

Abstract
The ERK cascade is a central signaling pathway that regulates a wide variety of cellular processes including proliferation, differentiation, learning and memory, development, and synaptic plasticity. A wide range of inputs travel from the membrane through different signaling pathway routes to reach activation of one set of output kinases, ERK1&2. The classical ERK activation pathway beings with growth factor activation of receptor tyrosine kinases. Numerous G-protein coupled receptors and ionotropic receptors also lead to ERK through increases in the second messengers calcium and cAMP. Though both types of pathways are present in diverse cell types, a key difference is that most stimuli to neurons, e.g. synaptic inputs, are transient, on the order of milliseconds to seconds, whereas many stimuli acting on non-neural tissue, e.g. growth factors, are longer duration. The ability to consolidate these inputs to regulate the activation of ERK in response to diverse signals raises the question of which factors influence the difference in ERK activation pathways. This review presents both experimental studies and computational models aimed at understanding the control of ERK activation and whether there are fundamental differences between neurons and other cells. Our main conclusion is that differences between cell types are quite subtle, often related to differences in expression pattern and quantity of some molecules such as Raf isoforms. In addition, the spatial location of ERK is critical, with regulation by scaffolding proteins producing differences due to colocalization of upstream molecules that may differ between neurons and other cells.
2. Introduction
The Extracellular signal-Regulated Kinase (ERK) cascade plays a critical role for numerous processes in all cell types. ERK is involved in cell proliferation and differentiation (Kao et al., 2001), regulates cell cycle entry by controlling the G1 to S-phase transition (Chambard et al., 2007), and is involved in apoptosis and autophagy (Cagnol and Chambard, 2010). A defect in the cascade leads to uncontrolled growth, a key step for the development of all cancers (Dhillon et al., 2007). In neurons, aside from its role in development, e.g., (Xing et al., 2016), ERK is required for learning and memory storage, and for synaptic plasticity, which is the change in strength of connections between neurons and a mechanism underlying memory storage. The long-lasting strengthening of synapses is called long term potentiation (LTP), and is subdivided into protein synthesis dependent (long lasting) and independent (early phase) types. ERK contributes to learning and memory through initiating local protein synthesis in dendrites and controlling transcription in the nucleus. ERK signaling is required for induction of LTP in striatum (Cerovic et al., 2015; Hawes et al., 2013) and hippocampus (Selcher et al., 2003), and plays a role in LTP expression by regulating AMPA receptor recruitment to synapses (Qin et al., 2005; Zhu et al., 2002) following LTP induction.
A key observation is that the dynamics of ERK activity (i.e. the spatiotemporal pattern of activity) can determine which of several responses a cell exhibits ( Kao et al., 2001; Sasagawa et al., 2005). For example, a long duration of ERK activation may cause differentiation, whereas a short duration of ERK activation may cause cell division. These ERK dynamics are controlled by various feedback loops involving numerous molecules along the ERK activation pathways. Diverse signaling pathways initiate the activation of ERK, including growth factor receptors (Ryu et al., 2015; Sasagawa et al., 2005) and also other transmembrane receptors ( Corson et al., 2003) including synaptic inputs to neurons.
A key difference between cell types are that synaptic inputs to neurons are extremely brief, ranging from milliseconds (ms) to seconds (sec), whereas during development and in many experimental protocols, external stimuli such as growth factors are present for prolonged times (minutes to hours). The difference in dynamics of inputs to neurons and non-neuronal cells coupled with the importance of ERK dynamics for selecting cell response suggests that the ERK cascade may differ between neurons and non-neuronal cells.
This review describes the diversity of pathways for activation of ERK, both demonstrated experimentally and included in computational models. We highlight which pathways appear to differ between cell types and also highlight key differences between the pathways and reactions suggested by experiments and those included in computational models. We attempt to answer the question of whether ERK activation pathways are structurally different between neurons and other cells, or whether they are functionally different due to different input durations recruiting different activation pathways.
3. The classical MAPK pathway: Ras, Raf, MEK and ERK
The classical ERK cascade is a set of biochemical reactions beginning with an activated GTP binding protein of the Ras family binding to a Raf family kinase and continuing with sequential phosphorylation and activation of MEK and ERK (Figure 1).
Figure 1: Schematic representation of the classical ERK cascade.
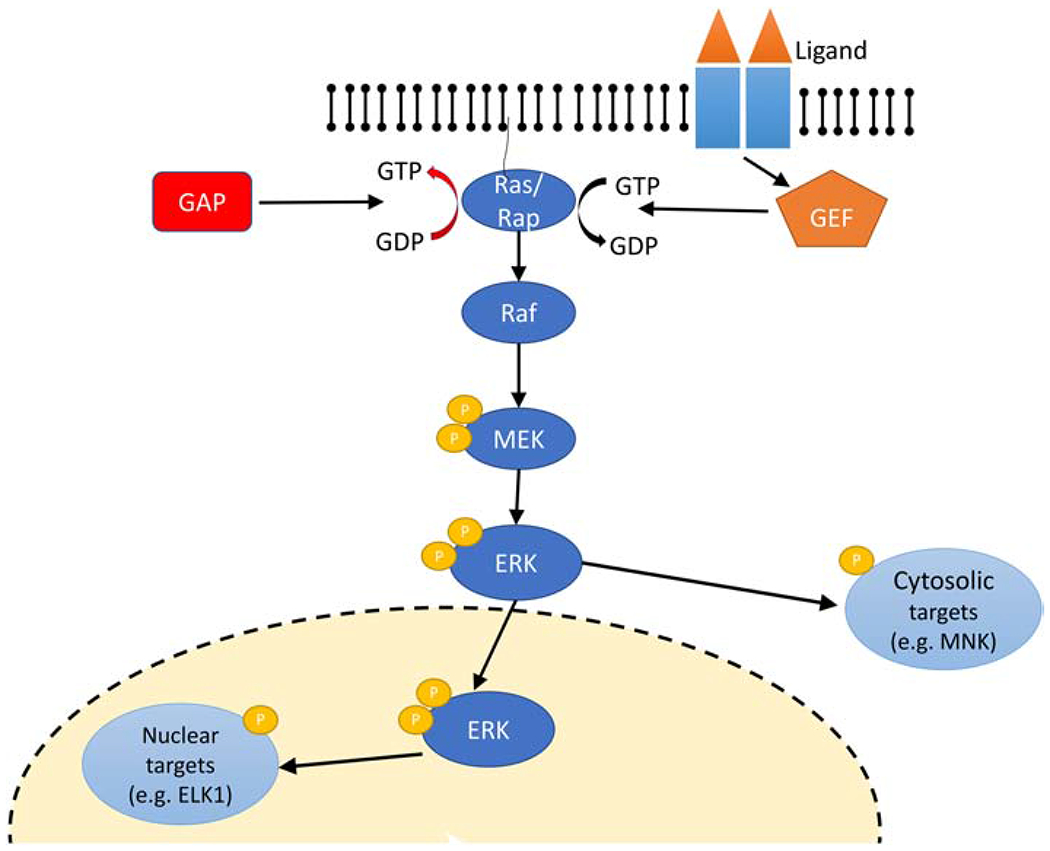
Activation of receptor upon ligand binding results in recruitment of the Guanosine Exchange Factor (GEF) that turns on the Ras family protein (exchange of GDP for GTP). Ras family proteins are turned off by GTPase Activating Proteins (GAP) that accelerate hydrolysis of GTP to GDP. Binding of RasGTP/RapGTP to Raf activates it and initiates the sequential phosphorylation steps of MEK which in turn activates ERK, which can phosphorylate either cytosolic or nuclear substrates.
3.1. ERK
ERK types 1 and 2 are also known as mitogen-activated protein kinases (MAPK), encoded by the genes MAPK3 and MAPK1, respectively (Table 1). They are serine/threonine protein kinases ubiquitously expressed, though not at the same level, in most tissues. ERK1 and ERK2 are 44 and 42-kDa kinases respectively, with about 90% amino acid identity (Buscà et al., 2016). Data on uniqueness versus redundancy of the two isoforms are conflicting. They appear to be redundant because they share the same substrate specificity in vitro ( a substrate that binds to ERK1 will also bind to ERK2 with the same efficiency) (Buscà et al., 2016; Frémin et al., 2015). On the other hand, ERK2 is essential for embryonic development (Hatano et al., 2003) whereas ERK1 disruptions are not embryonic lethal (Nekrasova et al., 2005). However, the lethality of the ERK2 knockout may be due to ERK2 quantity being greater than ERK1 coupled with the insufficient compensatory expression of ERK1 during development. This hypothesis is supported by recent experiments using transgenic mice in which ERK1 expression was controlled by the chicken β-actin promoter (Buscà et al., 2016; Frémin et al., 2015).
Table 1:
Representative RasGEF, RasGAP, and ERK core kinases protein and gene names as well as their human chromosome location number. Data were obtained from the NCBI gene resource website (https://www.ncbi.nlm.nih.gov/).
| Protein | Gene | Chromosome (human) |
|---|---|---|
| Ras GEF family | ||
| Epac1 | RAPGEF3 | 12 |
| Epac2 | RAPGEF4 | 2 |
| C3G | RAPGEF1 | 9 |
| REPAC | RAPGEF5 | 7 |
| PDZ-GEF1 | RAPGEF2 | 4 |
| PDZ-GEF2 | RAPGEF6 | 5 |
| GRF1 | RASGRF1 | 15 |
| GRF2 | RASGRF2 | 5 |
| CalDagGEFII | RASGRP1 | 15 |
| CalDagGEFI | RASGRP2 | 11 |
| GRP3 | RASGRP3 | 2 |
| RASGRFP4 | RASGRP4 | 19 |
| SOS1 | SOS1 | 2 |
| SOS2 | SOS2 | 14 |
| Kinases | ||
| ERK1 | MAPK3 | 22 |
| ERK2 | MAPK1 | 16 |
| MEK1 | MAP2K1 | 15 |
| MEK2 | MAP2K2 | 19 |
| bRaf | BRAF | 7 |
| cRaf | CRAF | 3 |
| aRaf | ARAF | X |
| Ras GAP family | ||
| Rap1GAP | RAP1GAP | 1 |
| Rap1GAP2 | RAP1GAP2 | 17 |
| RasGAP | RASA1 | 5 |
| synGAP | SYNGAP | 6 |
Overexpression of ERK1 was able to compensate for ERK2 knockout, suggesting that the total amount ERK must exceed some threshold. This experiment provides additional evidence that ERK1 and ERK2 exhibit functional redundancy, and that total quantity (magnitude and duration) is essential to achieve proper ERK function. Nonetheless, there are likely tissue-specific differences, as forebrain specific ERK2 knockout produces developmental deficits (Samuels et al., 2008), whereas forebrain specific postnatal knock out of ERK2 produces deficits in memory storage, but not LTP (Vithayathil et al., 2017). ERK2 specific deficits are also observed in sensitization of pain sensory neurons, whereas ERK1 and ERK2 are functionally redundant for survival of those same neurons (O’Brien et al., 2015).
Both ERK1 and ERK2 (referred to collectively as ERK) are activated by dual phosphorylation on Thr202/185 and Tyr204/187 (human sequences; Thr203/183 and Tyr205/185 rat sequence) mainly by MAPK/ERK Kinases (MEKs) (Seger et al., 1992). Upon phosphorylation, ERK can either homodimerize, heterodimerize or remain monomers (Khokhlatchev et al., 1998). The dimerization process enhances ERK activity but reduces the translocation of ERK monomers to the nucleus. The two phosphorylation steps seem to be distributed (Cirit et al., 2010; Kholodenko, 2006) which means MEK dissociates from ERK after each phosphorylation step. This process makes ERK ultrasensitive to the level of active MEK; i.e., ERK activation exhibits switch-like behavior.
The inactivation of ERK is mainly mediated by the removal of phosphate groups from either the Thr or the Tyr sites by one of several phosphatases. Dual specificity phosphatases (termed MAPK phosphatases (MKPs) or DUSPs) can dephosphorylate both Tyr and Ser/Thr residues. The isoforms differ in their specificity for ERK, and whether they are localized in the cytoplasm or nucleus. Regulation of their activity is via protein level as well as negative and positive feedback loops (discussed in other sections). ERK is also dephosphorylated by Ser/Thr phosphatases (PPs) and by protein Tyr phosphatases. The activity of many of the phosphatases is regulated by phosphorylation. For example, Striatally Enriched Tyrosine Phosphatase (STEP), is inactivated by PKA phosphorylation, whereas protein phosphatase 2A, is upregulated by PKA phosphorylation.
Once activated, ERK phosphorylates numerous substrates in all cellular compartments; at least 280 substrates have been identified so far (Buscà et al., 2016). Some of the target substrates colocalize with ERK in the cell cytoplasm, while others are in the nucleus (Yoon and Seger, 2006). Nuclear translocation of ERK is a critical step for inducing gene expression and cell cycle entry. PhosphoLipase A2 (PLA2), MAPK Interacting Kinase (MNK), Son of Sevenless (Sos), 90 kDa RiboSomal protein S6 Kinase (RSKs) are a few examples of cytosolic targets, while E twenty-six Like Tyrosine Kinase 1 (ELK1), Mitogen and Stress-activated protein Kinase (MSK), and RSKs are nuclear targets (Maik-Rachline et al., 2019; Yoon and Seger, 2006). Phosphorylation by ERK can either enhance or reduce activity of substrate molecules. For example, phosphorylation of ELK1 in the nucleus enhances DNA binding to ELK1 and increases transcriptional activity. In contrast, phosphorylation of Sos by ERK in the cytoplasm negatively regulates ERK activity by causing Soscomplex dissociation. Importantly, proliferation is mediated mainly by ERK activity in the nucleus, while other activities such as differentiation are correlated better with ERK activity in the cytoplasm (Michailovici et al., 2014).
In CA1 pyramidal neurons, ERK activation is required for induction of protein synthesis-dependent forms of LTP (Connor et al., 2012; Selcher et al., 2003) and structural LTP (Tang and Yasuda, 2017; Zhai et al., 2013). ERK also is required for LTP of glutamatergic synapses in striatum (Hawes et al., 2013), cingulate cortex (Toyoda et al., 2007) and amygdala (Schafe et al., 2008), and for LTD of cerebellar Purkinje neurons (Tanaka and Augustine, 2008). ERK contributes to LTP and memory by initiating local protein synthesis through phosphorylation of e.g ribosomal protein S6 kinase (Gobert et al., 2008; Kelleher et al., 2004) and controlling transcription through phosphorylation of CREB (Kanterewicz et al., 2000; Trifilieff et al., 2006), MSK1(McCoy et al., 2005) and Elk1 (Davis et al., 2000; Thiels et al., 2002). Aside from transcription and translation, in hippocampal CA1 pyramidal neurons, the pool of ERK scaffolded by KSR1 increases cell excitability by phosphorylates potassium channels. In CA1 pyramidal neurons, structural LTP is correlated with nuclear ERK signaling, which requires spatial and temporal integration of synaptic input on two or more dendritic branches (Zhai et al., 2013).
3.2. MEK
Mitogen Activated-Protein Kinase Kinase (MAP2K) or MEKs are cytoplasmic serine/threonine protein kinases that phosphorylate and activate ERK in the cytoplasm. Two isoforms of MEK, MEK1 and MEK2, have been identified so far. MEK1 and MEK2 show high homology in their kinase domains (Ussar and Voss, 2004), but diverge in the N terminus (ERK docking domain) and the proline-rich domain (which interacts with the activated Raf family kinase) sequences (Aoidi et al., 2016). Similar to ERK isoforms, MEK isoforms appear to be redundant because they share the same substrate specificity, yet knock-out experiments suggest otherwise. The MEK1 null-mutation is lethal during embryonic development whereas the MEK2 null-mutation is viable, suggesting that MEK1 but not MEK2 is critical in signaling transduction for growth and development (Bélanger et al., 2003; Giroux et al., 1999; Ussar and Voss, 2004). On the other hand, conditional deletion of MEK1 in the embryo, but not in extraembryonic tissue, generates viable mice (Bissonauth et al., 2006), suggesting functional redundancy of MEK isoforms for organ development. Recently, Aoidi et al., (2016) showed that a threshold concentration of MEK is required for proper growth and development, independent of MEK isoform, similar to the observed threshold quantity of ERK. In summary, it appears that MEK1 and MEK2 have overlapping functions, which are to activate indiscriminately their only substrate, ERK, by phosphorylation.
Both MEK1 and MEK2 are activated by Raf family kinases via phosphorylation of the two serine residues (Ser218/222, Ser217/221, mouse sequence) in their activation and catalytic loops. Dephosphorylation and inactivation of MEKs are mainly mediated by the removal of phosphate groups by Ser/Thr phosphatases (PPs) such as PP2A (Adams et al., 2005). In addition, double phosphorylated ERKs can mediate inhibitory feedback on MEK by phosphorylation of Thr292 (MEK1), which interferes with the binding of MEK1 to ERK (Hong et al., 2015).
3.3. Raf family kinases
Mitogen Activated-Protein Kinase kinase kinases (MAP3K) or Rafs are part of the family of cytoplasmic serine/threonine kinases that phosphorylate and activate MEKs, their only substrate. Two isoforms, C-Raf (also called Raf1) and B-Raf are the most relevant Raf kinases in the ERK signaling pathway. B-Raf has a higher affinity towards MEK than C-Raf (Desideri et al., 2015; Papin et al., 1998), but otherwise, their function in activation of MEK is similar.
A comprehensive review of Raf activation is found in (Matallanas et al., 2011; Terrell and Morrison, 2019); thus, only a summary will be provided here. In its inactive form, C-Raf is bound on both 14-3-3 binding N-terminal and C-terminal sites, (Ser259 and Ser621, mouse sequence) respectively, in the cytosol in a closed conformation (Desideri et al., 2015; Dhillon et al., 2002). Dephosphorylation at Ser259 by PP2A or PP1 (Emerson et al., 1995) produces the open inactive form, allowing membrane recruitment of C-Raf through interaction with lipids. This lipid associated C-Raf has very low activity. At the membrane, RasGTP or RapGTP, members of the Ras family of small monomeric G proteins, bind to C-Raf. Binding to RasGTP conveys activity to C-Raf, whereas binding to RapGTP prevents C-Raf activity. Subsequent to RasGTP binding, C-Raf can be phosphorylated by an Src family kinase (Tyr341, mouse sequence), or Protein Kinase C (PKC) (and possibly calcium-calmodulin dependent kinase type II (CaMKII)) on Ser338 (mouse sequence) of the N-terminal site to further increase C-Raf activity (Corbit et al., 2003; Salzano et al., 2012). Homodimerization or heterodimerization (with B-Raf or Kinase Suppressor of Ras (KSR)) is needed for full activation of C-Raf (Desideri et al., 2015; Garnett et al., 2005). It is unclear whether dimerization or N-terminal phosphorylation are dependent on the other. Once active, C-Raf phosphorylates to activate MEK. Dephosphorylation of C-Raf is mediated by protein phosphatase PP5 on Ser338 leading to re-phosphorylation of the 14-3-3 binding site on Ser259 returning C-Raf to the inactivate state (von Kriegsheim et al., 2006); alternatively, C-Raf is inactivated by dissociation of bound RasGTP. ERK can mediate an inhibitory feedback loop on C-Raf by phosphorylating five different sites to terminate its catalytic activity (Balan et al., 2006), which promotes dephosphorylation of C-Raf by PP2A or PP1 leading to re-phosphorylation of 14-3-3 on Ser259.
The mode of B-Raf activation is much simpler than C-Raf B-Raf activation requires two processes: unbinding from 14-3-3 protein at Ser365 (by PP2A or PP1) and binding to either RasGTP or RapGTP, both of which activate B-Raf (in contrast to RapGTP inhibition of C-Raf). B-Raf is constitutively phosphorylated at Ser445 (Desideri et al., 2015; Mason et al., 1999) eliminating the need for an additional phosphorylation step. This simpler activation may be the reason that B-Raf is the main ERK activator, and produces greater MEK activity than C-Raf in response to both EGF and NGF stimulation (Kao et al., 2001). ERK can mediate a feedback inhibition of B-Raf by phosphorylation at several different sites in the N-terminal of B-Raf (Brummer et al., 2003; Ritt et al., 2010). This ERK feedback loop can terminate B-Raf activity either by disrupting association with RasGTP or disrupting dimerization with C-Raf.
It is worth mentioning that both C-Raf and B-Raf are PKA substrates which exerts either a stimulatory or inhibitory effect while only C-Raf is a PKC substrate. These differences will be discussed in section 4 below.
3.4. Ras Family GTPase
The Ras family GTPases are monomeric GTP-binding proteins that have both a GDP/GTP binding site and GTPase properties (Colicelli, 2004). Two GTPases: Ras and Rap, are involved in ERK activation. The effector domain of both proteins is over 80% identical, suggesting that both proteins theoretically interact with the same effectors (Takai et al., 2001). They are regulated by Guanine nucleotide Exchange Factors (GEFs) and GTPase Activating Proteins (GAPs) that activate (via exchange of bound GDP for GTP) or inactivate (via hydrolysis of GTP to GDP), respectively, the Ras family GTPase.
Ras-Related protein (Rap) consists of Rap1 and Rap2 (Takai et al., 2001), and are activated by interaction with one of their GEFs. Once activated, RapGTP can bind to either C-Raf or B-Raf, with a higher affinity towards B-Raf (Hu et al., 1997; York et al., 1998). The RapGTP-B-Raf complex can phosphorylate MEK, causing sustained activation of ERK, and is considered to be the main ERK activator (Kao et al., 2001). In contrast, RapGTP binding to C-Raf forms an inactive complex because it cannot position C-Raf near the kinase that carries out the required phosphorylation at sites within the N-region (Carey et al., 2003). Thus, RapGTP inhibits C-Raf through competitive exclusion of RasGTP binding.
After activation of RasGDP proteins by one of its GEFs, RasGTP can bind and activate both C-Raf and B-Raf, though RasGTP has a higher affinity towards C-Raf than B-Raf. Both RasGTP-C-Raf and RasGTP-B-Raf complexes phosphorylate MEK which, in turn, phosphorylates and activates ERK. The RasGTP-C-Raf complex can dimerize with itself to form homodimers or with B-Raf or KSR to form heterodimers. Unlike RapGTP-B-Raf, the RasGTP-C-Raf complex causes a transient activation of ERK because of the negative feedback loop towards the Ras GEF, Son of Sevenless (Sos) (Kao et al., 2001). Double phosphorylated ERK catalyzes Sos phosphorylation which in turn leads to the Sos complex dissociation, thereby limiting the duration of RasGTP production.
Another difference between Ras and Rap is in their subcellular localization. Both Ras and Rap are membrane associated molecules, but they may be located on different membranes. Ras is localized and activated on the cytoplasmic face of the plasma membrane while Rap is on intracellular membranes (Mochizuki et al., 2001; Ohba et al., 2003). In PC12 cells, RapGDP activation is initiated inside the cell body (i.e., on intracellular membranes) and spreads towards the cell surface (Mochizuki et al, 2001). In cultured neurons, the induction of synaptic plasticity by Ras and Rap use different subcellular microdomains (Zhang et al., 2018). Ras localized to lipid rafts activates ERK to increase AMPA-type glutamate receptors while Rap localized to the bulk membrane is involved in the removal of AMPA-type glutamate receptors.
3.5. Scaffold proteins
Biochemical reactions such as phosphorylation and GDP-GTP exchange require that the substrate and enzyme molecules must encounter each other in the complex environment of the cell. An approach to increase the efficiency of such reactions is to colocalize molecules that work together. Such is the function of scaffolding or anchoring proteins, which play important roles in regulating ERK activation (Langeberg and Scott, 2015). In other words, scaffolding proteins bind multiple proteins that interact by interconnecting them into a stable complex. This effect enables the rapid transmission of the signal within a cascade of biochemical reactions. Another role is to sequester sets of interacting proteins to limit interactions with other proteins and minimize crosstalk between pathways that may share some components. With respect to ERK, anchoring proteins play a role in the spatial regulation of ERK, by controlling subcellular location (Zhang et al., 2018). Thus, scaffold proteins regulate the kinetics, amplitude and localization of ERK signaling. Several scaffold proteins that regulate ERK have been identified such as KSR, β-Arrestin, paxillin, IQGAP1, and more (Figure 2). We will briefly address some of them.
Figure 2: Scaffolding proteins in ERK signaling pathways:

In the cytosol, upon stimulation KSR translocates to the membrane and scaffolds a pool of Raf/MEK/ERK that is activated by PKC. GPCRs activate ERK through either G-proteins (not shown) or β-arrestin with different temporal dynamics. IQGAP1 scaffolds Raf/MEK/ERK in dendritic spines, and binding of calmodulin causes dissociation from actin. ppERK can either dimerize in the cytoplasm or translocate to the nucleus, though ERK scaffolded by β-arrestin does not translocate to the nucleus.
Kinase Suppressor of Ras (KSR) is one of the best-characterized scaffold protein in the ERK cascade. KSR has several distinct domains through which it can interact with C-Raf, MEK and ERK (Nguyen et al., 2002; Therrien et al., 1995), and a kinase domain of questionable function (Langeberg and Scott, 2015). The scaffolding function of KSR seems to be independent of its kinase activity. Two isoforms of KSR (1 and 2) interact with the protein phosphatase PP2A, scaffolding proteins such as 14-3-3, and components of the core ERK cascade, such as MEK (Dougherty et al., 2009) Cell stimulation, e.g. with PDGF in Cos cells, results in dephosphorylation of KSR at Ser392 and unbinding of 14-3-3 (Müller et al., 2001; Ory et al., 2003) allowing the translocation of KSR to the membrane. At the membrane, KSR scaffolds three kinases, C-Raf, MEK and ERK which facilitates signal transmission (Langeberg and Scott, 2015) from RasGTP to ERK. In neurons KSR1 segregates pools of ERK activated by PKC from pools of ERK activated by PKA (Shalin et al., 2006), and KSR1 null mutants have reduced LTP and ERK activation in the membranes (but not the cytosol) in response to Gq coupled receptor activation. KSR2 associates with the calcium dependent protein phosphatase calcineurin (Dougherty et al., 2009). This latter association allows translocation of KSR2 and ERK to the membrane in response to calcium stimulation.
G-protein coupled receptors (GPCRs) can activate ERK via a mechanism involving the heteromeric G-protein stimulated production of second messenger-dependent kinases, such as PKC and PKA (described below), or a β-arrestin-dependent pathway, β-arrestins are cytosolic proteins involved in desensitizing GPCR signaling, primarily through receptor internalization (Lefkowitz and Shenoy, 2005). In addition to this role in receptor inactivation, β-arrestin scaffolds the ERK cascade by binding to C-Raf, MEK and ERK, thus assembling the core components for ERK activation (Bourquard et al., 2015; DeFea et al., 2000) to enhance activation. Recruitment of β-Arrestins is regulated by G protein Receptor Kinases (GRKs) (Heitzler et al., 2012). Interestingly, the ERK signaling facilitated by β-arrestin is G protein independent, does not propagate to the nucleus (DeFea et al., 2000; Shenoy and Lefkowitz, 2011), and has a delayed activation (slow onset) but with longer duration of activity than G protein dependent ERK signaling.
IQ motif containing GTPase activating protein 1 (IQGAP1) is a multidomain molecule that contains several protein-interacting motifs. The name is derived from the presence of IQ motif (calmodulin binding) domains and a region similar to the catalytic domain of GAPs. IQGAP1, a ubiquitously expressed protein, regulates activity of many signaling pathways including the ERK cascade (McNulty et al., 2011). It scaffolds Raf, (Renet al., 2007), MEK (Roy et al., 2005), ERK (Roy et al., 2004) and calcium-calmodulin (Ho et al., 1999; Mateer et al., 2002). Interaction of calcium with IQGAP enhances the IQGAP scaffolding with B-Raf, while calmodulin interaction with IQGAP attenuates B-Raf binding to IQGAP (Ren et al., 2008). Both cadherin (Schrick et al., 2007) and calmodulin (Mateer et al. 2002) binding to IQGAP1 controls its interaction with the actin cytoskeleton within dendritic spines. In HeLa cells, IQGAP interacts with the Epidermal Growth Factor receptor (EGFR). An EGF stimulus induces phosphorylation of IQGAP1 Ser1443 (mouse sequence) via PKC activation (McNulty et al., 2011), enhancing EGFR activity through a positive feedback loop. In the hippocampus, IQGAP1 knock out impairs ERK activation in cultured neurons and in response to fear condition (Gao et al., 2011). Interestingly, either increasing or decreasing IQGAP1 by 50% or more considerably affects the ability of EGF to activate MEK/ERK (McNulty et al., 2011). Thus, quantitative balance between the kinases and IQGAP1 is essential for efficient signal production.
3.6. Models of Ras/RAF/MEK/ERK
Recent studies of signaling pathways have demonstrated that intracellular signals do not propagate in a linear fashion. Instead, they interact with one another non-linearly, with both cross-talk and feedback loops, to regulate multiple functions in the cell in a complicated system. Therefore, computational modeling approaches become necessary to perform systematic analyses of these complex interactions and to predict functions that can be validated by experiment. Given the importance of ERK, there are numerous models of ERK signaling pathways. The models differ by the molecules and interactions that are incorporated, as well as the scientific question addressed. Table 2 summarizes the main message, the key aspect, and the core ERK cascade protein isomers. It is worth noting that all models include Ras and C-Raf molecules, but only a subset of models also simulate Rap or B-Raf.
Table 2:
Summary of some key ERK cascade models in different cell types. Model type lists input and cell type (where specified). Main message summarizes the most important findings from the model. Key components list some unique mechanisms to that model. ERK feedback is negative unless otherwise specified. Only models with RTK activation of ERK are included here.
| Model name | Model type | Main message | Key components | GTPase | Raf Kinases | ERK Feedback |
|---|---|---|---|---|---|---|
| Arkun and Yasemi,2018 | EGFR | Dual phosphorylation-dephosphorylation of MEK and ERK is required for bistability, negative feedback loop causes ERK oscillation | ERK translocates to nucleus; positive feedback from RasGTP to Sos; Nuclear targets form positive and negative feedback loop to the ligand and receptor | Ras | C-Raf | Sos & EGFR |
| Brightman and Fell, 2000 | EGFR in PC12 | Negative feedback inhibition onto GEF is most important factor determining signal duration | Receptor internalization | Ras | C-Raf | Sos |
| Cirit et al., 2010 | PDGF in NIH 3T3 | ERK inhibition of Raf is the main ERK feedback loop | Feedforward inhibition of Sos via PI3K; ERK mediated upregulation of MKPs | Ras & Rap | C-Raf & B-Raf | Sos & C-Raf |
| Hornberg et al., 2005 | EGFR in HeLa | ERK activation dynamics is mainly controlled by Raf activity | Control analysis of an updated version of Schoeberl et al., 2002; Receptor degradation | Ras | C-Raf | none |
| Jain and Bhalla, 2014 | BDNF and Calcium in hippocampal Neurons | Quantity of several key molecules, including ERK, CaMKIV and mTORC, in the nucleus generates unique patterns in response to different LTP and LTD stimuli | ERK (via MSK and RSK), TORC, and CaMKIV phosphorylate CREB; PP1 dephosphorylates CREB | Ras & Rap | C-Raf & B-Raf | Sos & C-Raf |
| Orton et al., 2009 | NGF and EGF via EGFR in PC12 | Transient and sustained ERK activation in response to EGF and NGF are the result of receptor degradation instead of negative feedback to Sos | EGFR activates both Ras and Rap; feedforward inhibition of Raf1 via PI3K and Akt | Ras & Rap | C-Raf & B-Raf | Sos & C-Raf |
| Sasagawa et al., 2005 | TrkA and EGFR in PC12 | Rapid increase of NGF and EGF cause transient ERK while sustained ERK is dependent on NGF concentration and acts through Rap | Both TrkA and EGFR activate both Ras and Rap; feedforward inhibition of Ras through enhanced RasGAP; receptor internalization | Ras & Rap | C-Raf & B-Raf | Sos |
| Schoeberl et al., 2002 | EGFR in HeLa | Timing of ERK activation is a better readout of receptor activation than ERK amplitude | Shc-dependent and Shc-independent of Ras activation | Ras | C-Raf | none |
| Xu et al., 2010 | EGFR in PC12 and HEK293 | Best fit to experimental data requires EGFR activation of both Rap and Ras | Uses Bayesian Uses Bayesian inference to rank different pathway hypotheses based on experimental data; Receptor desensitization | Ras & Rap | C-Raf & B-Raf | Sos |
| Yoon and Deisboeck, 2009 | EGFR in PC12 | Raf and Ras activities are the main controlling factor for ERK controlling sustained or transient | Parameter sensitivity analysis of Brightman and Fell (2000) | Ras | C-Raf | Sos |
The main topic of ERK models in non-neuronal cells is to determine factors controlling ERK dynamics, such as sustained versus transient ERK activation in response to EGF and/or NGF stimulation. Most computational models use single Raf and Ras isomers; however, Xu et al. (2010) demonstrate the requirement of both isoforms (Ras and Rap) for full ERK activation in response to EGFR stimulation in PC12 and HEK293 cells. One key mechanism to produce transient ERK is the feedback loop from ERK to Sos (Sasagawa et al. 2005; Brightman and Fell, 2000) or C-Raf (Cirit et al., 2010). In contrast, Orton et al. (2009) suggest that receptor degradation and not the feedback loop is the key factor. Note that none of these models are spatial and thus do not discriminate different pools of ERK.
3.7. Differences between cell types
The reactions involved in Ras/Raf/MEK/ERK activation appear to be the same in all cell types. RasGTP/RapGTP starts the sequential phosphorylation of the ERK cascade by activating Raf which activates MEK, which then activates ERK leading to the initiation of gene transcription, protein synthesis, or synaptic plasticity. However, a difference is seen within the ERK negative feedback loop towards Raf. Phosphorylated ERK exerts a temporal control on C-Raf activity through inhibitory phosphorylation in all cell types, e.g. NIH 3T3, and PC12 cells. However, the inhibitory phosphorylation of B-Raf kinase has not been demonstrated in experiments with neurons. Though RasGTP/RapGTP binding and activation of Raf is similar in all cell types, a difference is observed in their upstream activation pathways, discussed in the next section.
4. Diverse pathways to activation of Ras and Rap
ERK responds to numerous membrane signals (Figure 3), mediated by several types of transmembrane receptors including receptor tyrosine kinases, ionotropic receptors and G-protein-coupled receptors. Stimulation of the ERK cascade via receptor tyrosine kinases is the classical pathway. The steps downstream of the receptor tyrosine kinase to ERK are less variable and most understood. In contrast, GPCR pathways leading to ERK phosphorylation are quite diverse. GPCRs interact differentially with heterotrimeric G protein isoforms, effectors and second messengers. Of relevance to ERK activation are the intracellular molecules cAMP, diacylglycerol and calcium, typically a consequence of either ionotropic receptor or GPCR stimulation.
Figure 3: Numerous signaling pathways contribute to ERK activation in CA1 pyramidal neurons:
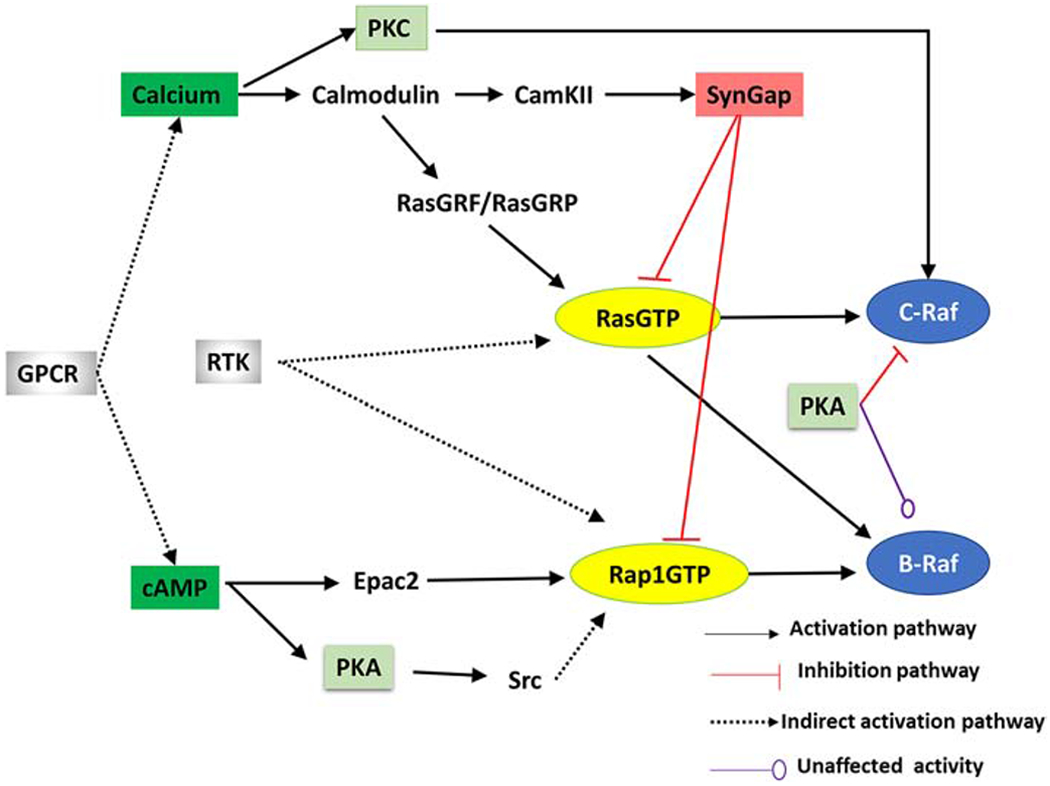
GPCRs stimulate the intracellular production of cAMP and calcium in both dendrites and dendritic spines. cAMP contributes to ERK through activation of PKA and RapGEFs (e.g. Epac). Calcium in both dendrites and dendritic spines leads to ERK by activating molecules such as PKC (which enhances C-Raf activity by phosphorylation) and RasGEFs (e g. RasGRF). Calcium activation of CaMKII modulates SynGap activity in dendritic spines. RTKs, such as TrkA, TrkB and EGFR, activate Ras family proteins through intermediate molecules. Note that all molecules, except for SynGap, are found in both dendrites and dendritic spines. Most of these molecules also are found in principal neurons of striatum, cerebellum and neocortex.
4.1. Classical pathways: Receptor Tyrosine Kinases
Receptor tyrosine kinases (RTKs) are part of the superfamily of transmembrane receptors that function to activate ERK in response to a wide array of growth factors. Several steps are required for RTK activation and recruitment of a GEF that activates Ras or Rap (Sasagawa et al., 2005): (i) binding of a specific ligand (e.g. growth factor) to its RTK; (ii) dimerization of two bound RTKs, (iii) autophosphorylation by the tyrosine kinase domain, (iv) binding of adaptor proteins to the phosphorylated RTK, (v) recruitment of GEFs to the adaptor proteins to form an active complex, (vi) GEF binding to RasGDP or RapGDP allowing exchange of GDP for GTP. Table 3 summarizes this process and gives two examples, one producing RasGTP and one producing RapGTP.
Table 3:
Summary of the signaling pathways activated by receptor tyrosine kinases and activating a Ras family GTPase. Note that some RTKs can activate both Ras and Rap.
| RTK | EGFR | TrkA/B |
| Growth factor | EGF | NGF, BDNF |
| Adaptor | Shc, Grb2 | Cbl, Crk |
| GEF | Sos | C3G |
| GTPase | Ras | Rap |
Epidermal Growth Factor (EGF) is the prototypical extracellular ligand that binds EGFR. Two EGF-EGFR complexes dimerize, which allows trans-phosphorylation of specific tyrosine residues on the intracellular C-terminal domain. Phospho-tyrosine residues allow binding of adaptor proteins. For example, the adaptor protein Shc binds to the receptor, then recruits an additional adaptor protein, Growth factor receptor-bound protein (Grb2) via its SH2 domain which recruits the GEF Sos through the SH3 domain of GRB2 (Lowenstein et al., 1992). The Soscomplex (Shc-Grb2-Sos) binds the membrane-associated RasGDP and catalyzes the exchange of GDP for GTP. As described above, RasGTP starts the sequential phosphorylation of the Raf/MEK/ERK cascade.
Nerves Growth Factor (NGF) is the prototypical extracellular ligand that binds Tyrosine kinase A (TrkA). Similar to EGF, NGF induces dimerization of TrkA, followed by trans-phosphorylation of tyrosine residues. Phospho-tyrosine residues on TrkA recruit a different set of adaptor proteins. For TrkA receptors, the adaptor protein c-Cbl recruits an additional adaptor protein Crk through its SH2 domain, which recruits the GEF C3G via the SH3 domain of Crk. The C3G complex (Cbl-Crk-C3G) allows the GEF C3G to bind to RapGDP and catalyzes the exchange of GDP for GTP, carrying the NGF stimulus downstream to sequential phosphorylation of the ERK cascade. In addition to NGF activating RapGTP instead of RasGTP, this pathway differs from the EGF pathway in that it can be bypassed by PKA through the phosphorylation of the protein kinase Src (discussed under PKA section).
Brain-Derived Neurotropic Factor (BDNF), a member of the neurotrophin family of growth factor that is prominent in the brain, is related to the canonical NGF. BDNF binds to TrkB leading to its dimerization and activation of the tyrosine kinase activity, which carries the stimulus downstream to ERK. Aside from its role in development, BDNF-TrkB plays an important role in learning and memory in adolescent and adult rodents. It is required for long term potentiation induced by theta-burst, but not tetanic stimulation in the hippocampus (Aarse et al., 2016; Chen et al., 1999, p. 199; Korte et al., 1995; Patterson et al., 2001; Zakharenko et al., 2003). Whenever tested, BDNF-TrkB was shown to be required for LTP of striatum spiny projection neurons (Jia et al., 2010; Park et al., 2014; Plotkin et al., 2014). BDNF is also required for various learning tasks, including amygdala dependent fear conditioning (Ou et al., 2010), hippocampal dependent spatial object recognition (Aarse et al., 2016; Mizuno et al., 2000) and insular cortex dependent conditioned taste aversion (Martínez-Moreno et al., 2011).
4.2. G-Protein couple receptors pathways
G-protein coupled receptors (GPCR) are the largest group of cell surface receptors. In response to extracellular stimuli, GPCRs change their structural conformation to catalyze the exchange of GDP for GTP of heterotrimeric GTP binding proteins, which allows the dissociation of the GαGTP and Gβγ subunits. The signaling pathways activated by GPRCs include the second messengers Cyclic Adenosine MonoPhosphate (cAMP) and calcium, and the phospholipid DiAcylGlycerol (DAG), all of which can activate the ERK cascade (Figure 4).
Figure 4: Schematic diagram of GPCR activation of ERK as well as feedback mechanisms.
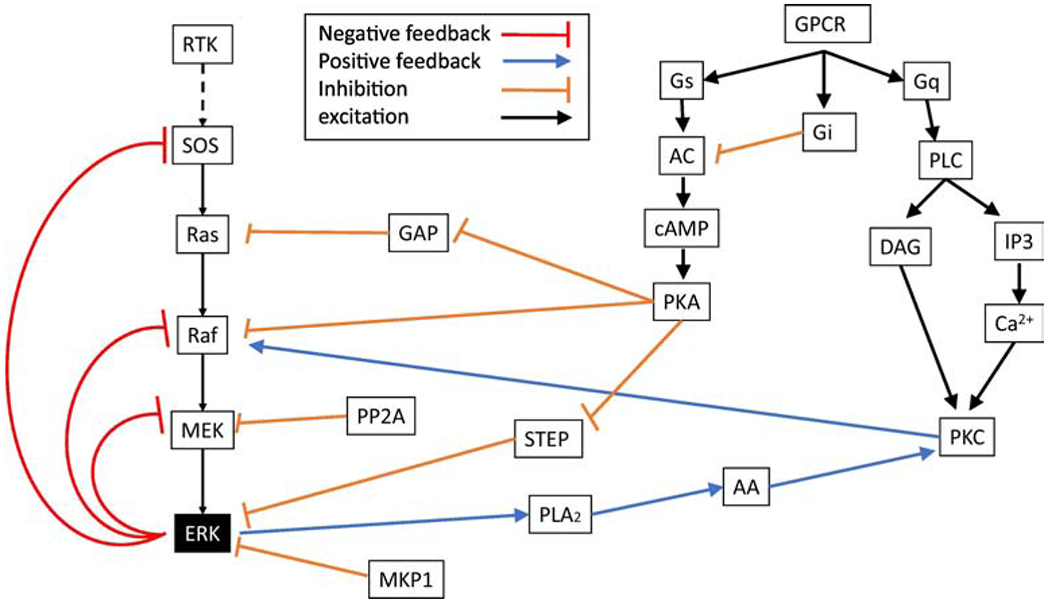
GPCRs influence cAMP and calcium through three major groups of heterotrimeric GTP binding proteins: Gs, Gi and Gq. Some feedback mechanisms are confined within ERK pathways, whereas others involve kinases activated by GPCRs.
4.2.1. Second messenger pathway cAMP
One important second messenger molecule for activation of ERK is cAMP, which is produced by the enzyme adenylyl cyclase from ATP and hydrolyzed (inactivated) by phosphodiesterases. A commonality to all eight isoforms of adenylyl cyclase is their activation by the heterotrimeric stimulatory GTP binding protein (Gs) and inhibition by the Gi subtype of GTP binding protein, both of which are activated by diverse metabotropic receptors (Hanoune and Defer, 2001). In addition, several isoforms of adenylyl cyclase are synergistically activated by calcium-bound calmodulin. After synthesis, cAMP diffuses through the cytoplasm to reach two key molecules upstream of ERK activation: protein kinase A(PKA) and exchange factor activated by cAMP (Epac).
4.2.1.1. PKA
PKA, also known as cAMP-dependent protein kinase, is an enzyme that plays a key role in several cellular processes through protein phosphorylation. In its inactivated state, PKA exists as a tetrameric complex of two catalytic subunits and two regulatory subunits (Zhang et al., 2012). cAMP binds to two sites on each of the regulatory subunits, which allows the dissociation of the regulatory and catalytic subunits. The catalytic subunits are constitutively active and thus after dissociation are free to phosphorylate specific target proteins. PKA regulates ERK activation by phosphorylation of several proteins in the ERK activation pathway, including Src, Rap1GDP, C-Raf and B-Raf, and can either decrease or increase ERK activation.
PKA phosphorylates a set of non-receptor tyrosine kinases called Src Family Kinases (SFK), of which Src is the prototype. Src leads to the activation of ERK through the GEF C3G and subsequent activation of RapGDP (Schmitt and Stork, 2002). This pathway has been demonstrated downstream of Gs coupled receptors in HEK293, CHO, adipocytes, NIH3T3 (Schmitt and Stork, 2002). PKA mediated phosphorylation of Src at Ser17 (mouse sequence) triggers the activation and autophosphorylation of Src (Brown and Cooper, 1996; Roth et al., 1983). Phosphorylated Src mediates the phosphorylation of Cbl (Schmitt and Stork, 2002), or alternatively may transactivate RTKs (Sasi et al., 2017), leading to the activation of RasGDP as in the NGF pathway. Regardless of whether RTKs or phosphorylated Src initiates this biochemical cascade, C3G activates Rap (Schmitt and Stork, 2002), which initiates the Raf/MEK/ERK cascade. In the nervous system and in lymphocytes, Cbl phosphorylation and Crk/C3G recruitment is mostly mediated by Fyn, a member of SFKs (Jin et al., 2019). In the striatum, Fyn is activated by the dopamine D1-receptor (Jin et al., 2019), whereas in the hippocampus and motor neurons, Fyn is activated by adenosine A2A receptors (Sasi et al., 2017). In addition, β-Arrestins can scaffold Src (Bourquard et al., 2015), suggesting a more direct pathway from PKA phosphorylated Src to ERK subsequent to GPCR activation.
RapGTP itself is a PKA substrate, with two phosphorylation sites. Phosphorylation of RapGTP by PKA within its C-terminus causes its dissociation from the membrane to the cytoplasm (Takahashi et al., 2013) and accelerates its inactivation by GAPs. This reaction serves to limit the duration, but not the peak amplitude of RapGTP, suggesting that PKA does not phosphorylate the inactivate RapGDP form. In contrast to PKA inhibition of C-Raf, B-Raf phosphorylation by PKA permits the indirect binding of B-Raf to RapGTP (Altschuler and Lapetina, 1993; Takahashi et al., 2013) to maintain its activity, as described below.
The phosphorylation of Ser365 (human sequence) of B-Raf by PKA disrupts its interaction with RapGTP in both neuronal and non-neuronal cell (Takahashi et al., 2017). Though phosphorylation inhibits B-Raf binding to RapGTP, it does not affect its ability to activate the ERK cascade. This counter-intuitive result has recently been explained (Takahashi et al., 2017) and is mediated by the scaffolding protein KSR. Despite the inhibition of RapGTP binding to B-Raf, phospho Ser179 (mouse sequence) RapGTP indirectly binds to phospho Ser297 (mouse sequence) on the KSR dimer with B-Raf, independent of interaction through the Ras-binding domain of B-Raf. Thus, the net effect of PKA phosphorylation of B-Raf is no change in activity.
PKA also phosphorylates C-Raf and inhibits its activity by preventing phosphorylated C-Raf from binding RasGTP. PKA phosphorylation of Ser259 (human sequence) (at the 14-3-3 protein binding site) inhibits C-Raf activity (in NIH3T3, HEK293 and fibroblasts) by preventing C-Raf phosphorylation at Ser 338 (human sequence) (Häfner et al., 1994; Li et al., 2016; Wu et al., 1993), which is required for binding to RasGTP. PKA phosphorylation of C-Raf at Ser259 keeps the kinase in its closed inactive form, and dephosphorylation is required to expose the catalytic site (Häfner et al., 1994; Li et al., 2016). Thus, PKA acts as a negative regulator by attenuating C-Raf/RasGTP-binding.
4.2.1.2. Epac
The exchange factor directly activated by cAMP (Epac) is a direct target for cAMP and a guanine-nucleotide exchange factor (GEF) for the small GTPase Rap (Enserink et al., 2002). Two isoforms of Epac have been found so far: Epac1 is ubiquitously distributed while Epac2 is mostly expressed in the brain and the adrenal gland (Kawasaki et al., 1998). Both Epac isoforms (RAPGEF3 and RAPGEF4, Table 1) share homologous sequences except that Epac2 has an extra CBD domain, and has higher affinity to RapGDP than Epac1 (de Rooij et al., 2000). The mechanism of ERK activation through Epac is quite direct: a single cAMP binds to Epac, which exposes the Rap-binding site; RapGDP binds to Epac-cAMP, allowing the exchange of GDP to GTP; RapGTP then starts the sequential phosphorylation of the ERK cascade. Epac plays various roles in synaptic plasticity such as converting a short lasting LTP into long lasting LTP (Cheung et al., 2006; Gelinas et al., 2008). In non-neuronal cells, Epac is important in cell survival and apoptotic gene stimulation (Calderón-Sánchez et al., 2016), human chorionic gonadotropin gene expression (Maymó et al., 2012), inducing histone deacetylase in lung cancer (Lim and Juhnn, 2016), among others.
4.2.1.3. Models of cAMP activation of ERK
Given the importance of PKA, many computational models include PKA pathways (Table 4). PKA activation of ERK is important in neurons because many synaptic inputs produce elevations in cAMP, either through Gs-coupled metabotropic receptors, such as the dopamine D1 or β-adrenergic receptors (O’Dell et al., 2015), or through elevations in calcium caused by activation of calcium permeable glutamate receptors or voltage dependent calcium channels (Park et al., 2016). In most neuron models, PKA activates ERK through phosphorylation of Src and subsequent activation of B-Raf (Jain and Bhalla, 2014; Neves et al.,2008; Sasagawa et al., 2005). The dominance of B-Raf in the nervous system (Mercer and Pritchard, 2003) may explain why computational models of CA1 hippocampal neurons, striatal spiny projection neurons but not cardiac, or HEK293 cells, include PKA activation of B-Raf Some models also include PKA inhibition of ERK phosphatases (Neves et al., 2008), or PKA inhibition of C-Raf (Xu et al., 2010). Though not included in most models, PKA can increase ERK activation indirectly, by phosphorylation of calcium and NMDA channels to increase calcium influx, which enhances ERK activation through PKC and CaMKII, as described in the next section.
Table 4:
Summary of some key ERK models which include cAMP pathways. Main message summarizes the most important findings regarding cAMP activation of ERK. Key components summarize unique aspects of cAMP activation of ERK. Sole cAMP target is PKA unless specified.
| Model name | Main message | Key components | PKA targets |
|---|---|---|---|
| Neves et al., 2008 (Hippocampal) | Spatial patterns of activity require feedforward excitation and thin dendritic diameter | cAMP produced by β-AR; No Ras activation of B-Raf | Inhibitory: PTP Excitatory: B-Raf, PDE4 |
| Xu et al., 2010 (PC12 and HEK293) | EGFR and Epac activate ERK while PKA slightly inhibits ERK through inhibition of C-Raf | cAMP input which activates PKA and Epac; Epac activates Rap | Inhibitory: C-Raf |
| Jain and Bhalla, 2014 (hippocampal) | mRNA synthesis is the result of the synergistic activity of ERK, CaMKIV and mTORC kinases | Adenylyl cyclase, activated by calcium, synthesizes cAMP from ATP | Inhibitory: PP1 (via Inhibitor 1); CaMKIV (via CaMKK) Excitatory: Rap (via Src and RapGAP), mTORC |
| Gutierrez-Arenas et al., 2014 (Striatum) | STEP activity dictates the dynamics of ERK in response to glutamate and dopamine inputs | cAMP produced by the dopamine D1 receptor; ERK dephosphorylation by STEP, which is regulated by PKA | Inhibitory: STEP, PP1 Excitatory: calcium influx through NMDAR |
| Khalilimeybodi et al., 2018 (Cardiac cells) | Gi signaling pathway to ERK activation facilitates hypertrophy gene expression through regulation of the hypertrophic factor GATA4 | PKA mediated switching of βAR from Gα to Gi, which then recruits ERK through Src | Inhibitory: β2AR Excitatory: ERK (through Gi) |
| Song et al., 2013 (Striatal neurons) | ERK inhibition of PDE4 during dopamine signaling modulates neuronal excitability by adjusting AMPAR internalization | Positive feedback loop to cAMP mediated by ERK inhibition of PDE4 | Inhibitory: PP1 (through I1 and DARPP32) Excitatory: B-Raf, PDE4 |
4.2.1.4. Differences between cell types
The cAMP-dependent pathways to ERK are quite diverse, both with respect to the effect on ERK (increase versus decrease) and with respect to intermediate molecules. In most experiments, cAMP activation enhances ERK activity (Clason et al., 2016; Emery et al., 2013; Grewal et al., 2000; Morozov et al., 2003), though there are reports of reductions in ERK activity (Häfner et al., 1994; Li et al., 2016; Wu et al., 1993). Though PKA phosphorylation of both B-Raf and C-Raf has been found in various cell types (Li et al., 2016; Takahashi et al., 2017, 2013), such as NIH3T3, HEK293, and PC 12 cells, the effect PKA phosphorylation differs, and in some cases PKA activates B-Raf indirectly through Src phosphorylation. Additional diversity stems from several cAMP activated molecules, such as Epac, or RapGEF2 (Emery et al., 2013) transducing cAMP into RapGTP activation.
4.2.2. Second messenger pathway: Calcium
The phosphorylation of ERK by calcium-activated pathways is important because of the role played by calcium in synaptic plasticity. Nearly all stimulation protocols that induce synaptic plasticity in neurons produce elevations in calcium and phosphorylation of ERK. In some neurons (e.g. hippocampal and neocortical pyramidal neurons), calcium leads to cAMP production, providing two pathways (through PKA and Epac) to ERK activation. However, in other neuron types (e.g. cerebellar Purkinje cells and striatal projection neurons) calcium does not lead to cAMP production. At least two types calcium-activated molecules can lead to ERK phosphorylation: a set of GEFS, including RasGRF and RasGRP (Table 1), and protein kinase C.
4.2.2.1. GEFs: RasGRF and RasGRP
Ras guanine nucleotide–releasing factor (RasGRF) constitute of a family of guanine nucleotide exchange factors (GEFs). The two main isoforms, GRF1 and GRF2, have 2 GEF domains that enable them to activate Ras in response to signals from a variety of neurotransmitter receptors (Farnsworth et al., 1995). GRF1 and GRF2 proteins are found predominantly in adult neurons of the central nervous system (Renata et al., 1997). RasGRF is activated by binding to calcium-bound calmodulin (CaMCa4) through an IQ motif Once activated, RasGRF binds to RasGDP and allows the exchange of GDP to GTP. Though present in small amounts in non-neuronal tissues, RasGRF plays a role in ERK signaling pathways, for example in lymphatic leukemia (Liao et al., 2016). With respect to learning and synaptic plasticity, RasGRF enhances LTP in the hippocampus (Darcy et al., 2014).
Ras guanine nucleotide-releasing proteins (RasGRPs), expressed in all cell types, are another family of GEFs that initiate the activation of ERK through either Ras or Rap activation (Madani et al., 2004). There are four isoforms of RasGRP (also called CalDagGEF): RasGRP1, RasGRP2, RasGRP3 and RasGRP4. Similar to Sos, RasGRPs catalytic domain is composed of a CDC25 domain (Ras activator) and Ras exchange motif that interact with both RasGDP and RapGDP. RasGRP also exhibits about 50% similarity to RasGRF (Farnsworth et al., 1995). In addition to the catalytic domain, RasGRPs have a pair of atypical EF-hands that bind to calcium and C1 domains functionally resembling the diacylglycerol (DAG)-binding domains of PKC. Thus, either calcium and DAG (for RasGRP2) or DAG (for RasGRP1) directly activate RasGRPs (Stefanini and Bergmeier, 2010), which then activate RasGDP or RapGDP to initiate the Raf-MEK-ERK kinase cascade. Because DAG is a membrane molecule, activation of RasGRP involves a membrane recruitment mechanism.
4.2.2.2. PKC
The protein kinase C (PKC) family of serine/threonine protein kinase enzymes are key mediators of several signaling transduction pathways and are activated in response to a variety of extracellular stimuli. PKC, distributed in all tissue but highly concentrated in the brain, contains 3 functional protein types, “conventional-activated by calcium and DAG”, “novel-activated by DAG only”, and “atypical-not activated by calcium or DAG”. Depending on the isoform, PKC may contain functional C1 and/or C2 regulatory domains, which bind DAG or calcium, respectively. When extracellular signals activate appropriate GPCRs, they release the Gq subunits which bind and activate phospholipase Cβ (PLCβ), which produces IP3 and DAG. Alternatively, PLCγ can be activated by RTKs. IP3 binds to its receptor in the endoplasmic reticulum, releasing calcium, which together with DAG activate PKC. Once PKC is activated, it phosphorylates C-Raf to either enhance its activity or activate it in the absence of RasGTP, thus causing ERK activation. Phosphorylated ERK can phosphorylate phospholipase A2 (CPLA2) in the cytoplasm leading to its activation and production of arachidonic acid that acts synergistically with DAG to cause a persistent activation of PKC. This positive feedback loop between PKC and ERK activation is involved in the induction of synaptic plasticity in some brain regions (Tanaka and Augustine, 2008).
4.2.2.3. Models of calcium activation of ERK
The secondary messenger calcium is important for cellular signaling, as it exerts regulatory effects on many enzymes. Given the importance of calcium, many computational models (Table 5) include calcium activation of ERK. In most of these models, intracellular increases of calcium activate PKC which not only activates but sustains ERK activation via the positive feedback loop via the ERK-PLA2-PKC-C-Raf loop (Ajay and Bhalla, 2007; Antunes and Schutter, 2012). Gallimore et al., (2018), shows how presynaptic production of nitric oxide enhances the positive feedback loop by preventing phosphatase inhibition of PLA2. Though RasGRF and RasGRP are widely distributed, only one computational model (Gutierrez-Arenas et al., 2014) incorporates calcium activation of RasGRF. That model of a dendritic spine in the striatum demonstrates the requirement of both cAMP though PKA and calcium through RasGRF for ERK activation.
Table 5:
Summary of key ERK models which include PKC.Main message summarizes the most important findings in PKC models. Key aspects summarize unique aspects of PKC and its activation of ERK.
| Model name | Main message | Key aspects | PKC contribution |
|---|---|---|---|
| Ajay and Bhalla, 2007 (Hippocampal) | Propagation of ERK activation along the dendrite is the result of both electrical and biochemical activities | Spatial model, positive feedback loop of PKC makes the model bistable | Positive feedback loop by PLA2/PKC/ERK, phosphorylation of C-Raf |
| Antunes and De Schutter, 2012 (Cerebellar) | Both LTD induction and ERK activation are bistable and probabilistic in a single spine | Stochastic simulation; calcium activation of PKC; PKC mediates AMPAR removal from synapse; PP2A dephosphorylates AMPAR, MEK and inhibits PLA2 | Positive feedback loop by PLA2/PKC/ERK |
| Gallimore et al., 2018 (Cerebellar) | Calcium below threshold produces LTD; calcium above threshold, which activates CaMKII, is needed to turn on PKC loop for LTD. NO facilitates the PKC loop. | PLC contributes post-synaptically to PKC and pre-synaptically to NO; PKC feedback loop activity is sensitive to CaMKII activity. Both NO and CaMKII work through PP2A inhibition | Positive feedback loop by PLA2/PKC/ERK, phosphorylation of C-Raf and inhibition of NO |
| Heitzler et al., 2012 (Cerebellar) | Competition between GRK2/3 and GRK5/6 for receptor phosphorylation; GRK2/3 inhibits β-arrestin signaling pathway | Gq coupled receptor activates PKC through PLC; GRK5/6 phosphorylation of receptor leads to β-arrestin recruitment and G protein independent ERK phosphorylation | Simplified, direct activation of ERK by PKC |
| Hepburn et al., 2017 (Cerebellar) | The probability of LTD induction is influenced by the number of PKC molecules; AMPAR binding to GRIP improves model robustness and decreases fluctuations of some molecules | Update of Antunes and De Schutter (2012) to include RKIP inhibition of Raf and MEK, relieved by PKC phosphorylation of RKIP; AMPAR binding to GRIP | Positive feedback loop by PLA2/PKC/ERK |
| Wang et al., 2008, 2007 (Lung cancer cell) | EGF stimulus in the presence of glucose and oxygen impact the migration pattern. PKC-PLC pathway is the most sensitive and critical | EGFR does not recruit GEF; it recruits PLCγ. Migration vs proliferation determined by PLC and ERK | Phosphorylation of Raf by PKC |
4.2.2.4. Differences between cell types
The pathway for PKC activation of ERK seems to be the same in all cells; namely, PKC phosphorylates and enhances C-Raf activity. However, PKC isoforms differ between tissue types, and between types of cancer cells. For example, PKCγ is expressed solely in neurons while PKCα and PKCβ are expressed in many cell types, including neurons (Ito et al., 1990). Given the diversity in PKC isoforms, activation of PKC may differ between cell types. A key difference in models that incorporate PKC activation of ERK, is that neuron models (hippocampal pyramidal cells, cerebellar Purkinje cells, and striatal spiny projection neurons) incorporate the ERK-PLA2-PKC feedback loop, which is absent in other models. It is unclear whether feedback loops in other cells such as PC12, lung cells (Wang et al., 2008, 2007) are not needed for prolonged ERK activation due to typically prolonged stimuli of growth factors.
5. Inactivation of ERK by GAPs
The Raf/MEK/ERK pathways are terminated by dephosphorylation of their components by several phosphatases such as PP2A and MKP, which were discussed above. Upstream of Raf, RasGTP inactivation is mediated by GTPase Activating Proteins (GAPs). GAPs accelerate the inactivation of the G protein’s activity by catalyzing the hydrolysis of GTP to GDP. GAPs are needed because the intrinsic GTPase activity of the small GTP-binding proteins is quite low. As with GEFs, there are often multiple GAPs that function on a given Ras family protein.
5.1. Classical GAPs: RasGAP and Rap1GAP
Several GAPs, encoded by different genes (Table 1; reviewed in Bos et al., 2007) have been characterized as regulating ERK activation. RasGAP inactivates RasGTP by stabilizing the position of Gln61 (human sequence) of Ras (Bos et al., 2007; Lu et al., 2016), which speeds up the GTP hydrolysis reaction. Rap1GAP accelerates RapGTP hydrolysis with the same mechanism as RasGAP but stabilizes the position of thr61 of Rap1 (Bos et al, 2007). Though Rap1GAP is constitutively active, it is downregulated by PKA (McAvoy et al., 2009), providing another mechanism whereby PKA can enhance ERK activity. Phosphorylation of Rap1GAP by PKA on Ser441 and Ser499 (human sequence) inhibits GAP activity, as demonstrated by increased RapGTP (McAvoy et al., 2009).
5.2. SynGAP
SynGAP is a synaptic RasGAP selectively expressed in the brain and found at higher concentrations specifically at excitatory synapses in the mammalian forebrain (Kim et al., 1998). It is localized to the postsynaptic density by binding to the PDZ domain of PSD-95 (Walkup IV et al., 2016) and exhibits GTPase activity toward Ras and Rap. SynGAP is critical during neuronal development since mice lacking SynGAP die postnatally. Furthermore, heterozygous null SynGAP mutant mice exhibit reduced LTP and perform poorly in spatial memory tasks (Kim et al., 2003). SynGAP is phosphorylated by CaMKII at Ser773 and Ser802 (rat sequence) (Oh et al., 2004; Walkup IV et al., 2015), which increases its activity toward both RasGTP and RapGTP (Walkup et al., 2015). CaMKII phosphorylation also causes SynGAP dispersion from the synapses during LTP, and thus attenuates GAP activity in the spine, which allows a long-lasting spine enlargement (Araki et al., 2015). Thus, the overall CaMKII impact on SynGAP during or after LTP induction is an increase in RasGTP, which facilitates LTP through enhanced ERK activation.
5.3. Differences between cell types
RasGTP/RapGTP serve as important molecules which start the sequential phosphorylation of the ERK cascade. Thus, regulation of their activity is crucial for proper function. Cell type specific differences in their regulation by the classical GAPs have not been noted. On the other hand, GAP activity appears to be unique in pyramidal neurons due to the high quantity of SynGAP in dendritic spines and its regulation by CaMKII.
6. Conclusion
In summary, ERK pathways do not differ structurally between neurons and non-neuronal cells, in that molecule pathways seem to be identical. The difference seems to be in which molecules are expressed and their quantity. For example, SynGAP is not expressed in non-neuronal cells, but in neurons is important for calcium regulation of ERK activation. Another difference is the role of PKA, which inhibits ERK in non-neuronal cells but not in neurons, likely because B-Raf activation, which is not inhibited by PKA, contributes more to ERK activation than C-Raf in neurons.
Computational models slightly diverge from experimental observations in that not all past or recent discoveries have been incorporated into the computational models. Several of these experimentally observed molecule interactions may need to be included in models to better understand ERK dynamics and its role in synaptic plasticity. For example, Raf dimerization has not been included in computational models to date, which may account for the faster than observed ERK activation in most computational (e.g. Jain and Bhalla, 2014; Schoeberl et al., 2002). Including C-Raf dimerization in computational models will delay the Raf activity and thereby delay ERK activation, more aligned with in vivo experiments. In addition, models of the ERK cascade do not include RapGTP binding with C-Raf, which forms an inactive complex. Including this competitive (and inhibitory) reaction may both reduce C-Raf activity (due to binding with RapGTP) and B-Raf activity (due to competition with C-Raf for RapGTP), and thus affect the magnitude and duration of ERK activity. Another key difference between models and experiments is the lack of Epac in most models. Despite its role in LTP (Gelinas et al. 2008) and cell survival (Calderón-Sánchez et al., 2016), Epac2 has not been included in enough computational models to evaluate differences. Including this RapGEF directly activated by cAMP in computational models may enhance basal ERK activation by GPCRs coupled to cAMP. Another difference between experiment and model is the regulation of RasGTP/RapGTP. Even though SynGAP is a key molecule connecting CaMKII to ERK, neuron modeling papers have not yet incorporated this molecule. Incorporating the calcium control of SynGAP will mimic better the modulation of GAP activity by in vivo activity.
Most computational models of the ERK cascade in both neurons and non-neuronal cells assume a single, well-stirred compartment in which molecule interactions are not limited by spatial encounters. Very few models are spatial because of the difficulty and complexity of spatial modeling. All spatial models are of neurons (Ajay and Bhalla, 2007; Neves et al., 2008), likely because a key information processing feature of neurons is their spatially extended dendrites. Even in cell types that are morphologically simple, research demonstrates that the scaffolding of the core kinases of the ERK cascade both enhance ERK activity and regulates the spatial location of phosphorylated ERK with consequences for downstream targets. Including this aspect of ERK activation requires spatial models and might provide insight into the flow of information from receptor to ERK then to cytosolic or nuclear targets.
Highlights.
Some functions of ERK1 and ERK2 are redundant, whereas other ERK2 functions are unique
ERK scaffolds both enhance ERK activation and segregates pools of ERK
BDNF is required for long term potentiation in response to weak stimulation protocols
G protein coupled receptors activate ERK through diverse, cell type dependent pathways
Acknowledgements:
This work was supported through the joint NIH-NSF CRCNS program through NSF grant 1515686 and NIAAA R01 AA016022
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors declare no competing financial interests.
References
- Aarse J, Herlitze S, Manahan-Vaughan D, 2016. The requirement of BDNF for hippocampal synaptic plasticity is experience-dependent. Hippocampus 26, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams DG, Coffee RL, Zhang H, Pelech S, Strack S, Wadzinski BE, 2005. Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J. Biol. Chem 280, 42644–42654. [DOI] [PubMed] [Google Scholar]
- Ajay SM, Bhalla US, 2007. A propagating ERKII switch forms zones of elevated dendritic activation correlated with plasticity. HFSP J. 1, 49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuler D, Lapetina EG, 1993. Analysis of the CAMP-dependent Protein Kinase-mediated Phosphorylation Site of Raplb. J. Biol. Chem 268, 7527–7631. [PubMed] [Google Scholar]
- Antunes G, Schutter ED, 2012. A Stochastic Signaling Network Mediates the Probabilistic Induction of Cerebellar Long-Term Depression. J. Neurosci 32, 9288–9300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoidi R, Maltais A, Charron J, 2016. Functional redundancy of the kinases MEK1 and MEK2: Rescue of the Mek1 mutant phenotype by Mek2 knock-in reveals a protein threshold effect. Sci. Signal 9. [DOI] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, Huganir RL, 2015. Rapid Dispersion of SynGAP from Synaptic Spines Triggers AMPA Receptor Insertion and Spine Enlargement during LTP. Neuron 85, 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balan V, Leicht DT, Zhu I, Balan K, Kaplun A, Singh-Gupta V, Qin J, Ruan H, Comb MJ, Tzivion G, 2006. Identification of Novel In Vivo Raf-1 Phosphorylation Sites Mediating Positive Feedback Raf-1 Regulation by Extracellular Signal-regulated Kinase. Mol. Biol. Cell 17, 1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger L-F, Roy S, Tremblay M, Brott B, Stefi A-M, Mourad W, Hugo P, Erikson R, Charron J, 2003. Mek2 Is Dispensable for Mouse Growth and Development. Mol. Cell. Biol 23, 4778–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonauth V, Roy S, Gravel M, Guillemette S, Charron J, 2006. Requirement for Map2k1 (Mek1) in extra-embryonic ectoderm during placentogenesis. Development 133, 3429–3440. [DOI] [PubMed] [Google Scholar]
- Bos JL, Rehmann H, Wittinghofer A, 2007. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 129, 865–877. [DOI] [PubMed] [Google Scholar]
- Bourquard T, Landomiel F, Reiter E, Crépieux P, Ritchie DW, Azé J, Poupon A, 2015. Unraveling the molecular architecture of a G protein-coupled receptor/β-arrestin/Erk module complex. Sci. Rep 5, 10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MT, Cooper JA, 1996. Regulation, substrates and functions of src. Biochim. Biophys. Acta 1287, 121–149. [DOI] [PubMed] [Google Scholar]
- Brummer T, Naegele H, Reth M, Misawa Y, 2003. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of B-Raf. Oncogene 22, 8823. [DOI] [PubMed] [Google Scholar]
- Buscà R, Pouysségur J, Lenormand P, 2016. ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy? Front. Cell Dev. Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnol S, Chambard J-C, 2010. ERK and cell death: Mechanisms of ERK-induced cell death -apoptosis, autophagy and senescence. FEBS J. 277, 2–21. [DOI] [PubMed] [Google Scholar]
- Calderón-Sánchez E, Díaz I, Ordóñez A, Smani T, 2016. Urocortin-1 Mediated Cardioprotection Involves XIAP and CD40-Ligand Recovery: Role of EPAC2 and ERK 1/2. PLOS ONE 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey KD, Watson RT, Pessin JE, Stork PJS, 2003. The Requirement of Specific Membrane Domains for Raf-1 Phosphorylation and Activation. J. Biol. Chem 278, 3185–3196. [DOI] [PubMed] [Google Scholar]
- Caunt CJ, Keyse SM, 2013. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 280, 489–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerovic M, Bagetta V, Pendolino V, Ghiglieri V, Fasano S, Morelia F, Hardingham N, Heuer A, Papale A, Marchisella F, Giampa C, Calabresi P, Picconi B, Brambilla R, 2015. Derangement of Ras-Guanine Nucleotide-Releasing Factor 1 (Ras-GRFl) and Extracellular Signal-Regulated Kinase (ERK) Dependent Striatal Plasticity in L-DOPA-Induced Dyskinesia. Biol. Psychiatry, Schizophrenia and Neurodevelopment 77, 106–115. [DOI] [PubMed] [Google Scholar]
- Chambard J-C, Lefloch R, Pouyssegur J, Lenormand P, 2007. ERK implication in cell cycle regulation. Biochim. Biophys. Acta, Mitogen-Activated Protein Kinases: New Insights on Regulation, Function and Role in Human Disease 1773, 1299–1310. [DOI] [PubMed] [Google Scholar]
- Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A, 1999. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J. Neurosci. Off J. Soc. Neurosci 19, 7983–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung U, Atwood HL, Zucker RS, 2006. Presynaptic effectors contributing to cAMP-induced synaptic potentiation inDrosophila. J. Neurobiol 66, 273–280. [DOI] [PubMed] [Google Scholar]
- Cirit M, Wang C-C,Haugh JM, 2010. Systematic Quantification of Negative Feedback Mechanisms in the Extracellular Signal-regulated Kinase (ERK) Signaling Network. J. Biol. Chem 285, 36736–36744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clason TA, Girard BM, May V, Parsons RL, 2016. Activation ofMEK/ERK Signaling by PACAP in Guinea Pig Cardiac Neurons. J. Mol. Neurosci. MN 59, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicelli J, 2004. Human RAS Superfamily Proteins and Related GTPases. Sci. Signal 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor SA, Maity S, Roy B, Ali DW, Nguyen PV, 2012. Conversion of short-term potentiation to long-term potentiation in mouse CA1 by coactivation of β-adrenergic and muscarinic receptors. Leam. Mem 19, 535–542. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Trakul N,Eves EM, Diaz B, Marshall M, Rosner MR, 2003. Activation of Raf-1 Signaling by Protein Kinase C through a Mechanism Involving Raf Kinase Inhibitory Protein. J. Biol. Chem 278, 13061–13068. [DOI] [PubMed] [Google Scholar]
- Corson LB, Yamanaka Y, Lai K-MV, Rossant J, 2003. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development 130, 4527–4537. [DOI] [PubMed] [Google Scholar]
- Darcy MJ, Trouche S, Jin S-X,Feig LA, 2014. Ras-GRF2 Mediates LTP, Survival and Response to an Enriched Environment of Newborn Neurons in the Hippocampus. Hippocampus 24, 1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S,2000. The MAPK/ERK Cascade Targets Both Elk-1 and cAMP Response Element-Binding Protein to Control Long-Term Potentiation-Dependent Gene Expression in the Dentate Gyms In Vivo. J. Neurosci 20, 4563–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL, 2000. Mechanism of Regulation of the Epac Family of cAMP-dependent RapGEFs. J. Biol. Chem 275, 20829–20836. [DOI] [PubMed] [Google Scholar]
- DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW,2000. β-Arrestin-Dependent Endocytosis of Proteinase-Activated Receptor 2 Is Required for Intracellular Targeting of Activated Erkl/2. J. Cell Biol 148, 1267–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desideri E, Cavallo AL, Baccarini M, 2015. Alike but Different: RAF Paralogs and Their Signaling Outputs. Cell 161, 967–970. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, Kolch W, 2007. MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W, 2002. Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J. 21, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty MK, Ritt DA, Zhou M, Specht SI, Monson DM, Veenstra TD, Morrison DK, 2009. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol. Cell 34, 652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebisuya M, Kondoh K, Nishida E, 2005. The duration, magnitude and compartmentalization of ERK MAP kinase activity: mechanisms for providing signaling specificity. J. Cell Sci 118,2997–3002. [DOI] [PubMed] [Google Scholar]
- Emerson SD, Madison VS, Palermo RE.,Waugh DS, Schelfler JE, Tsao K-L, Kiefer SE, Liu SP, Fry DC, 1995. Solution Structure of the Ras-Binding Domain of c-Raf-1 and Identification of Its Ras Interaction Surface. Biochemistry 34,6911–6918. [DOI] [PubMed] [Google Scholar]
- Emery AC, Eiden MV, Mustafa T, Eiden LE, 2013, Rapget2 connects GPCR-mediated cAMP signals to ERK activation in neuronal and endocrine cells. Sci. Signal 6, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, deRooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL, 2002. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol 4, 901–906. [DOI] [PubMed] [Google Scholar]
- Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA, 1995. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 376, 524–7. [DOI] [PubMed] [Google Scholar]
- Frémin C, Saba-El-Leil MK, Levesque K,Ang S-L, Meloche S, 2015. Functional Redundancy of ERK 1 and ERK2 MAP Kinases during Development. Cell Rep. 12, 913–921. [DOI] [PubMed] [Google Scholar]
- Gallimore AR, Kim T, Tanaka-Yamamoto K, De Schutter E, 2018. Switching On Depression and Potentiation in the Cerebellum. Cell Rep. 22, 722–733. [DOI] [PubMed] [Google Scholar]
- Gao C,Frausto SF, Guedea AL, Tronson NC, Jovasevic V, Leaderbrand K, Corcoran KA, Guzman YF, Swanson GT, Radulovic J, 2011. IQ GAP 1 regulates NR2A signaling, spine density, and cognitive processes. J. Neurosci. Off J. Soc. Neurosci 31, 8533–8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas JN, Banko JL, Peters MM, Klann E, Weeber EJ, Nguyen PV, 2008. Activation of exchange protein activated by cyclic-AMP enhances long-lasting synaptic potentiation in the hippocampus. Leam. Mem 15,403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry L, Samatar AA, Der CJ, 2013. Inhibitors of the ERK Mitogen-Activated Protein Kinase Cascade for Targeting RAS Mutant Cancers, in: The Enzymes. Elsevier, pp. 67–106. [DOI] [PubMed] [Google Scholar]
- Giroux S, Tremblay M, Bernard D, Cardin-Girard J-F, Aubry S, Larouche L, Rousseau S, Huot J, Landry J, Jeannotte L, Charron J, 1999. Embryonic death of Mekl-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr. Biol 9, 369–376. [DOI] [PubMed] [Google Scholar]
- Gobert D, Topolnik L, Azzi M, Huang L, Badeaux F, DesGroseillers L, Sossin WS, Lacaille J-C, 2008. Forskolin induction of late-LTP and up-regulation of 5’TOP mRNAs translation via mTOR, ERK, and PI3K in hippocampal pyramidal cells. J. Neurochem 106, 1160–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SS, Horgan AM, York RD, Withers GS, Banker GA, Stork PJS, 2000. Neuronal Calcium Activates aRapl and B-Raf Signaling Pathway via the Cyclic Adenosine Monophosphate-dependent Protein Kinase. J. Biol. Chem 275, 3722–3728. [DOI] [PubMed] [Google Scholar]
- Häfner S, Adler HS, Mischak H, Janosch P, Heidecker G, Wolfrnan A, Pippig S, Lohse M, Ueffing M, Kolch W, 1994. Mechanism of inhibition of Raf-1 by protein kinase A. Mol. Cell. Biol 14, 6696–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoune J, Defer N, 2001. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol 41, 145–174. [DOI] [PubMed] [Google Scholar]
- Hatano N, Mori Y, Oh- hora M, Kosugi A, Fujikawa T, Nakai N, Niwa H, Miyazaki J, Hamaoka T, Ogata M, 2003. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 8, 847–856. [DOI] [PubMed] [Google Scholar]
- Hawes SL, Gillani F, Evans RC, Benkert EA, Blackwell KT, 2013. Sensitivity to theta-burst timing permits LTP in dorsal striatal adult brain slice. J. Neurophysiol 110, 2027–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzler D, Durand G, Gallay N, Rizk A, Ahn S, Kim J, Violin JD, Dupuy L, Gauthier C, Piketty V, Crepieux P, Poupon A, Clement F, Fages F, Lefkowitz RJ, Reiter E, 2012. Competing G protein-coupled receptor kinases balance G protein and β-arrestin signaling. Mol. Syst. Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman M, Fischer A, Wennogle LP, Wang YK, Campbell SL, Rapp UR, 2005. Novel C-Raf phosphorylation sites: serine 296 and 301 participate in Raf regulation. FEBS Lett. 579, 464–468. [DOI] [PubMed] [Google Scholar]
- Ho Y-D, Joyal JL, Li Z, Sacks DB, 1999. IQGAP1 Integrates Ca2+/Calmodulin and Cdc42 Signaling. J. Biol. Chem 274, 464–470. [DOI] [PubMed] [Google Scholar]
- Hong S-Κ, Wu P-Κ, Karkhanis M, Park J-I, 2015. ERK1/2 can feedback-regulate cellular MEK1/2 levels. Cell. Signal 27, 1939–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C-D, Kariya K,Kotani G, Shirouzu M, Yokoyama S, Kataoka T, 1997. Coassociation of Rap 1A and Ha-Ras with Raf-1 N-terminal Region Interferes with Ras-dependent Activation of Raf-1. J. Biol. Chem 272, 11702–11705. [DOI] [PubMed] [Google Scholar]
- Ito A, Saito N, Hirata M, Kose A, Tsujino T, Yoshihara C, Ogita K, Kishimoto A, Nishizuka Y, Tanaka C, 1990. Immunocytochemical localization of the alpha subspecies of protein kinase C in rat brain. Proc. Natl. Acad. Sci. U. S. A 87, 3195–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P, Bhalla US, 2014. Transcription Control Pathways Decode Patterned Synaptic Inputs into Diverse mRNA Expression Profiles. PLoS ONE 9, e95154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Gall CM, Lynch G, 2010. Presynaptic BDNF Promotes Postsynaptic Long-Term Potentiation in the Dorsal Striatum. J. Neurosci 30, 14440–14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D-Z, Mao L-M, Wang JQ, 2019. Amphetamine activates non-receptor tyrosine kinase Fyn and stimulates ERK phosphorylation in the rat striatum in vivo. Eur. J. Pharmacol 843, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanterewicz BI, Urban NN, McMahon DB, Norman ED, Giffen LJ, Favata MF, Scherle PA, Trzskos JM, Barrionuevo G, Klann E, 2000. The extracellular signal-regulated kinase cascade is required for NMDA receptor-independent LTP in area CA1 but not area CA3 of the hippocampus. J. Neurosci. Off J. Soc. Neurosci 20, 3057–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao S, Jaiswal RK, Kolch W, Landreth GE, 2001. Identification of the Mechanisms Regulating the Differential Activation of the MAPK Cascade by Epidermal Growth Factor and Nerve Growth Factor in PC12 Cells. J. Biol. Chem 276, 18169–18177. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman E, Graybiel AM, 1998. A Family of cAMP-Binding Proteins That Directly Activate Rapl. Science 282, 2275–2279. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, Govindarajan A, Jung H-Y, Kang H, Tonegawa S, 2004. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116, 467–479. [DOI] [PubMed] [Google Scholar]
- Khokhlatchev AV, Canagarajah B, Wilsbacher J, Robinson M, Atkinson M, Goldsmith E, Cobb MH, 1998. Phosphorylation of the MAP Kinase ERK2 Promotes Its Homodimerization and Nuclear Translocation. Cell 93, 605–615. [DOI] [PubMed] [Google Scholar]
- Kholodenko BN, 2006. Cell Signaling Dynamics in Time and Space. Nat. Rev. Mol. Cell Biol 7, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee H-K, Takamiya K, Huganir RL, 2003. The Role of Synaptic GTPase-Activating Protein in Neuronal Development and Synaptic Plasticity. J. Neurosci 23, 1119–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau L-F, Huganir RL, 1998. SynGAP: a Synaptic RasGAP that Associates with the PSD-95/SAP90 Protein Family. Neuron 20, 683–691. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wol E, Brem G, Thoenen H, Bonhoeffer T, 1995. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc. Natl. Acad. Sci 92, 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake D, Corrêa SAL, Müller J, 2016. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci 73,4397–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langeberg LK, Scott JD, 2015. Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol 16, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK, 2005. Transduction of Receptor Signals by β-Arrestins. Science 308, 512–517. [DOI] [PubMed] [Google Scholar]
- Li Y, Dillon TJ, Takahashi M, Earley KT, Stork PJS, 2016. Protein Kinase A-independent Ras Protein Activation Cooperates with Rapl Protein to Mediate Activation of the Extracellular Signal-regulated Kinases (ERK) by cAMP. J. Biol. Chem 291, 21584–21595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Sharma S, Liao W, Sharma S, 2016. Modulation of B-cell receptor and microenvironment signaling by a guanine exchange factor in B-cell malignancies. Cancer Biol. Med 13, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JA, Juhnn Y-S, 2016. Isoproterenol increases histone deacetylase 6 expression and cell migration by inhibiting ERK signaling via PKA and Epac pathways in human lung cancer cells. Exp. Mol. Med 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullrich A, Skolnik EY, Bar-Sagi D, Schlessinger J, 1992. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70, 431–442. [DOI] [PubMed] [Google Scholar]
- Madani S, Hichami A, Charkaoui-Malki M, Khan NA, 2004. Diacylglycerols Containing Omega 3 and Omega 6 Fatty Acids Bind to RasGRP and Modulate MAP Kinase Activation. J. Biol. Chem 279,1176–1183. [DOI] [PubMed] [Google Scholar]
- Maik-Rachline G, Hacohen-Lev-Ran A, Seger R, 2019. Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int. J. Mol. Sci 20, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Moreno A, Rodriguez-Duran LF, Escobar ML, 2011. Late Protein Synthesis-Dependent Phases in CTA Long-Term Memory: BDNF Requirement. Front. Behav. Neurosci 5,61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R, 1999. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 18, 2137–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, Kolch W, 2011. Raf Family Kinases: Old Dogs Have Learned New Tricks. Genes Cancer 2, 232–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateer SC, McDaniel AE, Nicolas V, Habermacher GM, Lin M-JS, Cromer DA, King ME, Bloom GS, 2002. The Mechanism for Regulation of the F-actin Binding Activity of IQGAP1 by Calcium/Calmodulin. J. Biol. Chem 277, 12324–12333. [DOI] [PubMed] [Google Scholar]
- Maymó JL, Pérez Pérez A, Maskin B, Dueñas JL, Calvo JC, Sánchez Margalet V, Varone CL, 2012. The Alternative Epac/cAMP Pathway and the MAPK Pathway Mediate hCG Induction of Leptin in Placental Cells. PLoS ONE 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAvoy T, Zhou M, Greengard P, Naim AC, 2009. Phosphorylation of Rap1GAP, a striatally enriched protein, by protein kinase A controls Rapl activity and dendritic spine morphology. Proc. Natl. Acad. Sci 106, 3531–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy CE, Campbell DG, Deak M, Bloomberg GB, Arthur JSC, 2005. MSK1 activity is controlled by multiple phosphorylation sites. Biochem. J 387, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer KE, Pritchard CA, 2003. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta 1653, 25–40. [DOI] [PubMed] [Google Scholar]
- Michailovici I, Harrington HA, Azogui HH, Yahalom-Ronen Y, Plotnikov A, Ching S, Stumpf MPH, Klein OD, Seger R, Tzahor E, 2014. Nuclear to cytoplasmic shuttling of ERK promotes differentiation of muscle stem/progenitor cells. Development 141, 2611–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, Olariu A, Nawa H, Nabeshima T, 2000. Involvement of Brain-Derived Neurotrophic Factor in Spatial Memory Formation and Maintenance in a Radial Arm Maze Test in Rats. J. Neurosci 20, 7116–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, Matsuda M, 2001. Spatio-temporal images of growth-factor-induced activation of Ras and Rapl. Nature 411, 1065. [DOI] [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER, 2003. Rapl couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 39, 309–325. [DOI] [PubMed] [Google Scholar]
- Müller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK, 2001. C-TAK1 Regulates Ras Signaling by Phosphorylating the MAPK Scaffold, KSR1. Mol. Cell 8, 983–993. [DOI] [PubMed] [Google Scholar]
- Nekrasova T, Shive C,Gao Y, Kawamura K, Guardia R, Landreth G, Forsthuber T, 2005. ERK 1-Deficient Mice Show Normal T Cell Effector Function and Are Highly Susceptible to Experimental Autoimmune Encephalomyelitis. J. Immunol 175,2374–2380. [DOI] [PubMed] [Google Scholar]
- Neves SR, Tsokas P, Sarkar A, Grace EA, Rangamani P, Taubenfeld SM, Alberini CM, Schaff JC, Blitzer RD, Moraru II, Iyengar R, 2008. Cell Shape and Negative Links in Regulatory Motifs Together Control Spatial Information Flow in Signaling Networks. Cell 133, 666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A, Burack WR, Stock JL, Kortum R, Chaika OV, Afkarian M, Muller WJ, Murphy KM, Morrison DK, Lewis RE., McNeish J, Shaw AS, 2002. Kinase Suppressor of Ras (KSR) Is a Scaffold Which Facilitates Mitogen-Activated Protein Kinase Activation In Vivo. Mol. Cell. Biol 22,3035–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien DE, Alter BJ, Satomoto M, Morgan CD, Davidson S,Vogt SK, Norman ME, Gereau GB, Demaro JA, Landreth GE, Golden JP, Gereau RW, 2015. ERK2 Alone Drives Inflammatory Pain But Cooperates with ERK1 in Sensory Neuron Survival. J. Neurosci. Off J. Soc. Neurosci 35, 9491–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dell TJ, Connor SA, Guglietta R, Nguyen PV, 2015. b-Adrenergic receptor signaling and modulation of long-term potentiation in the mammalian hippocampus. Cold Spring Harb. Lab. Press 22, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JS, Manzerra P, Kennedy MB, 2004. Regulation of the Neuron-specific Ras GTPase-activating Protein, synGAP, by Calcium /Calmodulin-dependent Protein Kinase II. J. Biol. Chem 279, 17980–17988. [DOI] [PubMed] [Google Scholar]
- Ohba Y, Kurokawa K,Matsuda M, 2003. Mechanism of the spatio-temporal regulation of Ras and Rapl. EMBO J. 22, 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK, 2003. Protein Phosphatase 2A Positively Regulates Ras Signaling by Dephosphorylating KSR1 and Raf-1 on Critical 14-3-3 Binding Sites. Curr. Biol 13, 1356–1364. [DOI] [PubMed] [Google Scholar]
- Ou L-C, Yeh S-H, Gean P-W, 2010. Late expression of brain-derived neurotrophic factor in the amygdala is required for persistence of fear memory. Neurobiol. Learn. Mem 93, 372–382. [DOI] [PubMed] [Google Scholar]
- Papin C, Denouel-Galy A, Laugier D, Calothy G, Eychène A, 1998. Modulation of Kinase Activity and Oncogenic Properties by Alternative Splicing Reveals a Novel Regulatory Mechanism for B-Raf J. Biol. Chem 273, 24939–24947. [DOI] [PubMed] [Google Scholar]
- Park H, Popescu A, Poo M, 2014. Essential Role of Presynaptic NMDA Receptors in Activity-Dependent BDNF Secretion and Corticostriatal LTP. Neuron 84, 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P, Sanderson TM, Amici M, Choi S-L, Bortolotto ZA, Zhuo M, Kaang B-K, Collingridge GL, 2016. Calcium-Permeable AMPA Receptors Mediate the Induction of the Protein Kinase A-Dependent Component of Long-Term Potentiation in the Hippocampus. J. Neurosci 36,622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER, 2001. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron 32, 123–140. [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Peterson JD, Xie Z, Kress GJ, Rafalovich I, Kondapalli J, Gertler TS, Flajolet M, Greengard P, Stavarache M, Kaplitt MG, Rosinski J, Chan CS, Surmeier DJ, 2014. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron 83, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Zhu Y, Baumgart JP, Stometta RL, Seidenman K,Mack V, van Aelst L, Zhu JJ, 2005. State-dependent Ras signaling and AMPA receptor trafficking. Genes Dev. 19, 2000–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J-G, Li Z, Sacks DB, 2008. IQGAP1 Integrates Ca2+/Calmodulin and B-Raf Signaling. J. Biol. Chem 283, 22972–22982. 10.1074/jbc.M804626200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J-G, Li Z, Sacks DB, 2007. IQGAP1 modulates activation of B-Raf. Cell Biol. 104, 10465–10469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renata Z, Nerina G, Noa M-L, Enzo M, Daniella S, Zvi V, Emmapaola S, 1997. Ras-GRF, the activator ofRas, is expressed preferentially in mature neurons of the central nervous system. Mol. Brain Res 48, 140–144. [DOI] [PubMed] [Google Scholar]
- Resat H, Straatsma TP, Dixon DA, Miller JH, 2001. The arginine linger of RasGAP helps Gln-61 align the nucleophilic water in GAP-stimulated hydrolysis of GTP. Proc. Natl. Acad. Sci 98, 6033–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritt DA, Monson DM, Specht SI, Morrison DK, 2010. Impact of Feedback Phosphorylation and Raf Heterodimerization on Normal and Mutant B-Raf Signaling. Mol. Cell. Biol 30, 806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth CW, Richert ND, Pastan I, 1983. Cyclic AMP Treatment of Rous Sarcoma Virus-transformed Chinese Hamster Ovary Cells Increases Phosphorylation of pp60SRC and Increases pp60SRC Kinase Activity. J. Biol. Chem 258, 10768–10773. [PubMed] [Google Scholar]
- Roy M, Li Z, Sacks DB, 2005. IQGAP1 Is a Scaffold for Mitogen-Activated Protein Kinase Signaling. Mol. Cell. Biol 25, 7940–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Li Z, Sacks DB, 2004. IQGAP1 Binds ERK2 and Modulates Its Activity. J. Biol. Chem 279, 17329–17337. [DOI] [PubMed] [Google Scholar]
- Ryu H, Chung M, Dobrzyhski M, Fey D, Blum Y, Lee SS, Peter M, Kholodenko BN, Jeon NL, Pertz O, 2015. Frequency modulation of ERK activation dynamics rewires cell fate. Mol. Syst. Biol 11, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzano M, Rusciano MR, Russo E, Bifulco M, Postiglione L, Vitale M, 2012. Calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylates Raf-1 at serine 338 and mediates Ras-stimulated Raf-1 activation. Cell Cycle 11,2100–2106. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE, 2008. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J. Neurosci. Off J. Soc. Neurosci 28, 6983–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasagawa S, Ozaki Y, Fujita K, Kuroda S, 2005. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat. Cell Biol 7,365–373. [DOI] [PubMed] [Google Scholar]
- Sasi M, Vignoli B, Canossa M, Blum R, 2017. Neurobiology of local and intercellular BDNF signaling. Pflugers Arch. 469, 593–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Swank MW, Rodrigues SM, Debiec J, Doyere V, 2008. Phosphorylation of ERK/MAP kinase is required for long-term potentiation in anatomically restricted regions of the lateral amygdala in vivo. Learn. Mem 15, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt JM, Stork PJS, 2002. PKA Phosphorylation of Src Mediates cAMP’s Inhibition of Cell Growth via Rapl. Mol. Cell 9, 85–94. [DOI] [PubMed] [Google Scholar]
- Schrick C, Fischer A, Srivastava DP, Tronson NC, Penzes P, Radulovic J, 2007. N-Cadherin Regulates Cytoskeletally-Associated IQGAP1/ERK Signaling and Memory Formation. Neuron 55, 786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seger R, Ahnl NG, MunarB E, JensenS AM, Cooper JA, Cobb MH, 1992. Purification and Characterization Of Mitogen-Activated Protein Kinase Activator(s) from Epidermal Growth Factor-Stimulate Ad431 Cells. J. Biol. Chem 267, 14373–14381. [PubMed] [Google Scholar]
- Selcher JC, Weeber EJ, Christian J, Nekrasova T, Landreth GE, Sweatt JD, 2003. A Role for ERK MAP Kinase in Physiologic Temporal Integration in Hippocampal Area CA1. Learn. Mem 10,26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD, 2006. Kinase Suppressor of Rasl Compartmentalizes Hippocampal Signal Transduction and Subserves Synaptic Plasticity and Memory Formation. Neuron 50, 765–779. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Leikowitz RJ, 2011. β-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci 32, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanini L, Bergmeier W, 2010. CalDAG-GEFI and platelet activation. Platelets 21,239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Dillon TJ, Liu C, Kariya Y, Wang Z, Stork PJS, 2013. Protein Kinase A-dependent Phosphorylation of Rap 1 Regulates Its Membrane Localization and Cell Migration. J. Biol. Chem 288, 27712–27723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Li Y, Dillon TJ, Stork PJS, 2017. Phosphorylation of Rapl by cAMP-dependent Protein Kinase (PKA) Creates a Binding Site for KSR to Sustain ERK Activation by cAMP. J. Biol. Chem 292, 1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T, 2001. Small GTP-Binding Proteins. Physiol. Rev 81, 56. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Augustine GJ, 2008. A positive feedback signal transduction loop determines timing of cerebellar long-term depression. Neuron 59, 608–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Yasuda R, 2017. Imaging ERK and PKA Activation in Single Dendritic Spines during Structural Plasticity. Neuron 93, 1315–1324.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrell EM, Morrison DK, 2019. Ras-Mediated Activation of the Raf Family Kinases. Cold Spring Harb. Perspect. Med. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therrien M, Chang HC, Solomon NM, Karim FD, Wassarman DA, Rubin GM, 1995. KSR, a Novel Protein Kinase Required for RAS Signal Transduction. Cell 83, 870–888. [DOI] [PubMed] [Google Scholar]
- Thiels E, Kanterewicz BI, Norman ED, Trzaskos JM, Klann E, 2002. Long-term depression in the adult hippocampus in vivo involves activation of extracellular signal-regulated kinase and phosphorylation of Elk-1. J. Neurosci. Off J. Soc. Neurosci 22, 2054–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Zhao M-G, Xu H, Wu L-J, Ren M, Zhuo M, 2007. Requirement of extracellular signal-regulated kinase/mitogen-activated protein kinase for long-term potentiation in adult mouse anterior cingulate cortex. Mol. Pain 3, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilieff P, Herry C, Vanhoutte P, Caboche J, Desmedt A, Riedel G, Mons N, Micheau J, 2006. Foreground contextual fear memory consolidation requires two independent phases of hippocampal ERK/CREB activation. Leam. Mem. Cold Spring Harb. N 13, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S, Voss T, 2004. MEK1 and MEK2, Different Regulators of the G 1 /S Transition. J. Biol. Chem 279, 43861–43869. [DOI] [PubMed] [Google Scholar]
- Vithayathil J, Pucilowska J, Friel D, Landreth GE, 2017. Chronic impairment of ERK signaling in glutamatergic neurons of the forebrain does not affect spatial memory retention and LTP in the same manner as acute blockade of the ERK pathway. Hippocampus 27, 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kriegsheim A, Pitt A, Grindlay GJ, Kolch W, Dhillon AS, 2006. Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat. Cell Biol 8, 1011–1016. [DOI] [PubMed] [Google Scholar]
- Walkup WG IV, Mastro TL, Schenker LT, Vielmetter J, Hu R, Iancu A, Reghunathan M, Bannon BD, Kennedy MB, 2016. A model for regulation by SynGAP-al of binding of synaptic proteins to PDZ-domain ‘Slots’ in the postsynaptic density. Elife 5, el6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup IV WG, Washburn L, Sweredoski MJ, Carlisle HJ, Graham RL, Hess S, Kennedy MB, 2015. Phosphorylation of Synaptic GlPase-activating Protein (synGAP) by Ca 2+/Calmodulin-dependent Protein Kinase II (CaMKII) and Cyclin-dependent Kinase 5 (CDK5) Alters the Ratio of Its GAP Activity toward Ras and Rap GTPases. J. Biol. Chem 290, 4908–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Birch CM, Deisboeck TS, 2008. Cross-scale sensitivity analysis of a non-small cell lung cancer model: Linking molecular signaling properties to cellular behavior. Biosystems 92, 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang L, Sagotsky J, Deisboeck TS, 2007. Simulating non-small cell lung cancer with a multiscale agent-based model. Theor. Biol. Med. Model 4, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartmann M, Hofer P, Turowski P, Saltiel AR, Hynes NE, 1997. Negative Modulation of Membrane Localization of the Raf-1 Protein Kinase by Hyperphosphorylation. J. Biol. Chem 272, 3915–3923. [DOI] [PubMed] [Google Scholar]
- Wu J, Dent P, Jelinek T, Wolfman A, Weber MJ, Sturgill TW, 1993. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3’,5’-monophosphate. Science 262, 1065–1069. [DOI] [PubMed] [Google Scholar]
- Xing L, Larsen RS, Bjorklund GR, Li X, Wu Y, Philpot BD, Snider WD, Newbem JM, 2016. Layer specific and general requirements for ERK/MAPK signaling in the developing neocortex. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T-R, Vyshemirsky V, Gormand A, von Kriegsheim A, Girolami M, Baillie GS, Ketley D, Dunlop AJ, Milligan G, Houslay MD, Kolch W, 2010. Inferring Signaling Pathway Topologies from Multiple Perturbation Measurements of Specific Biochemical Species. Sci. Signal 3. [PubMed] [Google Scholar]
- Yoon S, Seger R, 2006. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44. [DOI] [PubMed] [Google Scholar]
- York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJS, 1998. Rapl mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392, 622–626. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A, 2003. Presynaptic BDNF Required for aPresynaptic but Not Postsynaptic Component of LTP at Hippocampal CA1-CA3 Synapses. Neuron 39, 975–990. [DOI] [PubMed] [Google Scholar]
- Zhai S, Ark ED, Parra-Bueno P, Yasuda R, 2013. Long-Distance Integration of Nuclear ERK Signaling Triggered by Activation of a Few Dendritic Spines. Science 342, 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang P,Wang G, Zhang H, Zhang Y, Yu Y, Zhang M, Xiao J, Crespo P, Hell JW, Lin L, Huganir RL, Zhu JJ, 2018. Ras and Rap Signal Bidirectional Synaptic Plasticity via Distinct Subcellular Microdomains. Neuron 98, 783–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Smith-Nguyen EV, Keshwani MM, Deal MS, Kornev AP, Taylor SS, 2012. Structure and Allostery of the PKA RIIβ Tetrameric Holoenzyme. Science 335,712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R, 2002. Ras and Rap Control AMPA Receptor Trafficking during Synaptic Plasticity. Cell 110,443–455. [DOI] [PubMed] [Google Scholar]