

Short abstract
http://aasldpubs.onlinelibrary.wiley.com/hub/journal/10.1002/(ISSN)2046-2484/video/15-3-reading-chiang a video presentation of this article
http://aasldpubs.onlinelibrary.wiley.com/hub/journal/10.1002/(ISSN)2046-2484/video/15-3-interview-chiang an interview with the author
Abbreviations
- ASBT
apical sodium‐dependent bile salt transporter
- BSEP
bile salt export pump
- BSH
bile salt hydrolase
- C4
7α‐hydroxy‐4‐cholesten‐3‐one
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- CYP27A1
sterol 27‐hydroxylase
- CYP7A1
cholesterol 7α‐hydroxylase
- CYP7B1
oxysterol 7α‐hydroxylase
- CYP8B1
sterol 12α‐hydroxylase
- DCA
deoxycholic acid
- FGF
fibroblast growth factor
- FGFR4
FGF receptor 4
- FXR
farnesoid X receptor
- GLP‐1
glucagon‐like peptide 1
- ICP
intrahepatic cholestasis of pregnancy
- LCA
lithocholic acid
- MDR3
multidrug‐resistant protein 3
- MRP
MDR resistance‐associated protein
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NTCP
Na+‐dependent taurocholate cotransporting peptide
- OST
organic solute transporter
- PBC
primary biliary cirrhosis
- PFIC
progressive familial intrahepatic cholestasis
- PSC
primary sclerosing cholangitis
- SHP
small heterodimer partner
- T2D
type 2 diabetes
- TGR5
Takeda G protein‐coupled receptor 5
- TJP2
tight junction protein 2
- UDCA
ursodeoxycholic acid
Bile acids are synthesized from cholesterol exclusively in the liver and function as physiological detergents that facilitate biliary cholesterol excretion. Bile acids are stored in the gallbladder as bile salts. After meal intake, bile acids are released into the gastrointestinal tract to aid in absorption of nutrients, dietary fats, steroids, vitamins, and drugs. Recent advances in basic research have identified bile acids as nutrient sensors and metabolic integrators that activate farnesoid X receptor (FXR) and Takeda G protein‐coupled receptor 5 (TGR5) to regulate lipid, glucose, and energy metabolism, and maintain metabolic homeostasis.1 This review will briefly cover bile acid physiology and synthesis, pathophysiology of cholestatic liver diseases and nonalcoholic fatty liver disease (NAFLD), and bile acid–based therapy for liver‐related diseases.
There are two major bile acid synthesis pathways in the liver.2 The classic pathway is initiated by cholesterol 7α‐hydroxylase (CYP7A1) and synthesizes the two primary bile acids in humans, chenodeoxycholic acid (CDCA) and cholic acid (CA), the latter of which requires sterol 12α‐hydroxylase (CYP8B1) (Fig. 1). Serum 7α‐hydroxy‐4‐cholesten‐3‐one (C4), a common precursor for CA and CDCA, is used as an indicator of bile acid synthesis rate. The alternative pathway is initiated by sterol 27‐hydroxylase (CYP27A1), which synthesizes oxidized sterols, followed by oxysterol 7α‐hydroxylase (CYP7B1). Bile acids are conjugated to taurine or glycine to increase solubility for biliary secretion. In the colon, gut bacterial bile salt hydrolase (BSH) deconjugates bile acids, and 7α‐dehydroxylase removes the 7α‐HO‐group from CA and CDCA to form the secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA), respectively. Conjugated CA, CDCA, and DCA are secreted into portal blood circulation and reabsorbed into the liver to inhibit bile acid synthesis.
Figure 1.
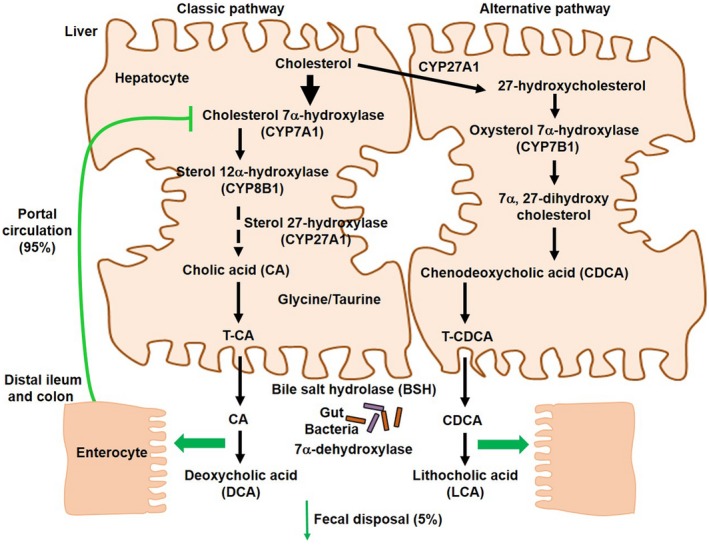
Bile acid synthesis. Bile acids are synthesized from cholesterol in the liver. A total of 17 enzymes located in the cytosol, endoplasmic reticulum, mitochondria, and peroxisomes of hepatocytes are involved in bile acid synthesis. The classic pathway is initiated by the rate‐limiting enzyme, CYP7A1, to synthesize the two primary bile acids in humans, CA and CDCA. CYP8B1 is required for CA synthesis, and mitochondrial CYP27A1 catalyzes a steroid side‐chain oxidation. The alternative pathway is initiated by CYP27A1, followed by CYP7B1. A common precursor of CA and CDCA, C4, is used as a serum marker for bile acid synthesis rate. After synthesis, bile acids are conjugated to the amino acids taurine or glycine for biliary secretion. In the distal ileum and colon, gut bacterial BSH deconjugates the conjugated bile acids, and bacterial 7α‐dehydroxylase removes the 7α‐hydroxyl group to convert CA and CDCA to the secondary bile acids DCA and LCA, respectively. The classic pathway is the major pathway for daily synthesis of about 80% of the bile acids in humans, whereas the alternative pathway synthesizes about 20%. Most bile acids (~95%) are reabsorbed in the ileum and transported via portal blood to the liver to inhibit bile acid synthesis. A small amount of bile acids (~5%) lost in feces is replenished by de novo synthesis.
Emerging research in bile acid metabolism in the past three decades has contributed significantly to our current understanding of the roles of FXR and TGR5 in the pathophysiology of liver‐related diseases. FXR is highly expressed in the gastrointestinal system and plays a central role in the regulation of enterohepatic circulation of bile acids. Bile acids activate FXR to induce bile salt export pump (BSEP), which secretes bile acids into bile canaliculi, forming mixed micelles with cholesterol and phospholipids (Fig. 2). In the ileum, most conjugated bile acids are reabsorbed by apical sodium‐dependent bile salt transporter (ASBT). FXR also induces sinusoidal organic solute transporter α/β (OSTα/OSTβ) to secrete bile acids into the portal circulation. Bile acids are reabsorbed from portal blood into hepatocytes by sodium‐dependent taurocholate transporting peptide (NTCP). Bile acid synthesis is tightly regulated by complex mechanisms to maintain low levels of bile acids in the liver. In the liver, FXR inhibits CYP7A1 gene transcription indirectly via inducing the negative nuclear receptor small heterodimer partner (SHP). In the intestine, FXR induces fibroblast growth factor 19 (FGF19), which activates liver FGF receptor 4 (FGFR4) to inhibit CYP7A1 via extracellular signal‐regulated kinase 1/2 signaling. TGR5 is expressed in many tissues, except hepatocytes. Activation of TGR5 by secondary bile acids in enteroendocrine cells stimulates secretion of glucagon‐like peptide 1 (GLP‐1), which promotes insulin secretion in the pancreas to improve insulin sensitivity. TGR5 signaling also promotes adipose tissue browning and energy metabolism to reduce weight.1
Figure 2.
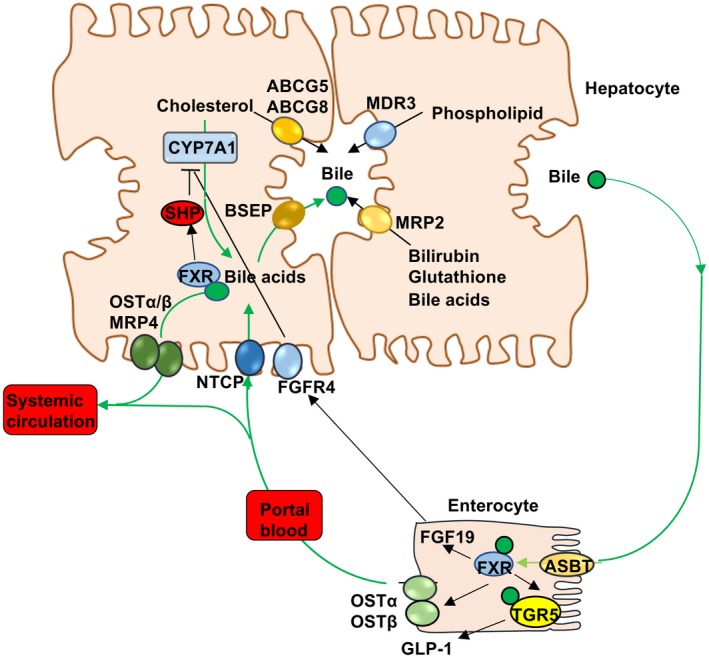
FXR regulation of bile acid synthesis and enterohepatic circulation of bile acids. In hepatocytes, FXR induces SHP, which inhibits transcription of CYP7A1 and bile acid synthesis. FXR induces BSEP, which actively excretes bile acids into bile. Bile acids also facilitate biliary cholesterol secretion via ATP binding cassette family G5 and G8 transporters (ABCG5, ABCG8). MDR3 (ABCB4) transports phospholipids into bile, whereas MRP2 transports bilirubin, glutathione, and unconjugated bile acids. In the ileum, bile acids are reabsorbed into enterocytes via ASBT. Intestinal FXR induces sinusoidal OSTα/OSTβ heterodimer to efflux bile acids into the portal blood circulation to the liver, where NTCP takes up bile acids into hepatocytes. Intestinal FXR induces FGF19, which circulates to the liver and binds to hepatic FGFR4, inhibiting bile acid synthesis. Bile acids also activate FXR to induce OSTα/OSTβ and MRP4 to efflux bile acids as an adaptive response to cholestasis. FXR induces TGR5 expression in enteroendocrine cells to stimulate secretion of glucagon‐like peptide 1 (GLP‐1), which stimulates insulin secretion from the pancreas and improves insulin sensitivity, and also promotes adipose tissue browning and energy metabolism.
The global epidemic of obesity and type 2 diabetes (T2D) has contributed to the increased prevalence of NAFLD, which is about 25% of the adult population in the world. NAFLD is a spectrum of chronic liver diseases ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) fibrosis and cirrhosis. NAFLD has surpassed viral hepatitis as the leading cause of hepatocellular carcinoma and liver transplant.3 Dysregulation of bile acid metabolism contributes to dyslipidemia, hyperglycemia, and insulin resistance in T2D.4 The gut‐to‐liver axis plays a critical role in determining circulating bile acid pool size and bile acid composition, which control gut bacteria overgrowth. Obesity, T2D, and NASH fibrosis are associated with an altered gut microbiome. Bariatric surgery alters the gut microbiome to increase serum bile acids, FGF19, and GLP‐1, and contributes to rapidly improved insulin sensitivity and glycemic control in overly obese patients.5
In inborn errors of bile acid synthesis, accumulation of toxic bile acid intermediates cause neonatal cholestasis, fibrosis, and hepatitis.1 Obstruction of bile flow by stones, tumors, or inflammation of the intrahepatic small bile duct or extrahepatic common bile duct increases toxic bile acids and intermediates in the liver, leading to cholestatic liver injury and fibrosis. Progressive familial intrahepatic cholestasis (PFIC) is a collection of cholestatic liver diseases of varying severity caused by deficiencies in bile formation and secretion. Mutations of BSEP (ABCB11), multidrug‐resistant protein 3 (MDR3, ABCB4), FXR, tight junction protein 2 (TJP2), and myosin 5B (MYO5B) have been identified in patients with PFIC6 (Table 1). Primary biliary cirrhosis (PBC) involves autoimmune destruction of the small bile ducts, portal infiltration, and fibrosis with high prevalence in middle‐aged women, whereas primary sclerosing cholangitis (PSC) causes fibrosis in bile ducts and obstruction of bile flow with higher prevalence in men. Intrahepatic cholestasis of pregnancy (ICP) is common in the third trimester of pregnancy.
Table 1.
Cholestatic Liver Diseases
| PFIC | Mutations of bile acid transporters (BSEP, ABCB11), phospholipid transporters (ATP8B1, MDR3), FXR, TJP2, or MYO5B |
| PBC | Autoimmune destruction of the small bile ducts, portal infiltration, and fibrosis |
| PSC | Injury and fibrosis of both intrahepatic and extrahepatic bile ducts, biliary strictures, and obstructed bile flow |
| ICP | Reversible and typically resolves after delivery |
Basic research in bile acid metabolism has been translated to bile acid–based therapies for cholestasis and NASH (Table 2). CA (Cholbam) and CDCA (Chenodiol) are used as bile acid replacement therapies for inborn errors of bile acid synthesis. Ursodeoxycholic acid (UDCA; ursodiol [Actigall]) has been used for gallstone dissolution. UDCA is used to treat PBC, whereas NorUDCA, a side‐chain‐shortened C23 UDCA homologue, is used to treat PSC.7 Bile acid sequestrants reduce the bile acid pool and result in CYP7A1 induction, increased bile acid synthesis, and reduced serum cholesterol. Sequestrants also reduce FGF19 and stimulate TGR5‐mediated GLP‐1 secretion to improve insulin sensitivity and white adipose tissue browning.8 There is no approved drug therapy for NASH. Recently, obeticholic acid, a synthetic bile acid and potent FXR agonist, has been used for PBC treatment9 and is in phase III clinical trials for NASH fibrosis. NGM282, a nontumorigenic FGF19 analogue, has been used in clinical trials to treat PSC and PBC, and improves NASH fibrosis.10 However, bile acid–based drugs may have undesirable side effects such as pruritus, diarrhea, and hypercholesterolemia. It is anticipated that bile acid–based drug therapies will be approved for NASH treatment in the near future.
Table 2.
Bile Acid–Based Therapies
| Bile acids |
|
|
|
| Bile acid sequestrants |
|
| FXR agonists: anti‐inflammation, cholestasis, NASH fibrosis, diabetes, obesity |
|
|
| FXR antagonists (intestine): PBC; reduces weight, improves diabetes |
|
|
This study was supported by National Institutes of Health grants DK44442 and DK58379.
Potential conflict of interest: Nothing to report.
References
- 1. Chiang JYL, Ferrell JM. Bile acids as metabolic regulators and nutrient sensors. Annu Rev Nutr. Available at: 10.1146/annurev-nutr-082018-124344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res 2009;50:1955‐1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hardy T, Oakley F, Anstee QM, et al. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol 2016;11:451‐496. [DOI] [PubMed] [Google Scholar]
- 4. Haeusler RA, Astiarraga B, Camastra S, et al. Human insulin resistance is associated with increased plasma levels of 12alpha‐hydroxylated bile acids. Diabetes 2013;62:4184‐4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bozadjieva N, Heppner KM, Seeley RJ. Targeting FXR and FGF19 to treat metabolic diseases‐lessons learned from bariatric surgery. Diabetes 2018;67:1720‐1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bull LN, Thompson RJ. Progressive familial intrahepatic cholestasis. Clin Liver Dis 2018;22:657‐669. [DOI] [PubMed] [Google Scholar]
- 7. Fickert P, Hirschfield GM, Denk G, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol 2017;67:549‐558. [DOI] [PubMed] [Google Scholar]
- 8. Hansen M, Sonne DP, Mikkelsen KH, et al. Bile acid sequestrants for glycemic control in patients with type 2 diabetes: a systematic review with meta‐analysis of randomized controlled trials. J Diabetes Complications 2017;31:918‐927. [DOI] [PubMed] [Google Scholar]
- 9. Kowdley KV, Luketic V, Chapman R, et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018;67:1890‐1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harrison SA, Rossi SJ, Paredes AH, et al. NGM282 improves liver fibrosis and histology in 12 weeks in patients with nonalcoholic steatohepatitis. Hepatology. Available at: 10.1002/hep.30590. [DOI] [PMC free article] [PubMed] [Google Scholar]