

Short abstract
http://aasldpubs.onlinelibrary.wiley.com/hub/journal/10.1002/(ISSN)2046-2484/video/15-3-reading-gochanour a video presentation of this article
http://aasldpubs.onlinelibrary.wiley.com/hub/journal/10.1002/(ISSN)2046-2484/video/15-3-interview-kowdley an interview with the author
Abbreviations
- ERC
endoscopic retrograde cholangiography
- IBD
inflammatory bowel disease
- MRC
magnetic resonance cholangiography
- PSC
primary sclerosing cholangitis
Primary sclerosing cholangitis (PSC) is a rare, idiopathic, and progressive biliary tract disease that can lead to liver fibrosis and cirrhosis. PSC is characterized by inflammation and destruction of intrahepatic and extrahepatic bile ducts, leading to progressive hepatic fibrosis.1 The etiology of PSC is unknown, although several genes are associated with its development and clinical course.2, 3 There is a strong association between inflammatory bowel disease (IBD) of the colon and PSC.1, 2 An overlap syndrome of autoimmune hepatitis and PSC is an additional, well‐established presentation, particularly in younger patients. The diagnosis of PSC relies heavily on characteristic cholangiographic features in the setting of cholestatic liver enzyme abnormalities.2 Patients with PSC are at increased risk for cholangiocarcinoma and gallbladder cancer, as well as colon cancer in those with concurrent IBD.4 Screening for malignancies involving the gallbladder, bile ducts, and colon is therefore a critical component of PSC management. Liver transplantation is by far the most effective treatment, although a posttransplant recurrence rate of approximately 20% is reported. This review aims to highlight the most salient aspects of identifying and managing this unique cholestatic liver disease.
Epidemiology
PSC is a rare disorder with regional variation in prevalence.3 The prevalence in the United States is estimated to be 1 to 16 per 100,000.1 An estimated 80% of patients diagnosed with PSC have concurrent IBD.2 There is a 2:1 male predominance when PSC is associated with IBD and a slight female predominance in the absence of IBD (Table 1).1 Average age at diagnosis is typically 30 to 40 years old.1 In patients with IBD, the prevalence rate of PSC is estimated to be approximately 5%.3
Table 1.
PSC Epidemiology
| Characteristics |
|---|
| Sex: Men (2:1) |
| Age at Diagnosis: Fourth to fifth decade of life |
| IBD in PSC: 80% |
| PSC in IBD: 5% |
Genetics
The pathogenesis of PSC remains unclear, although multiple genes have been implicated in its development.3 An immunologically mediated process is supported by known associations with human leukocyte antigens.3 Another theory involves bacterial translocation to the biliary tree based on an animal model of bacterial overgrowth and link with ulcerative colitis.3, 4 Certain gene mutations including cystic fibrosis transmembrane receptor (CFTR) mutations have also been associated with the development of PSC.3, 4
Diagnosis
Patients with PSC are often asymptomatic early in the disease course; usually they are identified through a persistent cholestatic pattern of elevated liver enzymes.2 Fatigue and pruritus are common symptoms, but many patients are asymptomatic. Biliary tract obstruction may lead to acute pruritus and/or cholangitis, presenting with jaundice, fever, and abdominal pain. Patients with PSC‐IBD may present with abnormal bowel habits, abdominal pain, and intestinal bleeding, with concurrent liver enzyme abnormalities.
The differential diagnosis of PSC includes secondary sclerosing cholangitis, immunoglobulin G4–associated cholangitis, autoimmune hepatitis, primary biliary cholangitis, ischemic cholangiopathy, papillary tumors, cholangiocarcinoma, cholangiolithiasis, and HIV cholangiopathy.3 These causative factors should be evaluated during the diagnostic workup.
Liver biopsy is usually unnecessary in diagnosing PSC, but it is helpful to evaluate PSC‐autoimmune hepatitis overlap syndrome and small‐duct PSC.3 Characteristic features of PSC on histology include concentric periductal “onion skinning” fibrosis, although this distinctive feature is insensitive.3 Similarly, there are no known sensitive serological assays for PSC.
The Role of Imaging in PSC
Given the limitations of liver histology and absence of sensitive serological assays, it is essential to perform cholangiography when PSC is suspected. Magnetic resonance cholangiography (MRC) is now recommended by all major professional societies as the initial diagnostic modality of choice2, 3, 5, 6 (Table 2).
Table 2.
Diagnosing PSC
| MRCP | ERCP | Liver Biopsy |
|---|---|---|
| Noninvasive | Greater sensitivity | Used to rule out overlapping conditions |
| Less expensive | Allows for therapeutic intervention | |
| No radiation exposure | ||
| Eliminates risk for pancreatitis |
Even though MRC is highly sensitive and specific (0.86 and 0.94, respectively) in detecting the multifocal biliary stricturing and peripheral pruning phenotype of large‐duct PSC, indeterminate cases warrant consideration of endoscopic retrograde cholangiography (ERC), which remains the reference standard for cholangiography (Fig. 1). Isolated involvement of peripheral ductules (small‐duct PSC) is seen in less than 10% of cases, necessitating liver biopsy because both MRC or ERC are insensitive for small‐duct changes.7 Although useful for diagnosis, neither baseline nor serial cholangiographic features predict disease progression or prognosis in PSC.
Figure 1.
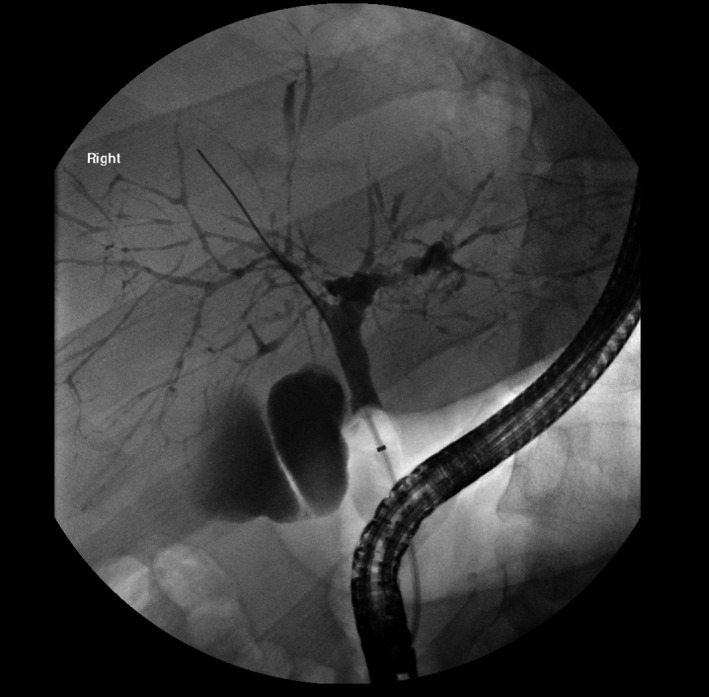
Endoscopic retrograde cholangiography demonstrating focal strictures and dilations creating characteristic “beading” appearance of intrahepatic bile ducts.
The increased risk for clinically significant biliary obstruction, cholelithiasis, hepatolithiasis, cholangiocellular dysplasia and cholangiocarcinoma, hepatocellular carcinoma, polyps and cancer of the gallbladder, and colon cancer drives the vital role of regular imaging in the long‐term management of PSC (Table 3).
Table 3.
Complications of PSC
| Cholangitis |
| Gastrointestinal malignancies |
| Fat‐soluble vitamin deficiencies |
| Bone diseases |
| Cirrhosis and portal hypertension |
Screening for Malignancy and Premalignant States
Despite the modest positive predictive values, most professional societies and expert opinion support screening for cholangiocarcinoma with ultrasonography, MRC, and ERC, with or without serum CA 19‐9 every 6 to 12 months.2, 3, 5 Annual ultrasonography is the recommended modality for gallbladder polyp and cancer screening. As with patients without PSC, the presence of advanced (stage 3) liver fibrosis or cirrhosis warrants hepatocellular carcinoma screening every 6 months, with ultrasonography, computed tomography, or magnetic resonance imaging, with or without serum alpha‐fetoprotein. Elastography modalities (e.g., FibroScan, magnetic resonance elastography) for fibrosis staging in PSC require further validation. Even in asymptomatic individuals, colonoscopy to screen for IBD is recommended in all patients at PSC diagnosis. Presence of concurrent PSC and IBD warrants colon cancer screening with colonoscopy every 1 to 2 years.2, 5 European guidelines additionally recommend colonoscopy with random colonic biopsies for IBD screening every 5 years in patients with PSC who do not have concurrent IBD.5
Imaging Directed by Clinical Features
Differentiating malignant and benign dominant biliary strictures is difficult. Nevertheless, the development of dominant biliary strictures should prompt an exhaustive evaluation for cholangiocarcinoma. Despite very high specificity, sensitivity for cholangiocarcinoma of brush cytology obtained on ERC is only 43%.8 The addition of fluorescent in situ hybridization increases sensitivity to 68%, although specificity decreases to 70%.8, 9 Pooled data from small studies of endoscopic retrograde cholangioscopy with targeted biopsy demonstrate a 65% sensitivity and 97% specificity.10
Current Management
There are no approved pharmacological therapies for PSC. Ursodeoxycholic acid is the best studied, although the role of this therapy in PSC remains unclear.3 High‐dose ursodeoxycholic acid (>28 mg/kg/day) is associated with increased adverse clinical outcomes and should be avoided.11 Despite enthusiasm about its use as a possible therapy for PSC, recent studies with oral vancomycin have not shown significant benefit, and randomized clinical trials are needed.3 Patients with decompensated cirrhosis caused by PSC or with refractory cholangitis may be best treated with liver transplantation. Liver transplantation has shown success for selected patients with hilar cholangiocarcinoma caused by PSC.12 Recurrent PSC is estimated to occur in approximately 20% of patients.3
As with other cholestatic liver diseases, osteopenia may be a complication of PSC, and bone density screening at diagnosis and at 2‐ to 4‐year intervals is suggested.3 Fat‐soluble vitamin deficiencies may occur.3
Prognosis
The disease course of PSC is highly variable. Serum alkaline phosphatase levels <1.5× upper limit of normal have been associated with improved survival in patients with PSC,13 whereas patients with alkaline phosphatase levels ≥2.4× upper limit of normal are more likely to die or require liver transplantation.14
Conclusion
PSC is a rare idiopathic cholestatic liver disease with no approved treatment, although a number of agents are currently showing promise in clinical trials. The disease course is variable, and management is focused on mitigating several life‐threatening complications, including hepatic and extrahepatic malignancies, with emphasis on early consideration of liver transplantation in appropriately selected patients; liver transplantation is an effective treatment for end‐stage liver disease. Disease recurrence has been reported but does not reduce favorable long‐term outcomes.
Potential conflict of interest: K.K. consults for, is the on the speakers' bureau for, and received grants from Gilead. He advises, is on the speakers' bureau for, and received grants from Intercept. He consults for and received grants from HighTide.
References
- 1. Hirschfield GM, Karlsen TH, Lindor KD, et al. Primary sclerosing cholangitis. Lancet 2013;382:1587‐1599. [DOI] [PubMed] [Google Scholar]
- 2. Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010;51:660‐678. [DOI] [PubMed] [Google Scholar]
- 3. Lindor KD, Kowdley KV, Harrison ME. ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol 2015;110:646‐659. [DOI] [PubMed] [Google Scholar]
- 4. Karlsen TH, Folseraas T, Thorburn D, et al. Primary sclerosing cholangitis: a comprehensive review. J Hepatol 2017;67:1298‐1323. [DOI] [PubMed] [Google Scholar]
- 5. Aabakken L, Karlsen TH, Albert J, et al. Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) Clinical Guideline. J Hepatol 2017;66:1265‐1281. [DOI] [PubMed] [Google Scholar]
- 6. Schramm C, Eaton J, Ringe KI, et al; MRI Working Group of the IPSCSG . Recommendations on the use of magnetic resonance imaging in PSC‐A position statement from the International PSC Study Group. Hepatology 2017;66:1675‐1688. [DOI] [PubMed] [Google Scholar]
- 7. Dave M, Elmunzer BJ, Dwamena BA, et al. Primary sclerosing cholangitis: meta‐analysis of diagnostic performance of MR cholangiopancreatography. Radiology 2010;256:387‐396. [DOI] [PubMed] [Google Scholar]
- 8. Trikudanathan G, Navaneethan U, Njei B, et al. Diagnostic yield of bile duct brushings for cholangiocarcinoma in primary sclerosing cholangitis: a systematic review and meta‐analysis. Gastrointest Endosc 2014;79:783‐789. [DOI] [PubMed] [Google Scholar]
- 9. Navaneethan U, Njei B, Venkatesh PGK, et al. Fluorescence in situ hybridization for diagnosis of cholangiocarcinoma in primary sclerosing cholangitis: a systematic review and meta‐analysis. Gastrointest Endosc 2014;79:943‐950.e3. [DOI] [PubMed] [Google Scholar]
- 10. Njei B, McCarty TR, Varadarajulu S, et al. Systematic review with meta‐analysis: endoscopic retrograde cholangiopancreatography‐based modalities for the diagnosis of cholangiocarcinoma in primary sclerosing cholangitis. Aliment Pharmacol Ther 2016;44:1139‐1151. [DOI] [PubMed] [Google Scholar]
- 11. Lindor KD, Kowdley KV, Luketic VA, et al. High‐dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50:808‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sapisochín G, Fernández de Sevilla E, Echeverri J, et al. Liver transplantation for cholangiocarcinoma: current status and new insights. World J Hepatol 2015;7:2396‐2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al Mamari S, Djordjevic J, Halliday JS, et al. Improvement of serum alkaline phosphatase to < 1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol 2013;58:329‐334. [DOI] [PubMed] [Google Scholar]
- 14. Goode EC, Clark AB, Mells GF. Factors associated with outcomes of patients with primary sclerosing cholangitis and development and validation of a risk scoring system. Hepatology 2019;69:2120‐2135. [DOI] [PMC free article] [PubMed] [Google Scholar]