA sequencing data is available via GEO: GSE136553. Processed mass spectrometry data and RNA sequencing data are available in this article’s supplemental tables.
Supplementary Material
Table S1. Intermediate filament expression in WT and vimentin KO NSCs. Related to Figure 2.
Table S2. Proteins co-precipitated by vimentin-mNeon in either a DMSO or MG132 treated condition, followed by LC-MS/MS (attached as excel file). Related to Figure 2.
Table S3. Total RNA sequencing data from untreated WT and vimentin KO NSCs (attached as excel file). Related to Figure 2.
Highlights.
Mammalian NSCs utilize vimentin-caged aggresomes to recover proteostasis
Vimentin is required for proteasome localization to the aggresome in NSCs
Quiescent NSCs upregulate vimentin protein and utilize aggresomes during activation
Vimentin knockout in NSCs impairs proteostasis recovery and delays quiescence exit
Acknowledgments:
We thank S. Jessberger, M. Knobloch, and X. Zhao for comments on the manuscript; the UW-Madison flow cytometry core (P30 CA014520 and 1S10RR025483-01); The UW-Madison Analytical Instrumentation Center for performing the LC-MS/MS; X. Zhao for Nestin-GFP mice; A. Roopra for assistance with analysis; R. Taylor and E. Dent for their mNeon plasmid; M. Blackmore for the BFP plasmid; A. Webb for the mCherry-GFP-LC3 plasmid; H. Kampinga for the Htt plasmid; members of the Moore lab and UW-Madison community for their input. We thank our funding sources: NIH T32 T32GM008688 (to C.S.M.), SciMed Graduate Research Fellowship (to T.J.P.), NIH T32 T32AG000213 (to T.J.P.), AFAR Young Investigator Award (to D.L.M.), the Sloan Foundation Fellowship (to D.L.M.), and a DP2 NIH New Innovator Award (to D.L.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
References
- AUDESSE AJ, DHAKAL S, HASSELL LA, GARDELL Z, NEMTSOVA Y & WEBB AE 2019. FOXO3 directly regulates an autophagy network to functionally regulate proteostasis in adult neural stem cells. PLoS Genet, 15, e1008097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEN ABDALLAH NMB, SLOMIANKA L, VYSSOTSKI AL & LIPP HP 2010. Early age-related changes in adult hippocampal neurogenesis in C57 mice. Neurobiology of Aging, 31, 151–161. [DOI] [PubMed] [Google Scholar]
- BENNETT EJ, BENCE NF, JAYAKUMAR R & KOPITO RR 2005. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell, 17, 351–65. [DOI] [PubMed] [Google Scholar]
- BISKOU O, CASANOVA V, HOOPER KM, KEMP S, WRIGHT GP, SATSANGI J, BARLOW PG & STEVENS C 2019. The type III intermediate filament vimentin regulates organelle distribution and modulates autophagy. PLoS One, 14, e0209665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUFALINO MR, DEVEALE B & VAN DER KOOY D 2013. The asymmetric segregation of damaged proteins is stem cell-type dependent. J Cell Biol, 201, 523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN M, PUSCHMANN TB, MARASEK P, INAGAKI M, PEKNA M, WILHELMSSON U & PEKNY M 2018. Increased Neuronal Differentiation of Neural Progenitor Cells Derived from Phosphovimentin-Deficient Mice. Mol Neurobiol, 55, 5478–5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG F, SHEN Y, MOHANASUNDARAM P, LINDSTROM M, IVASKA J, NY T & ERIKSSON JE 2016. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-beta-Slug signaling. Proc Natl Acad Sci U S A, 113, E4320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHO RH, SIEBURG HB & MULLER-SIEBURG CE 2008. A new mechanism for the aging of hematopoietic stem cells: aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood, 111, 5553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARK PJ, KOHMAN RA, MILLER DS, BHATTACHARYA TK, BRZEZINSKA WJ & RHODES JS 2011. Genetic influences on exercise-induced adult hippocampal neurogenesis across 12 divergent mouse strains. Genes Brain Behav, 10, 345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLUCCI-GUYON E, PORTIER MM, DUNIA I, PAULIN D, POURNIN S & BABINET C 1994. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell, 79, 679–94. [DOI] [PubMed] [Google Scholar]
- DANTUMA NP, GROOTHUIS TA, SALOMONS FA & NEEFJES J 2006. A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol, 173, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FABUNMI RP, WIGLEY WC, THOMAS PJ & DEMARTINO GN 2000. Activity and regulation of the centrosome-associated proteasome. J Biol Chem, 275, 409–13. [DOI] [PubMed] [Google Scholar]
- FRAZEE AC, PERTEA G, JAFFE AE, LANGMEAD B, SALZBERG SL & LEEK JT 2015. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol, 33, 243–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALOU M, COLUCCI-GUYON E, ENSERGUEIX D, RIDET JL, GIMENEZ Y RIBOTTA M, PRIVAT A, BABINET C & DUPOUEY P 1996. Disrupted glial fibrillary acidic protein network in astrocytes from vimentin knockout mice. J Cell Biol, 133, 853–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARTHE A, BEHR J & KEMPERMANN G 2009. Adult-generated hippocampal neurons allow the flexible use of spatially precise learning strategies. PLoS One, 4, e5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONCALVES JT, SCHAFER ST & GAGE FH 2016. Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell, 167, 897–914. [DOI] [PubMed] [Google Scholar]
- HAO R, NANDURI P, RAO Y, PANICHELLI RS, ITO A, YOSHIDA M & YAO TP 2013. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol Cell, 51, 819–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSTON JA, WARD CL & KOPITO RR 1998. Aggresomes: a cellular response to misfolded proteins. J Cell Biol, 143, 1883–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOUNG J, KONERMANN S, GOOTENBERG JS, ABUDAYYEH OO, PLATT RJ, BRIGHAM MD, SANJANA NE & ZHANG F 2017. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc, 12, 828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAGANOVICH D, KOPITO R & FRYDMAN J 2008. Misfolded proteins partition between two distinct quality control compartments. Nature, 454, 1088–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KALAMAKIS G, BRUNE D, RAVICHANDRAN S, BOLZ J, FAN W, ZIEBELL F, STIEHL T, CATALA-MARTINEZ F, KUPKE J, ZHAO S, LLORENS-BOBADILLA E, BAUER K, LIMPERT S, BERGER B, CHRISTEN U, SCHMEZER P, MALLM JP, BERNINGER B, ANDERS S, DEL SOL A, MARCINIAK-CZOCHRA A & MARTIN-VILLALBA A 2019. Quiescence Modulates Stem Cell Maintenance and Regenerative Capacity in the Aging Brain. Cell, 176, 1407–1419 e14. [DOI] [PubMed] [Google Scholar]
- KAWAGUCHI Y, KOVACS JJ, MCLAURIN A, VANCE JM, ITO A & YAO TP 2003. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell, 115, 727–738. [DOI] [PubMed] [Google Scholar]
- KIM D, PERTEA G, TRAPNELL C, PIMENTEL H, KELLEY R & SALZBERG SL 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOBLOCH M, PILZ GA, GHESQUIERE B, KOVACS WJ, WEGLEITER T, MOORE DL, HRUZOVA M, ZAMBONI N, CARMELIET P & JESSBERGER S 2017. A Fatty Acid Oxidation-Dependent Metabolic Shift Regulates Adult Neural Stem Cell Activity. Cell Rep, 20, 2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOBLOCH M, VON SCHOULTZ C, ZURKIRCHEN L, BRAUN SM, VIDMAR M & JESSBERGER S 2014. SPOT14-positive neural stem/progenitor cells in the hippocampus respond dynamically to neurogenic regulators. Stem Cell Reports, 3, 735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGMEAD B & SALZBERG SL 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods, 9, 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEDUC C & ETIENNE-MANNEVILLE S 2017. Intermediate filaments join the action. Cell Cycle, 16, 1389–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEEMAN DS, HEBESTREIT K, RUETZ T, WEBB AE, MCKAY A, POLLINA EA, DULKEN BW, ZHAO X, YEO RW, HO TT, MAHMOUDI S, DEVARAJAN K, PASSEGUE E, RANDO TA, FRYDMAN J & BRUNET A 2018. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science, 359, 1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIVAK KJ & SCHMITTGEN TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25, 402–8. [DOI] [PubMed] [Google Scholar]
- LOPEZ-OTIN C, BLASCO MA, PARTRIDGE L, SERRANO M & KROEMER G 2013. The hallmarks of aging. Cell, 153, 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUKJANENKO L, KARAZ S, STUELSATZ P, GURRIARAN-RODRIGUEZ U, MICHAUD J, DAMMONE G, SIZZANO F, MASHINCHIAN O, ANCEL S, MIGLIAVACCA E, LIOT S, JACOT G, METAIRON S, RAYMOND F, DESCOMBES P, PALINI A, CHAZAUD B, RUDNICKI MA, BENTZINGER CF & FEIGE JN 2019. Aging Disrupts Muscle Stem Cell Function by Impairing Matricellular WISP1 Secretion from Fibro-Adipogenic Progenitors. Cell Stem Cell, 24, 433–446 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUNDKVIST A, REICHENBACH A, BETSHOLTZ C, CARMELIET P, WOLBURG H & PEKNY M 2004. Under stress, the absence of intermediate filaments from Muller cells in the retina has structural and functional consequences. J Cell Sci, 117, 3481–8. [DOI] [PubMed] [Google Scholar]
- MARTIN M 2011. Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMB - Bioinformatics in action, 17. [Google Scholar]
- MARTYNOGA B, MATEO JL, ZHOU B, ANDERSEN J, ACHIMASTOU A, URBAN N, VAN DEN BERG D, GEORGOPOULOU D, HADJUR S, WITTBRODT J, ETTWILLER L, PIPER M, GRONOSTAJSKI RM & GUILLEMOT F 2013. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev, 27, 1769–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIRA H, ANDREU Z, SUH H, LIE DC, JESSBERGER S, CONSIGLIO A, SAN EMETERIO J, HORTIGUELA R, MARQUES-TORREJON MA, NAKASHIMA K, COLAK D, GOTZ M, FARINAS I & GAGE FH 2010. Signaling through BMPR-IA regulates quiescence and long-term activity of neural stem cells in the adult hippocampus. Cell Stem Cell, 7, 78–89. [DOI] [PubMed] [Google Scholar]
- MOORE DL, BLACKMORE MG, HU Y, KAESTNER KH, BIXBY JL, LEMMON VP & GOLDBERG JL 2009. KLF family members regulate intrinsic axon regeneration ability. Science, 326, 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOORE DL & JESSBERGER S 2017. Creating Age Asymmetry: Consequences of Inheriting Damaged Goods in Mammalian Cells. Trends Cell Biol, 27, 82–92. [DOI] [PubMed] [Google Scholar]
- MOORE DL, PILZ GA, ARAUZO-BRAVO MJ, BARRAL Y & JESSBERGER S 2015. A mechanism for the segregation of age in mammalian neural stem cells. Science, 349, 1334–8. [DOI] [PubMed] [Google Scholar]
- OGRODNIK M, SALMONOWICZ H, BROWN R, TURKOWSKA J, SREDNIAWA W, PATTABIRAMAN S, AMEN T, ABRAHAM AC, EICHLER N, LYAKHOVETSKY R & KAGANOVICH D 2014. Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin. Proc Natl Acad Sci U S A, 111, 8049–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OUYANG H, ALI YO, RAVICHANDRAN M, DONG A, QIU W, MACKENZIE F, DHE-PAGANON S, ARROWSMITH CH & ZHAI RG 2012. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J Biol Chem, 287, 2317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARK D, XIANG AP, MAO FF, ZHANG L, DI CG, LIU XM, SHAO YA, MA BF, LEE JH, HA KS, WALTON N & LAHN BT 2010. Nestin Is Required for the Proper Self-Renewal of Neural Stem Cells. Stem Cells, 28, 2162–2171. [DOI] [PubMed] [Google Scholar]
- PERTEA M, PERTEA GM, ANTONESCU CM, CHANG TC, MENDELL JT & SALZBERG SL 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol, 33, 290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAN FA, HSU PD, WRIGHT J, AGARWALA V, SCOTT DA & ZHANG F 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc, 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERT A, HOOKWAY C & GELFAND VI 2016. Intermediate filament dynamics: What we can see now and why it matters. Bioessays, 38, 232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODRIGUEZ-FERNANDEZ IA, QI Y & JASPER H 2019. Loss of a proteostatic checkpoint in intestinal stem cells contributes to age-related epithelial dysfunction. Nat Commun, 10, 1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUJANO MA, BOSVELD F, SALOMONS FA, DIJK F, VAN WAARDE MA, VAN DER WANT JJ, DE VOS RA, BRUNT ER, SIBON OC & KAMPINGA HH 2006. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol, 4, e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHIPPER-KROM S, JUENEMANN K, JANSEN AH, WIEMHOEFER A, VAN DEN NIEUWENDIJK R, SMITH DL, HINK MA, BATES GP, OVERKLEEFT H, OVAA H & REITS E 2014. Dynamic recruitment of active proteasomes into polyglutamine initiated inclusion bodies. FEBS Lett, 588, 151–9. [DOI] [PubMed] [Google Scholar]
- SZKLARCZYK D, FRANCESCHINI A, WYDER S, FORSLUND K, HELLER D, HUERTA-CEPAS J, SIMONOVIC M, ROTH A, SANTOS A, TSAFOU KP, KUHN M, BORK P, JENSEN LJ & VON MERING C 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res, 43, D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERZI F, HENRION D, COLUCCI-GUYON E, FEDERICI P, BABINET C, LEVY BI, BRIAND P & FRIEDLANDER G 1997. Reduction of renal mass is lethal in mice lacking vimentin. Role of endothelin-nitric oxide imbalance. J Clin Invest, 100, 1520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERDOES M, FLOREA BI, MENENDEZ-BENITO V, MAYNARD CJ, WITTE MD, VAN DER LINDEN WA, VAN DEN NIEUWENDIJK AM, HOFMANN T, BERKERS CR, VAN LEEUWEN FW, GROOTHUIS TA, LEEUWENBURGH MA, OVAA H, NEEFJES JJ, FILIPPOV DV, VAN DER MAREL GA, DANTUMA NP & OVERKLEEFT HS 2006. A fluorescent broad-spectrum proteasome inhibitor for labeling proteasomes in vitro and in vivo. Chem Biol, 13, 1217–26. [DOI] [PubMed] [Google Scholar]
- VERHOEF LG, LINDSTEN K, MASUCCI MG & DANTUMA NP 2002. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum Mol Genet, 11, 2689–700. [DOI] [PubMed] [Google Scholar]
- VILABOA NE, GARCIA-BERMEJO L, PEREZ C, DE BLAS E, CALLE C & ALLER P 1997. Heat-shock and cadmium chloride increase the vimentin mRNA and protein levels in U-937 human promonocytic cells. J Cell Sci, 110 (Pt 2), 201–7. [DOI] [PubMed] [Google Scholar]
- VILCHEZ D, BOYER L, MORANTTE I, LUTZ M, MERKWIRTH C, JOYCE D, SPENCER B, PAGE L, MASLIAH E, BERGGREN WT, GAGE FH & DILLIN A 2012. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature, 489, 304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILCHEZ D, SIMIC MS & DILLIN A 2014. Proteostasis and aging of stem cells. Trends Cell Biol, 24, 161–70. [DOI] [PubMed] [Google Scholar]
- WIGLEY WC, FABUNMI RP, LEE MG, MARINO CR, MUALLEM S, DEMARTINO GN & THOMAS PJ 1999. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol, 145, 481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILHELMSSON U, POZO-RODRIGALVAREZ A, KALM M, DE PABLO Y, WIDESTRAND A, PEKNA M & PEKNY M 2019. The role of GFAP and vimentin in learning and memory. Biol Chem. [DOI] [PubMed] [Google Scholar]
- YAMAGUCHI M, SAITO H, SUZUKI M & MORI K 2000. Visualization of neurogenesis in the central nervous system using nestin promoter-GFP transgenic mice. Neuroreport, 11, 1991–6. [DOI] [PubMed] [Google Scholar]
- YANG C, TAN W, WHITTLE C, QIU L, CAO L, AKBARIAN S & XU Z 2010. The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS One, 5, e15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU L, CHEN Y & TOOZE SA 2018. Autophagy pathway: Cellular and molecular mechanisms. Autophagy, 14, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZIEBELL F, DEHLER S, MARTIN-VILLALBA A & MARCINIAK-CZOCHRA A 2018. Revealing age-related changes of adult hippocampal neurogenesis using mathematical models. Development, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Intermediate filament expression in WT and vimentin KO NSCs. Related to Figure 2.
Table S2. Proteins co-precipitated by vimentin-mNeon in either a DMSO or MG132 treated condition, followed by LC-MS/MS (attached as excel file). Related to Figure 2.
Table S3. Total RNA sequencing data from untreated WT and vimentin KO NSCs (attached as excel file). Related to Figure 2.
Data Availability Statement
RNA sequencing data is available via GEO: GSE136553. Processed mass spectrometry data and RNA sequencing data are available in this article’s supplemental tables.