

Abstract
Patients with the Loeys-Dietz syndrome (LDS) have mutations in the TGF-βR1, TGF-βR2, and SMAD3 genes. However, little is known about the redox homeostasis in the thoracic aortic aneurysms (TAA) they develop. Here, we evaluate the oxidant/antioxidant profile in the TAA tissue from LDS patients and compare it with that in nondamaged aortic tissue from control (C) subjects. We evaluate the enzymatic activities of glutathione peroxidase (GPx), glutathione S-transferase (GST), glutathione reductase (GR), catalase (CAT), superoxide dismutase (SOD) isoforms, and thioredoxin reductase (TrxR). We also analyze some antioxidants from a nonenzymatic system such as selenium (Se), glutathione (GSH), and total antioxidant capacity (TAC). Oxidative stress markers such as lipid peroxidation and carbonylation, as well as xanthine oxidase (ORX) and nuclear factor erythroid 2-related factor 2 (Nrf2) expressions, were also evaluated. TAA from LDS patients showed a decrease in GSH, Se, TAC, GPx, GST, CAT, and TrxR. The SOD activity and ORX expressions were increased, but the Nrf2 expression was decreased. The results suggest that the redox homeostasis is altered in the TAA from LDS patients, favoring ROS overproduction that contributes to the decrease in GSH and TAC and leads to LPO and carbonylation. The decrease in Se and Nrf2 alters the activity and/or expression of some antioxidant enzymes, thus favoring a positive feedback oxidative background that contributes to the TAA formation.
1. Introduction
The Loeys-Dietz syndrome (LDS) is a variant of the Marfan syndrome (MS), and in it, as in all complex genetic diseases, there is damage to different organs and systems. Bart Loeys and Harry Dietz described the LDS in 2005 [1]. This syndrome is an autosomal dominant disease due to mutations in several genes including the gene that encodes for the beta I receptor of transforming growth factor (TGF-βR1), which is located on chromosome 9q22.33; mutations in the TGF-βR2 gene, located on chromosome 3p24.1; and mutations in the SMAD3 gene located on chromosome 15q22.33. The LDS is characterized by unusual facies, dolichocephaly, craniosynostosis, hypertelorism, malar hypoplasia, arched palate, wide or bifid uvula, retrognathia, sternum deformity, scoliosis, joint laxity, dural ectasia, long fingers without bone growth, contractures, equine foot, translucent skin, and learning problems [1]. The patients with LDS show complications during pregnancy, such as rupture of the uterus and death [2]. The patients also have a strong predisposition to arterial tortuosity [3] and a high frequency in the appearance of thoracic aortic aneurysms (TAA) at an early age [4]. The TAA in LDS are characterized by a high risk of dissection or rupture, even when diameters are small (less than 5 cm). Up to 53% of patients also develop aneurysms in other aortic locations [5]. The first histological alterations described in TAA from LDS showed similarities with lesions in aortas from MS. In both syndromes, a loss of content of elastic fibers and their disordered disposition were found [5]. There was also an increase in the collagen deposition. However, in LDS, these changes are more aggressive than those in MS [6], resulting in a faster growth of the aneurysm (growing rate of 1.0 cm/year), that may lead to premature death [5]. At present, 5 different types of LDS have been reported each having different clinical characteristics. For example, the characteristics of type 2 LDS may not be easily recognized from those of MS, since in this form of LDS, there are some musculoskeletal criteria similar to those of MS that can be easily confused or lead to a late diagnosis of the disease [7].
In the development of the TAA in MS and other related diseases, there is a presence of endothelial dysfunction (ED) and oxidative stress (OS) [8]. The endothelium directly responds to the physical forces of blood flow losing the inflammatory/anti-inflammatory balance and leading to the release of mediators that participate in the regulation of the vascular tone. As an important part of this imbalance, there are alterations in the NO produced by eNOS [9].
Regarding OS, overproduction of ROS can contribute to arterial wall degeneration [9–11], as has been described in animal models and in humans with MS [12, 13]. ROS production is linked to ED. When the substrate in the synthesis of the NO is insufficient, the transfer of electrons is diverted from O2 to highly reactive substances such as superoxide (O2−) and hydrogen peroxide (H2O2). These products damage the lipids of the cell membrane, the proteins, and the DNA. They also inactivate antioxidant enzymes, increase the proinflammatory gene expression, and induce cell damage and apoptosis [14]. However, not only do reactive oxygen species (ROS) come from the NO synthesis pathway but also they originated from xanthine oxidase (ORX), NADPH dehydrogenase, cytochrome p450, and the mitochondria.
Antioxidant systems are also present in cells, and they constitute a compensatory mechanism, which are activated to reduce the damage caused by ROS and are capable of stabilizing and neutralizing the oxidative effects [15]. These antioxidant systems are divided into the enzymatic and nonenzymatic systems. Superoxide dismutase (SOD) isoforms, glutathione S-transferase (GST), catalase (CAT), glutathione reductase (GR), glutathione peroxidase (GPx), and thioredoxin reductase (TrxR) form part of the enzymatic antioxidant system [14]. The nonenzymatic system includes antioxidant substances such as selenium (Se), vitamins E and C, and the glutathione tripeptide (GSH) among others [15].
In studies on the role of OS in MS, the enzymatic and nonenzymatic systems show disequilibrium [14]. The activities of the enzymes that use GSH are decreased [14], and iNOS expression and the nitrate and nitrite ratio (NO3−/NO2−) are increased. These changes are associated with a decrease in the total antioxidant capacity (TAC) and an increase in LPO and carbonylation [16]. These changes contribute to a chronic inflammatory process [10], which may lead to the development of the aneurysm in the aortic root [17, 18]. Although there are several studies on the role of OS in TAA in MS, there are no reports showing the participation or presence of OS in the development of TAA in LDS. Therefore, the goal of this investigation was to analyze the oxidant/antioxidant profile in the TAA of LDS patients and to assess the participation of OS in this syndrome.
2. Material and Methods
2.1. Ethical Considerations
The research protocol was approved by the research and ethics committee of the National Institute of Cardiology “Ignacio Chávez” (protocol number 09654). An informed consent form was presented and signed by each LDS patient and control individual in accordance with the Helsinki declaration, as amended by the congress of Tokyo, Japan [19]. This was an observational and comparative study that was performed in a prospective cohort of patients that attended our institution and that were assisted to the aorta clinic of the National Institute of Cardiology “Ignacio Chávez” between March 2011 and March 2019.
Based on the inclusion criteria, 4 men with ages ranging from 13 to 46 years and 6 women with ages between 14 and 35 years were included. The patients with LDS were incorporated when they had been evaluated and diagnosed by an expert rheumatologist (M.E.S.) according to the criteria proposed by Ghent [20]. When the patients had other characteristics that suggested a diagnosis more of LDS than of MS, genetic tests were performed to unmask mutations in the FBN-1, TGF-BR2, and COL3A1 genes. In patients in whom LDS was diagnosed, there were hyperelasticity and hypermobility similar to those in the Ehlers-Danlos syndrome. However, nowadays there is no international consensus on diagnostic criteria of LDS. Patients were programmed for surgery, when they had ≥4.5 cm dilation, shown by magnetic resonance or computerized tomography, and had been previously presented and discussed in an interdisciplinary meeting with surgeons and the heart team (HT) or when they had attended the institute for the first time with aortic dissection and/or dilation. The surgical techniques employed were Bentall and Bono and David type 5 according to their arterial complication and the final decision by the HT [21].
The control (C) subjects included had no aortic damage and did not undergo surgery for aortic stenosis, having had no syndrome pathology diagnosed. In these patients, there was no suspicion of inflammatory disease or presence of degenerative disorders such as thyroid and autoimmune diseases, diabetes mellitus, and arterial hypertension. Medications that could interfere with the outcome of the study such as lipid-lowering drugs or NSAIDs were suspended in the perioperative period. Cases were dealt with caution, to avoid including patients undertaking treatment with allopurinol, antioxidants, or probable inhibitors of ROS production pathways. Warfarin, aspirin, clopidogrel, anticoagulant, and antiplatelet medications and other drugs were suspended.
In patients with LDS and C subjects, laboratory tests were made to determine acute-phase reactants, high-density lipoprotein (HDL), low-density lipoprotein (LDL), cholesterol (CT), and triglycerides (TG). Additionally, image studies by magnetic resonance echocardiography or computerized tomography were done to discard aortic damage in addition to valvular damage. We assessed the presence of congenital heart anomalies (CHA) including tetralogy of Fallot, atrial septal defect, patent ductus arteriosus, or bicuspid aortic valve based on echocardiographic images or institutional MR angiographies because in these patients, the CHA are related and support the clinical diagnosis.
Based on the exclusion criteria, we excluded two patients because the integral genetic study was not possible.
The Bentall technique consists in the replacement of the aortic valve root and ascending aorta with a Dacron tube, to which both coronary arteries are anastomosed on the lateral faces and at one of its ends to a valvular prosthesis. The difference between the Bentall technique and the David type 5 techniques is that in the latter, the native aortic valve and valve commissures are preserved and are reimplanted within the Dacron tube [21].
A segment of the TAA was taken on the surgery; the tissue was placed in LN2 and was kept at -70°C until used.
2.2. Thoracic Aortic Aneurysm Tissue Homogenization
The segment from the TAA was homogenized in LN2, according to the methods that were previously described by Soto et al. [16]. The Bradford method was utilized to determine the protein concentration in the homogenates [22]. All assays on biochemical variables (except for those in the native gels and western blot analysis) were made in duplicate.
2.3. Superoxide Dismutase and Catalase Activities in Native Gels
The measurement of the activity of the SOD isoforms and CAT was performed by electrophoresis in 10% polyacrylamide native gels. CAT and SOD isoforms were revealed according to the methods that were previously described by Pérez-Torres et al. [23]. Purified SOD from bovine erythrocytes with a specific activity of 112 U/mg of protein (Sigma-Aldrich, St. Louis, MO, USA) and purified CAT from a bovine liver having a specific activity of 60 U/mg (Sigma-Aldrich) were used as positive controls. The previously mentioned antioxidant enzyme activity determinations were performed according to the manufacturer's instructions. Samples were placed in a separate lane of the gel and run in parallel with the biological samples. The intensity of the signal from the controls was used as a reference to measure the enzymatic activity in the tissue samples. Therefore, the results are expressed as U of activity per mg of protein. The gels were analyzed by densitometry with image SigmaScan Pro 5.1 software (Systat Software, Inc., San Jose, CA, USA).
2.4. Enzymes That Use Glutathione and/or Oxidized Glutathione
For the GPx, GST, and GR activities, 100 μg of the protein of the TAA homogenate was utilized according to previously described methods [24, 25]. The GPx activity is expressed as nmol of NADPH oxidized/min/mg protein, with an extinction coefficient of 6220 M–1 cm–1 at 340 nm for NADPH. The GST activity is expressed as units of GS-TNB mol/min/mg protein with an extinction coefficient of 14150 M−1 cm−1. The GR activity is expressed as μmol of reduced GSSG/min/mg protein, with an extinction coefficient of 6220 M–1 cm–1.
2.5. Thioredoxin Reductase
The TrxR activity was determined indirectly by the amount of DTNB in the presence of NADPH to form 2 moles of TNB. The DTNB oxidation is monitored at 412 nm at 37°C for 6 min with an extinction coefficient of 13600 M−1 cm−1. 100 μg of TAA homogenate protein suspended in 3 ml of 0.1 mM phosphate buffer (KH2PO4, pH 7.0) was added to 0.2 mM NADPH, 1 mM EDTA, and 0.1 mg/ml bovine serum albumin. The samples were read in the presence of 20 μl of the specific TrxR inhibitor (10 μM auranofin) and together with a duplicate of the sample without the inhibitor [26].
2.6. Evaluation of Oxidative Stress Markers
For the measurement of the protein carbonylation, 1 mg of protein from the TAA homogenate was processed according to the method described by Pech et al. [27]. Absorbance was read at 370 nm, using air as the blank and a molar absorption coefficient of 22000 M−1 cm−1.
2.7. Lipid Peroxidation
1 mg of protein from the TAA homogenate was measured by the TBARS method that has been previously reported [23]. In this assay, the product malondialdehyde (MDA), which is a reactive aldehyde produced by lipid peroxidation (LPO) of polyunsaturated fatty acids that form a 1 : 2 adduct with thiobarbituric acid, is determined. The absorbance was read at 532 nm and is proportional to the concentration of MDA and the calibration curve obtained using tetraethoxypropane as the standard.
2.8. The Total Antioxidant Capacity
The total antioxidant capacity (TAC) of the nonenzymatic system was determined in 1 mg of protein from the TAA homogenate according to the method that was previously described by Benzie and Strain [28]. The absorbance was measured at 593 nm. The calibration curve was obtained using 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, which is an analogue of vitamin E, and it is commonly used as a positive control in antioxidant assays.
2.9. Quantification of GSH Concentrations
In 1 mg of protein from the TAA homogenate, GSH levels were measured by the Ellman reagent (5,5-dithiobis-2-nitrobenzoic), according to the method described by Ellman [29]. The absorbance was read at 412 nm. The calibration curve was determined using GSH at concentrations ranging from 5 to 25 μM.
2.10. Quantification of Vitamin C
1 mg of the protein from the TAA homogenate was mixed with C2HCl3O2 (20%) and was kept in an ice bath for 5 min and centrifuged at 5000 rpm for 5 min. The supernatant was recuperated and added to 200 μl of Folin-Ciocalteu reagent (0.20 mM) and incubated for 10 min. The absorbance was measured at 760 nm [30].
2.11. Selenium
The technique used was previously described by Pérez Ruiz et al. [31], with some adaptation and modification described by Rodríguez et al. [32] and with some modifications and adaptations in our laboratory. All solutions were made with tridistilled H2O and are only used for the assay and discarded. In brief, in new Corning sterile polypropylene centrifuge tubes, 250 μg of TAA homogenate protein and 500 μl of acid mixture (4 : 1 vol/vol of HNO3+HCl) plus 500 μl of 10% H2O2 were added and incubated in a sand bath at 120°C for 4 hours. After incubation, 100 μl of tridistilled H2O, 150 μl of 0.5 N NaOH, 200 μl of 30% formaldehyde, 200 μl of a mixture containing 0.5 N N2S and 0.5 N Na2SO3, plus 250 μl of 0.01 M EDTA (pH 10.2), and 300 μl of 4 mM toluidine blue were added. Samples were incubated for 15 min at 25°C. At the end of the incubation, they were centrifuged at 448 rcf for 2 min and the absorbance was read at 600 nm. The calibration curve was determined with 100 ng/ml Na2SeO3 which was treated under conditions similar to the experimental samples.
2.12. Western Blotting of eNOS, Nrf2, Cu/Zn-SOD, and ORX
50 μg of protein from the homogenate was run on 12% SDS-PAGE (bis-acrylamide-laemmli gel) and blotted onto a polyvinylidene difluoride (PVDF) membrane (0.22 μm, Millipore, Billerica, MA, USA) and then blocked 1 h at room temperature with Tris buffer solution-0.01% Tween (TBS-T 0.01%) plus 5% nonfat milk. The membranes were incubated overnight at 4°C with mouse primary monoclonal antibodies eNOS (sc-376751), Nrf2 (sc-365949), Cu/Zn-SOD (sc-101523), and ORX (sc-398548) from Santa Cruz Biotechnology, Santa Cruz, CA, USA, with 1 : 1000 dilution. After that, the membranes were incubated overnight at 4°C with a secondary antibody that is conjugated with horseradish peroxidase with 1 : 10000 dilutions (Santa Cruz Biotechnology, Santa Cruz, CA, USA). All the blots were incubated with an α-actin antibody as a load control. The protein was detected by the chemiluminescence assay (Clarity Western ECL Substrate, Bio-Rad Laboratories, Inc., Hercules, CA, USA). Chemiluminescence that was emitted in this process was detected in X-ray films (AGFA, Ortho CP-GU, Agfa HealthCare NV, Belgium). Images from each film were acquired with a GS-800 densitometer (including Quantity One software from Bio-Rad). The values of the density of each band are expressed as arbitrary units (AU).
2.13. Histology
A segment of 2 mm from the TAA was processed according to conventional histological techniques and stained with hematoxylin and eosin (HE) staining, Weigert's staining for elastic fibers, and Masson's trichrome staining. Histological sections were acquired according to the method and equipment described by Zúñiga-Muñoz et al. [14] and examined at a 40x magnification.
2.14. Assay Design and the HRMA Technique
High-resolution melting analysis (HRMA) was performed on the Bio-Rad Real-Time CFX96 PCR System, using precision melt super mix (Bio-Rad Laboratories, Hercules, CA, USA). Specific pairs of oligonucleotide primers (forward and reverse) were designed using Primer3 v4 (http://frodo.wi.mit.edu) to flank FBN-1 exons 14, 19, 28, and 42 and TGF-βR2 exon 6 (Table 1). PCR was performed in a 20 μl reaction volume containing 10 μM of each primer, 25 ng genomic DNA, and master mix high resolution. Cycling conditions were set as follows: an initial denaturation step of 95°C for 10 min, followed by 40 cycles of 95°C for 10 s, annealing T (according to each primer) for 30 s, and 72°C for 30 s. After completion of amplification, DNA was heated at 95°C for 30 s, kept at 50°C for 30 s, and then melted from 65 to 95°C (increment 0.2°C, dwell time 10 s). The results were analyzed using the Bio-Rad Precision Melt Analysis software. Melting profiles were normalized, grouped, and displayed as fluorescence-versus-temperature plots or subtractive difference plots (−df/dt vs. T). All samples with distinguished melting curves from wild type were confirmed by a duplicate study. Oligonucleotide primer sequences to carry out HRMA of the FBN-1 and TGF-βR2 coding regions are shown (Table 1). For the study of COL3A1, the molecular analysis was performed on genomic DNA by polymerase chain reaction amplification of all coding exons and the flanking intron regions. The amplicons were analyzed by the sequencing-by-synthesis (SBS) technology (MiSeq Personal Sequencer, Illumina, San Diego, California, United States). The gene panel including COL3A1, NOTCH1, MYH11, and SMAD3, but not all, was identified in these groups of patients. The presence of the variants was confirmed by Sanger sequencing.
Table 1.
Oligonucleotide primer sequences to the FBN-1 and TGF-βR2 coding regions.
| Gene | Exon | Forward primer | Reverse primer | Annealing temperature (°C) |
|---|---|---|---|---|
| FBN-1 | 14 | 5′-TGGCCGGATCTGCAATAATG-3′ | 5′-ACAGTTCTTCCCATCTCGTGT-3′ | 56 |
| FBN-1 | 19 | 5′-TGGTGCAGATATAAATGAATGTGC-3′ | 5′-GAAAATGGGTAAAACTTCTCACCA-3′ | 64 |
| FBN-1 | 28 | 5′-AGATCCTCTCCTATGCCGAG-3′ | 5′-GATACACGCGGAGATGTTGG-3′ | 57 |
| FBN-1 | 42 | 5′-GCATCACCAACCCTCCAATC-3′ | 5′-CACCTGTACTTGGGATGGGA-3′ | 56 |
| TGF-βR2 | 6 | 5′-ATGGGCCTCACTGTCTGTTT-3′ | 5′-CACAATGATGCTGGTCCACA-3′ | 55 |
2.15. Statistical Analysis
The SigmaPlot 14 program (Jendel Corporation, 1986-2017) was used to generate the graphs and to perform the statistical analyses. Statistical significance was determined by the Mann-Whitney rank sum test followed by the normality test (Shapiro-Wilk). The correlation was made with Spearman's test. The data are presented as the median and Min–Max range. Differences were considered statistically significant when p ≤ 0.05.
3. Results
3.1. General Characteristics
Out of the 10 LDS patients, 4 were men and 6 were women. The demographical characteristics of the patients are shown in Table 2.
Table 2.
Demographic characteristics of Loeys-Dietz syndrome patients.
| Sex | Age | MS family history | Clinical characteristics | Gene | Dao (mm) | Z score | Surgery type | Evolution over time |
|---|---|---|---|---|---|---|---|---|
| M | 15 | The father died at age 26 and was diagnosed with LDS | Dolichocephaly, malar hypoplasia, retrognathia, ogival palate, enlarged uvula, hypertelorism, low implantation of atrial pavilion, milia in the malar region, arm stroke ratio size > 1.06, stretch marks, pectus carinatum, scoliosis, equine foot varus. Steinberg and Walker-Murdoch positive, without ectopia lentis | FBN-1 exon 28 and TGF-βR2 exon 6 | 29 | 4.07 | Aortic root replacement by David's procedure (09/06/2013) | He died a year after the cardiovascular postoperative period, due to an acute cholecystitis complication |
| F | 22 | The father and sister died at 27 and 17 years ago with LDS diagnosis | Hypertelorism, ogival-type palate, dental overlap, uvula, widened millia, pectus carinatum, scoliosis, Steinberg and Walker-Murdoch, positive flat feet and valgus. Arm/height atio rgreater than 1.04, without ectopia lentis | FBN-1 exon 15 and TGF-βR2 exon 6 | 48 | 5.2 | Aortic root replacement by David's procedure (02/07/2013) | Satisfactory evolution currently in functional class 1 (NYHA) was evaluated in consultation in May 2019 |
| M | 13 | His mother was diagnosed with LDS, died at 22 due to aortic rupture, and he has a sister with LDS | Dolichocephaly, hypertelorism, ogival palate, uvula bifida, micrognathia, low implantation of atrial pavilion, pectus carinatum of right hemithorax (asymmetric), stretch marks, Steinberg sign and Walker-Murdoch positive, flat foot, contractures in eyes, scoliosis, and mitral valve prolapse | TGF-βR2 exon 6 and COL3A1 | 24 | 2.98 | Aortic surgery by David's procedure and he had mitral plasty (11/01/2013) | Since 11/11/2013, he has had dyskinetic and ballism that improved with anticonvulsive, the CT scan did not show aneurysms, he is hemodynamically stable, and he remains alive until 2019 |
| F | 18 | No history of MS or LDS | She showed dolichocephaly, hypertelorism, micrognathia, ogival palate and overlap, dental bifida tooth, arm reaction/height > 1.06 hyperelasticity, pectus carinatum, contractural arachnodactyly in feet and hands, flatfoot and varus Steinberg- and Walker Murdoch-positive scoliosis ecstasy. In her study of magnetic resonance, she had artificial tortuosity | FBN-I exon 28 and TGF-βR2 exon 6 | 46 | 9.5 | Aortic surgery by David's procedure with a 30 mm graft (04/10/2012) | Stable evolution, functional class 1. Last consultation July 2019 |
| M | 38 | Mother, one brother, and 7 cousins with MS, 1 cousin with LDS | Hypertelorism, ogival palate, dental overlap, bifida uvula, scoliosis, stretch marks. Without lens dislocation, size and stroke 1.98 and height 1.90 | FBN-1 exon 60+TGF-βR2 exon 6 | 48 | NA | Aortic surgery by David's procedure (20/01/2014) | Hemodynamically asymptomatically stable. In 2019, it was found in functional class 1 |
| F | 14 | Father with LDS | Hypertelorism, uvula bifid, scoliosis pectus carinatum | FBN-1 exon 28 and TGF-βR2 exon 6 | 54 | 4.30 | Aortic surgery by Bentall and De Bono (17/12/2018) | In 2019, he went to evaluation and was stable |
| F | 21 | The background is unknown | Hypertelorism, ogival palate, wide uvula, pectus carinatum valgus, foot without lens dislocation. Tortuous arteries | TGF-βR2 exon 6 | 25 | 3.47 | Aortic surgery by David's procedure (07/09/2015) | Satisfactory evolution. The last revision was in 2019 |
| M | 46 | The background is unknown | With hypertelorism, uvula bifida, scoliosis, flat feet | FBN-1 exon 28 and TGF-βR2 exon 6 | 63 | NA | Aortic surgery by Bentall and De Bono (09/09/2015) | He came to the office until 2019 |
| F | 25 | Her father died of acute aortic dissection at age 24 and was diagnosed with MS | Hypertelorism, millia in the malar region, pectus carinatum, normal uvula flat foot. By imaging, she had tortuous arteries | FBN-1 exon 42 and TGF-βR2 exon 6 | 46 | NA | Aortic surgery by Bentall and De Bono (18/12/2015) | She came to the hospital in 2019, and she was stable |
| F | 35 | The background is unknown | Hypertelorism, pectus carinatum, flat feet, tortuous arteries. Height 1.59 weight 64 | FBN-1 exon 28 and TGF-βR2 exon 6 | 58 | NA | Aortic surgery by Bentall and De Bono (06/04/2017) | She visited the hospital in 2019 |
Abbreviations: M = male; F = female; NYHA: New York Health Association; MS = Marfan syndrome; LDS = Loeys-Dietz syndrome; Dao = diameter aortic.
3.2. Mutations in LDS Patients
The percentage of positive mutations considering the mutation in any of the 4 exons of FBN-1 was of 80%. It was 100% for TGF-βR2 and 10% for COL3A1. Also, 90% of the patients shared mutations in all genes.
3.3. Correlations
The correlations between the LPO and aortic diameters and TAC and aortic diameters are shown in Table 3.
Table 3.
Correlations of LPO with TAC and aortic diameters.
| Total | C | LDS | ||||
|---|---|---|---|---|---|---|
| R 2 | p | R 2 | p | R 2 | p | |
| LPO/TAC | -0.59 | 0.008 | -0.55 | NS | -0.45 | NS |
| Carbonylation/TAC | -0.55 | 0.01 | -0.49 | NS | -0.32 | NS |
| VBD/LPO | -0.01 | 0.97 | 0.40 | NS | -0.75 | NS |
| SUD/LPO | 0.07 | 0.83 | 0.02 | NS | -0.52 | NS |
| VBD/TAC | -0.02 | 0.93 | -0.47 | NS | 0.74 | NS |
| SUD/TAC | -0.14 | 0.69 | -0.29 | NS | 0.82 | NS |
Abbreviations: LDS = Loeys-Dietz syndrome; LPO = lipid peroxidation; TAC = total activity antioxidant; VBD = Valsalva breast diameter; SUD = sinotubular union diameter.
3.4. General Variables
Table 4 shows the different general variables of both C subjects and patients with LDS. These are demographic variables and serum biochemistry; in this table, the results are expressed as median with the Min–Max values.
Table 4.
Demographic variables and serum biochemistry in C subjects and patients with LDS.
| C Median (Min–Max) |
LDS Median (Min–Max) |
p | |
|---|---|---|---|
| General characteristics | |||
| Age (range) | 62 (37–77) | 23 (14–46) | 0.001 |
| Size (m) | 1.65 (1.45–1.76) | 1.67 (1.58–1.77) | NS |
| Weight (kg) | 67 (56–105) | 57 (33–71.5) | 0.03 |
| BMI (range) | 27.8 (22.2–36.3) | 20.8 (12.7 – 25.3) | 0.005 |
| Serum biochemistry | |||
| CT (mg/dl) | 125 (102–191) | 129.5 (33–171) | NS |
| HDL (mg/dl) | 39 (32–54) | 48 (8.4–61) | NS |
| LDL (mg/dl) | 73.5 (50–143) | 84 (11.4–122) | NS |
| TG (mg/dl) | 97.5 (67–148) | 69 (57–118) | 0.09 |
Abbreviations: BMI=body mass index; CT = cholesterol; HDL = high-density lipoproteins; LDL = low-density lipoproteins; TG = triglycerides.
3.5. Enzymatic Activity and Expression
Table 5 shows the enzymatic activity and expression in the homogenate of the thoracic aortic aneurysms in both C subjects and patients with LDS.
Table 5.
Enzymatic activity and expression in the homogenate of the thoracic aortic aneurysms in both C subjects and patients with LDS.
| C Median (Min–Max) |
LDS Median (Min-Max) |
p | |
|---|---|---|---|
| Enzymatic activity | |||
| Mn-SOD (U/mg/protein) | 107.9 (93.3–117.8) | 118.5 (103.9–125.2) | 0.04 |
| Zn/Cu-SOD (U/mg protein) | 101.2 (90.9–122) | 123.4 (103.5–132.23) | 0.03 |
| CAT (μM/mg/protein) | 74.7 (45.9–82.4) | 40.2 (14.9–77.5) | 0.01 |
| GPx (nmol/mg/protein) | 0.23 (0.003–0.69) | 0.04 (0.01–0.06) | 0.02 |
| GST (μM/mg/protein) | 7 × 10−5 (2 × 10−5–4 × 10−4) | 1 × 10−5 (1 × 10−4–8 × 10−5) | 0.04 |
| TRx (μM/mg/protein) | 3.30 (1.8–7.5) | 2.1 (0.23–3.7) | 0.01 |
| GR (μM/mg/protein) | 0.32 (-1.1–0.71) | 0.86 (0.30–1.56) | 0.006 |
| Enzymatic expression | |||
| eNOS | 0.99 (0.48–1.13) | 0.87 (0.17–1.08) | NS |
| Nrf2 | 1.13 (0.98–1.35) | 0.95 (0.87–1.08) | 0.02 |
| ORX | 0.85 (0.54–0.97) | 1.00 (0.77–1.19) | 0.05 |
Mn-SOD = manganese superoxide dismutase; Zn/Cu-SOD = zinc copper superoxide dismutase; CAT = catalase; GPx = glutathione peroxidase; GST = glutathione reductase; TRx = thioredoxin reductase; GR = glutathione reductase; eNOS = endothelial nitric oxide synthase; Nrf2 = nuclear factor erythroid 2-related factor 2; ORX = xanthine reductase.
3.6. Enzymatic Activity
The Mn- and Cu/Zn-SOD isoforms showed a significant increase in their activities compared to the C group (p = 0.04 and p = 0.03, respectively) (Figures 1(a) and 1(b)). The enzymatic activities of CAT (p = 0.01), GPx (p = 0.02), GST (p = 0.04), and TrxR (p = 0.01) in patients with LDS were significantly decreased when compared to those in the C subjects (Figures 2(a)–2(d), respectively). The GR activity showed a significant increase (p = 0.006) in patients with LDS when compared to the C subjects (Figure 3).
Figure 1.

(a) Average activity of the Mn-SOD and (b) average activity of the Cu/Zn-SOD in LDS patients (n = 10) and C subjects (n = 9). Values expressed represent the median and Min–Max range. The image of the center is a representative gel of the electrophoresis of the SOD isoforms. Abbreviations: LDS = Loeys-Dietz syndrome; C = control subjects.
Figure 2.

(a) Average activity of CAT. The image below the graph is a representative native gel of the electrophoresis. (b) GPx activity, (c) GST activity, and (d) TrxR activity in LDS patients (n = 10) and C subjects (n = 9). Values are expressed as the median and Min–Max range. Abbreviations: LDS = Loeys-Dietz syndrome; C = control subjects.
Figure 3.
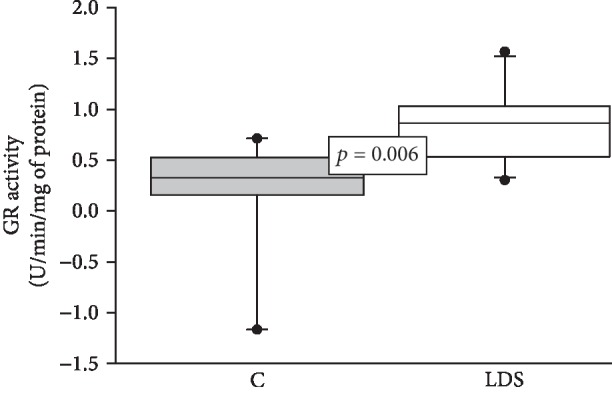
Average activity of GR in LDS patients (n = 10) and C subjects (n = 9). Values are expressed as the median and Min–Max range. Abbreviations: LDS = Loeys-Dietz syndrome; C = control subjects.
3.7. eNOS, Cu/Zn-SOD, ORX, and Nrf2 Expressions
The eNOS and Cu/Zn-SOD expressions did not show a significant difference in patients with LDS when compared to the C subjects (Figures 4(a) and 4(b)). The ORX and Nrf2 expressions showed significant increases and decreases, respectively, in patients with LDS when compared to the C subjects (p ≤ 0.05 and p = 0.02, respectively; Figures 4(c) and 4(d)).
Figure 4.

(a) Representative histograms of eNOS/α-actin expression. (b) Cu/Zn-SOD/α-actin expression, (c) ORX/α-actin expression, and (d) Nrf2/α-actin expression in LDS patients (n = 10) vs. C subjects (n = 9). Values are expressed as the median and Min–Max range. Abbreviations: LDS = Loeys-Dietz syndrome; C = control subjects.
3.8. Nonenzymatic Antioxidant System
The TAC levels, GSH, and vitamin C concentration showed a significant decrease (p = 0.006, p = 0.006, and p = 0.01, respectively) in patients with LDS when compared to the C subjects (Table 6). The protein carbonylation in patients with LDS showed a significant increase (p = 0.01, Table 6). The Se concentration in the TAA homogenate from patients with LDS was significantly decreased (p = 0.01, Table 6) when compared to that from the C subjects. However, the LPO index and NO3−/NO2− ratio tended to rise without reaching significance in patients with LDS compared to the C subjects (Table 6).
Table 6.
Redox biomarkers of the nonenzymatic system in the homogenate of the thoracic aortic aneurysm patients with LDS and C subjects.
| Parameters (mg/protein) | C Median (Min–Max) |
LDS Median (Min–Max) |
p |
|---|---|---|---|
| TAC (nmol Trolox) | 119.6 (35–220) | 15.09 (0.38–145) | 0.006 |
| LPO index (nmol MDA) | 2.4 (0.53–7.4) | 5.7 (2.3–15) | 0.04 |
| Carbonylation (ng carbonyls) | 5.5 (2.04–11.8) | 10.4 (4.3–21.1) | 0.01 |
| GSH (nM) | 9 × 10−2 (1 × 10−3–6 × 10−2) | 8 × 10−3 (3 × 10−3–1 × 10−2) | 0.006 |
| NO3−/NO2− (nM) | 2.6 (1.97–4.3) | 3.37 (2.14–7.8) | NS |
| Vitamin C (μM) | 0.03 (0.02–0.04) | 0.02 (0.01–0.04) | 0.01 |
| Se (pg) | 119.6 (35–220) | 15.09 (0.38–145) | 0.01 |
Abbreviations: TAC: total antioxidant capacity; LPO: lipid peroxidation; GSH: glutathione; Se: selenium; LDS: Loeys-Dietz syndrome; C: control subjects. The data are presented as the median and Min–Max range.
3.9. Histology
3.9.1. Hematoxylin-Eosin Staining
Figure 5(a) shows the representative photomicrograph of the aortic middle wall of a C subject, showing black nuclei of fibrocytes arranged in fascicles, red elastic fibers, and pink collagen fibers, without cystic necrosis. Figure 5(b) shows the representative photomicrograph of the middle aortic wall of a patient with LDS, showing blue nuclei of fibrocytes arranged in fascicles, in red elastic fibers, and in pink collagen fibers; the presence of cystic necrosis is observed in white.
Figure 5.

(a) Representative photomicrograph at 40x with hematoxylin-eosin staining of the aortic middle wall of a control subject. (A) Elastic fibers in deep pink. (B) Dark-colored fibrocyte nucleus. (b) Representative photomicrograph of the aortic middle wall of an LDS patient. (A) Cystic necrosis, with accumulation of amorphous material. (B) Elastic and collagen fibers in reddish and deep pink. (C) Blue-stained fibrocyte nucleus.
3.9.2. Masson's Trichrome Stain
Figure 6(a) shows the representative photomicrograph of the aortic middle wall of a C subject; elastic fibers are observed in red, collagen fibers are observed in blue, and no cystic necrosis is observed. Figure 6(b) shows the representative photomicrograph of the aortic middle wall of a patient with LDS; elastic fibers are observed in red, collagen fibers are observed in blue, and cystic necrosis is observed in white with the presence of amorphous material in faint blue.
Figure 6.

(a) Representative photomicrograph at 40x with Masson's trichrome staining of the aortic wall of a control subject. (A) Elastic fibers in red. (B) Collagen fibers in blue. (b) Representative photomicrograph of the aortic middle wall of an LDS patient. (A) Collagen fibers in deep blue color. (B) Cystic necrosis with the presence of amorphous material in faint blue color. (C) Elastic fibers in red color.
3.9.3. Weigert's Staining
Figure 7(a) shows the representative photomicrograph of the aortic middle wall of a C subject; elastic fibers are observed in black, collagen fibers are observed in brown-yellow, and cystic necrosis is not distinguished. Figure 7(b) shows the representative photomicrograph of the aortic middle wall of a patient with LDS; fragmented elastic fibers are observed in black, collagen fibers are observed in brown-yellow, and cystic necrosis is distinguished in white.
Figure 7.

(a) Representative photomicrograph at 40x with Weigert's staining of the aortic wall of a control subject. (A) Elastic fibers in black color. (B) Collagen and elastic fibers in pink and light brown color. (b) Representative photomicrograph of the aortic wall of an LDS patient. (A) Elastic fibers. (B) Cystic necrosis. (C) Collagen fibers.
4. Discussion
In the MS, there is an imbalance of the redox homeostasis that contributes to the alteration and development of the TAA [33]. However, in LDS which is a rare variant of the MS [5], the role of the imbalance in the redox homeostasis has not been evaluated. The main LDS characteristic is the presence of TAA with rapid progression that decreases the survival of the patients [3]. Therefore, the aim of this work was to analyze the oxidant/antioxidant profile in the TAA from patients with LDS. An imbalance similar to that found in MS with a positive feedback oxidizing background was found.
4.1. The FBN-1, TGF-βR2, and COL3A1 Coding Regions
The molecular testing in LDS is crucial to assess the diagnosis and management of the patients. We analyzed the mutations in the FBN-1, TGF-βR2, and COL3A1 genes. The percentage of positive mutations considering the mutation in any of the 4 exons of FBN-1 was of 80%. It was 100% for TGF-βR2 and 10% for COL3A1. 90% of the patients also shared mutations in all genes. The fact that these mutations were not found in the FBN-1 gene in all patients is due to the size of the gene which is greater than 200 k. The coding sequence is divided into 65 exons, and more than 800 mutations are known within it. This makes the complete study of the gene unavailable and expensive. The same happens for the TGF-βR2 gene. Therefore, we only examined those exons that have the highest frequency in this disease and mutations in the FBN-1 gene.
Most investigations on LDS focus on the study of mutations [34]. Mutations in FBN-1 are associated with 60-90% of the diagnosis [35]. In addition, mutations in the genes that encode for TGF-β1 and TGF-β2 have been identified in patients having this disease. TGFβ is involved in the development and maintenance of blood vessels and craniofacial growth [34]. In patients in whom no mutations in FBN-1 and TGFβ were found, homozygous deletions in the COL3A1 gene have been identified, and these mutations lead to structural alterations of the collagen that could cause aortic dissection [36]. Mutations affecting the intracellular kinase domain of this protein can disturb TGF-β signaling, which subsequently leads to features of LDS patients. In fact, TGF-βR2 mutations have been related to skeletal abnormalities, aortic dilation, and ED. Mutations of the FBN-1 gene have also been found in MS, and they produce a deconstructed protein product which cannot bind to the latent TGF-βR1 [4, 9, 10]. Activation of the latent TGF-βR1 by ROS could be associated with the decrease in GPx activity [37]. Therefore, an increase in the TGF-βR1 concentration in the TAA could be expected, and an increase in the TGF-βR1 and TGF-βR2 receptor expressions could upregulate the noncanonical and canonical TGF-β pathways which are increased in myocytes from TAA [9].
TGF-βR1 and TGF-βR2 can upregulate the expression of elastases and metalloproteinases (MMPs), which may enhance elastin degradation. MMPs participate in the elastic fiber disintegration, which is observed in the representative microphotographs of the patients with LDS. In these patients, there is a reduction of connective tissue elasticity that results in weakness of the aneurysm aortic wall [9, 16, 38]. These breakings in structure could be associated with decreases in GSH and GPx. ROS can affect the migration, differentiation, and proliferation of the myocyte cells which express MMPs that may be altered by the GPx that is dynamically regulated in the blood vessels [39]. They are the result of the TGF-βR1 and TGF-βR2 increases and ROS accumulation that contribute to the cystic necrosis of the aortic wall, vasomotor dysfunction, and ED, thus favoring the aneurysm formation [6, 9].
4.2. Antioxidant Enzymatic Profile in TAA from Patients with LDS
Several studies in MS mouse models have described vasomotor dysfunction in the TAA associated with ROS overproduction. This has been related to alterations in antioxidant enzymes such as SOD isoforms [9, 18] and to several diseases such as Turner syndrome, arterial hypertension, and some MS variants [16]. However, another investigation has shown a decrease in SOD isoforms due to the increase in O2− in the TAA in a mouse model of MS. This decrease was related to the deterioration of aortic function [10].
Our results suggest that the alteration of the activity of Cu/Zn- and Mn-SOD can be due to the overproduction of O2− through ORX and NADPH oxidase overexpression in tissue from the TAA [10]. An increase in ORX expression could increase the O2− concentration and stimulate Cu/Zn- and Mn-SOD activity, increasing the H2O2 concentration. This ROS is more harmful than others due to its capacity to diffuse through the cell membrane [40]. However, in our determinations, the expression of the Cu/Zn-SOD did not show significant changes. These results can be explained considering that the enzyme expression is not necessarily related to an increase in its activity since the expressed protein may be inactive or have a lower expression while having a greater activity. Thus, the degree of phosphorylation of the active site of SOD could increase its activity [41]. The increase in SOD activity in the patients with LDS suggests that the SOD isoforms try to counteract the O2− overproduction. However, this seems counterproductive since it results in H2O2 overproduction.
CAT is responsible for the H2O2 elimination, and its overexpression can decrease OS in the TAA [12]. CAT activity is altered in aortas of patients with MS through continued H2O2 exposure [10]. Our results show that CAT activity is significantly decreased in TAA of patients with LDS. This may be due to an H2O2 overproduction by the SOD isoforms [42, 43]. Also, the excessive O2− concentration can cause glucose-6-phosphate inhibition that catalyzes the formation of NADPH, which serves as an electron donor to CAT [44].
GPx is among the enzymes responsible for the H2O2 detoxification. It is more efficient than CAT when removing intracellular H2O2 and organic peroxides [15]. The GPx activity was decreased in patients with LDS, and this may be due to the low availability of GSH in these patients. The GPx has a Se atom in the form of selenocysteine in its catalytic site, and Se is a limiting factor of the antioxidant defense [45]. The deficiency of Se leads to cardiovascular pathologies [46] and diseases such as Kashin-Beck and Keshan [47–49]. In the patients with Keshan disease who have dilated cardiomyopathy, the Se deficiency decreases the GPx activity [49]. This suggests that selenoprotein expression or activity is regulated by the concentration of this trace element [50]. Our results show that the Se concentrations are decreased in patients with LDS, and this may impact on the activity of Se-dependent enzymes such as GPx and TrxR. Also, Se is absorbed in the form of organic compounds and in the presence of vitamins A, D, and E. Patients with LDS probably have an intestinal malabsorption syndrome which could cause the decrease in the Se [51]. Our results also showed a decrease in the concentration of vitamin C, which is important to regenerate vitamin E [15]. The decrease in both may affect the Se absorption.
GST is responsible for conjugating GSH with xenobiotics, peroxidized lipids, hydroperoxides, and H2O2 among other molecules favoring their elimination [15]. The substrates used by the GST are formed as a result of ROS modification of macromolecules [52]. A decrease in GST in aortas of patients with MS [14] and in patients with other pathologies that affect the cardiovascular system has been associated with OS and with an increase in LPO [16]. Our results show that the GST activity is decreased in patients with LDS. This suggests that the GST activity is compromised by the low availability of GSH. This contributes to the tendency to increase LPO. In addition, the increase in protein carbonylation may depend on the decrease in the TrxR activity, which would be reflected in a smaller reduction of thiol groups in proteins. TrxR is another Se-dependent protein possessing a selenocysteine in its catalytic site [53]. OS induces a compensatory increase in TrxR to reduce the oxidative modification of proteins present in several pathologies [53]. However, TrxR is decreased in pathologies with severe OS and metabolic disturbances [54]. The low TrxR activity in patients with LDS could increase the carbonylation and be associated with the OS present in this syndrome.
4.3. Antioxidant Nonenzymatic Profile in TAA from LDS Patients: Glutathione Levels and Glutathione Reductase
GSH is the main defense mechanism against ROS and electrophiles [55]. It is formed by γ-ι-glutamyl-ι-cysteinyl-glycine. GSH can also be a substrate for metabolic pathways such as the one mediated by the SOD isoforms and ORX. GSH homeostasis occurs mainly through the balance of consumption and supply [34]. When GSH reduces target molecules, electrophiles, or ROS, the thiol group of the cysteine is oxidized through the GPx activity. Excessive GSH oxidation by GPx might be due to chronic OS in patients with LDS. GSH is then reduced to GSSG by GR to restore GSH levels [55, 56]. GR requires NADPH consumption for its activity [40, 56]. A decrease in the GR activity will cause a decrease in the reduced GSH concentrations resulting in an increase in the level of the ROS. In several pathological processes, an alteration of the GR activity and in the GSH levels has been found, which is associated with an increased risk of OS [57]. Our results show that the GR activity was increased in patients with LDS, but despite this, the GSH values were not restored. This may be due to low levels of GSH which may stimulate the GR activity [58]. A low GSH level limits the activities of GPx and GST, and in turn, the low activities of GPX and GST may contribute less to the scavenging of H2O2 and O2− and this can favor and contribute to chronic OS in patients with LDS. It may also be due to a compensatory protective mechanism of the cells against oxidant agents [59]. Moreover, the GSH produced by GR may be oxidized and therefore be insufficient to compensate for the high production of ROS [60].
Another possible cause of the GSH decrease could be the alteration of the enzyme γ-glutamyl-cysteine synthase which is responsible for the GSH synthesis or for a deficient amount of the amino acids that constitute GSH [56]. In addition, the TAC results which include the GSH level show a decrease in the LDS patients. This confirms the presence of OS in the TAA of tissues from patients with this disease. Likewise, the LPO index showed a tendency to increase, and this could lead to an increase in prooxidant compounds such as 4-hydroxinonenal and MDA [61].
4.4. Expressions of Nrf2, Xanthine Oxidase, and Endothelial Nitric Oxide Synthase
The Nrf2 transcription factor is responsible for the regulation of the inducible expression of numerous antioxidant enzyme genes such as GPx, SOD isoforms, GST, and TrxR [62]. The low levels of Nrf2 expression in patients with LDS suggest that it can be responsible, in part, for the decrease in the activity of the antioxidant enzymes. Our results showed a tendency to decrease eNOS expression in the patients with LDS. Other studies have shown that the iNOS activity is increased and the activity of eNOS is decreased in the homogenate of TAA from patients with MS [63]. This result also suggests that the NO production mostly comes from the iNOS pathway and to a lesser extent from the eNOS [39]. Furthermore, in MS animal models, O2− overproduction by the NADPH oxidase and ORX pathway is associated with increase in NO production by iNOS and is involved in inflammatory processes [14, 18, 63]. This promotes the ONOO− formation that contributes to OS in the TAA [64]. High concentrations of ONOO− can lead to apoptosis in endothelial cells and may favor the activation of MMPs 1 and 2 in vascular smooth muscle cells degrading essential proteins to maintain the anatomical integrity of the aorta [64].
4.5. Histological Characteristics
The observation of the histological cuts from the TAA segments of patients with LDS showed cystic necrosis and accumulation of amorphous material. This suggests the development of fibrosis in the TAA that may be associated with chronic OS. The participation ROS in the formation of the abdominal aortic aneurysm was associated with the overexpression of the MMPs 2 and 9 [65]. The results with Weigert's staining showed fragmentation, thickening, and a smaller number of elastic fibers in the LDS group. This could be related to aortic stiffness. The elastic fiber fragmentation in the aorta is related to the interaction between OS, MMPs, and the TGF-β deregulation [66, 70]. Our results show a collagen increase in photomicrographs from the patients with LDS. This is probably due to deregulation in TGF-β signaling.
5. Conclusion
The antioxidant enzyme activities, including those of SOD isoforms, CAT, TrxR, and GST, decrease in the TAA of patients with LDS. These decreases in the enzyme activities favor the accumulation of ROS that contributes to GSH decrease and favor LPO and carbonylation. The decrease in Se and Nrf2 also impacts on the activity and/or expression of some of these antioxidant enzymes. The GR increase does not completely restore the GSH concentration, which is reflected in the decrease in the TAC and in the enzymes that use it which contributes to and favors ROS production. This leads to a positive feedback oxidizing background which contributes to the TAA formation.
5.1. Perspectives
The goal of the medical treatment in LDS is to delay the progression of aortic dilation to avoid catastrophic complications. This study proposes that the use of antioxidants together with the current treatments could help patients with LDS. The use of β blockers, as well as the use of angiotensin 1 antagonists, constitutes the first-line therapy to this day for LSD and MS [67]. However, the scientific evidence supporting these therapies is limited since it has been obtained from simple randomized trials with a restricted sample number [68]. In 2007, a meta-analysis of 6 studies that included 802 patients with MS showed no difference in the results of β blocker therapy in relation to mortality and morbidity [69].
The excessive activity of MMPs resulting in an excessive deposition of matrix elements such as proteoglycans and collagen which are organized in a random manner and fragmentation of elastic fibers is similar to that seen in atheromatous plaques, and therefore, the use of statins such as pravastatin at a dose of 40 mg/day might also be useful for the treatment of TAA in LDS and MS. Statins are inhibitors of HMG-CoA reductase and are used as CT-reducing agents with proven efficacy to reduce morbidity. Moreover, a study concluded that statins could have a similar effect to that of losartan in their capability to attenuate aortic root dilation in a mouse model with MS. Although these drugs reduced excessive protein manufacturing by vascular smooth muscle cells [70], inhibition of MMPs by these drugs may alter fibrillin-1 which is the component of the microfibrillins that provides a connection between elastin fibers and vascular smooth muscle cells [70, 71]. The structural homology between fibrillins and latent TGF-β binding proteins suggests an additional role for FBN-1 to direct TGF-β to the extracellular matrix and to keep it in an inactive state. Therefore, an FBN-1 deficiency would lead to excessive TGF-β signaling [71] which would not benefit LDS patients.
Deregulation of TGF-β signaling contributes to aortic and mitral valve pathology in mouse models with MS [72]. This is supported by the observation of a Marfan phenotype in humans with mutations in the TGF-β receptor [73]. Fibrillin-1 deficiency that leads to deregulation of TGF-β signaling causes vascular smooth muscle cells to reshape the extracellular matrix through the SMAD pathway [73]. However, other mechanisms such as redox status also importantly participate in the expansion of TAA in LDS together with the genetic predisposition, according to our findings, and therefore, therapeutics in this regard should be evaluated.
It is a very important state that during the evolution and progression of aneurysms, patients are typically asymptomatic and it is not until aortic dissection occurs, generating severe pain, that the doctor suspects the diagnosis. At that stage of the disease, it is too late for many therapeutic interventions and sudden death can happen in up to 50% of patients. Since it is clear that OS plays an important role in the development of aneurysms, OS should be monitored in clinical practice and aimed during treatment since diagnosis. The execution of randomized clinical trials in this regard should be addressed. Finally, it is possible that the use of a combination of antioxidants or antioxidants together with AT-1 inhibitors and statins may have an impact on the control of aortic dilation. Their beneficial effect could be monitored by various avant-garde imaging studies. In this sense, the treatment with antioxidant supplements such as N-acetylcysteine, a GSH donor, vitamins C and E, or combination of these or other antioxidants could contribute to the deceleration of the aggressive and lethal development of the TAA and increase the quality of life and survival in patients with LDS.
5.2. Limitations
The main limitation of this study was the obtainment of TAA fragments from patients with LDS. As described above, this disease is a variant of MS with unknown incidence that may be of around 1/10000. Only 10 patient samples could be obtained in a period of approximately 9 years. Another limitation was the improbability of having matched C subjects for gender and age. Although may there be a correlation of TAC with age, it is not possible to obtain aortic tissue from healthy subjects at specific ages. The only way to obtain these samples is finding subjects having a surgical indication where there is a possibility to ethically withdraw a small sample. This depends on the treatment applied and/or the surgical technique employed and on the obtainment of their informed consent. However, even if there are age differences in C subjects and patients with LDS, there is certainty that there were no comorbidities or aortic damage as shown in the photomicrographs of the C subjects.
Acknowledgments
We thank Benito Chávez Rentería for histology technical support and Alberto Guevara Flores, Héctor Vázquez Meza, and José de Jesús Martínez González for providing auranofin and the Department of Biochemistry of the Faculty of Medicine, UNAM.
Data Availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
All the authors declare that there are no competing interests regarding the publication of this paper.
References
- 1.Aalberts J. J. J., van den Berg M. P., Bergman J. E. H., et al. The many faces of aggressive aortic pathology: Loeys-Dietz syndrome. Netherlands Heart Journal. 2008;16(9):299–304. doi: 10.1007/bf03086168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bashari H., Brooks A., O’Brien O., Brennecke S., Zentner D. Maternal Loeys-Dietz syndrome (transforming growth factor ligand 2) in a twin pregnancy: case report and discussion. SAGE Open Medical Case Reports. 2019;7 doi: 10.1177/2050313X19852539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maleszewski J. J., Miller D. V., Lu J., Dietz H. C., Halushka M. K. Histopathologic findings in ascending aortas from individuals with Loeys-Dietz syndrome (LDS) The American Journal of Surgical Pathology. 2009;33(2):194–201. doi: 10.1097/PAS.0b013e31817f3661. [DOI] [PubMed] [Google Scholar]
- 4.Kuzmik G., Sang A., Elefteriandes J. Natural history of thoracic aortic aneurysms. Journal of Vascular Surgery. 2012;56(2):561–575. doi: 10.1016/j.jvs.2012.04.053. [DOI] [PubMed] [Google Scholar]
- 5.Meester J. A. N., Verstraeten A., Schepers D., Alaerts M., Van Laer L., Loeys B. L. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Annals of Cardiothoracic Surgery. 2017;6(6):582–594. doi: 10.21037/acs.2017.11.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loeys B. L., Chen J., Neptune E. R., et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature Genetics. 2005;37(3):275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 7.Takeda N., Yagi H., Hara H., et al. Pathophysiology and management of cardiovascular manifestations in Marfan and Loeys–Dietz Syndromes. International Heart Journal. 2016;57(3):271–277. doi: 10.1536/ihj.16-094. [DOI] [PubMed] [Google Scholar]
- 8.Branchetti E., Poggio P., Sainger R., et al. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovascular Research. 2013;100(2):316–324. doi: 10.1093/cvr/cvt205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soto M. E., Iturriaga Hernández A. V., Guarner Lans V., et al. Participation of oleic acid in the formation of the aortic aneurysm in Marfan syndrome patients. Prostaglandins and Other Lipid Mediators. 2016;123:46–55. doi: 10.1016/j.prostaglandins.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Yang H. H. C., van Breemen C., Chung A. W. Y. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vascular Pharmacology. 2010;52(1-2):37–45. doi: 10.1016/j.vph.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Xiong W., Mactaggart J., Knispel R., et al. Inhibition of reactive oxygen species attenuates aneurysm formation in a murine model. Atherosclerosis. 2009;202(1):128–134. doi: 10.1016/j.atherosclerosis.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parastatidis I., Weiss D., Joseph G., Taylor W. R. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(10):2389–2396. doi: 10.1161/ATVBAHA.113.302175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiellaro-Rafferty K., Weiss D., Joseph G., Wan W., Gleason R. L., Taylor W. R. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. American Journal of Physiology Heart and Circulatory Physiology. 2011;301(2):H355–H362. doi: 10.1152/ajpheart.00040.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zúñiga-Muñoz A. M., Pérez-Torres I., Guarner-Lans V., et al. Glutathione system participation in thoracic aneurysms from patients with Marfan syndrome. VASA. 2017;46(3):177–186. doi: 10.1024/0301-1526/a000609. [DOI] [PubMed] [Google Scholar]
- 15.Pérez-Torres I., Guarner-Lans V., Rubio-Ruiz M. E. Reductive stress in inflammation-associated diseases and the pro-oxidant. Effect of antioxidant agents. International Journal of Molecular Sciences. 2017;18(10):p. 2098. doi: 10.3390/ijms18102098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soto M. E., Soria-Castro E., Guarner Lans V., et al. Analysis of Oxidative Stress Enzymes and Structural and Functional Proteins on Human Aortic Tissue from Different Aortopathies. Oxidative Medicine and Cellular Longevity. 2014;2014:13. doi: 10.1155/2014/760694.760694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilton E., Bland M., Thompson M., Jahangiri M. Matrix metalloproteinase expression in the ascending aorta and aortic valve. Interactive Cardiovascular and Thoracic Surgery. 2008;7(1):37–40. doi: 10.1510/icvts.2007.163311. [DOI] [PubMed] [Google Scholar]
- 18.Soto M. E., Zuñiga-Muñoz A., Guarner Lans V., Duran-Hernández E. J., Pérez-Torres I. Infusion of hibiscus sabdariffa L. modulates oxidative stress in patients with Marfan syndrome. Mediators of Inflammation. 2016;2016:12. doi: 10.1155/2016/8625203.8625203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.World Medical Association. Declaration of Helsinki. Ethical principles for medical research involving human subjects. Journal of the American Medical Association. 2013;310(20):2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 20.Loeys B. L., Dietz H. C., Braverman A. C., et al. The revised Ghent nosology for the Marfan syndrome. Journal of Medical Genetics. 2010;47(7):476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 21.Bentall H., De Bono A. A technique for complete replacement of the ascending aorta. Thorax. 1968;23(4):338–339. doi: 10.1136/thx.23.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1976;72(1-2):248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 23.Pérez-Torres I., Roque P., El Hafidi M., Diaz-Diaz E., Baños G. Association of renal damage and oxidative stress in a rat model of metabolic syndrome. Influence of gender. Free Radical Research. 2009;43(8):761–771. doi: 10.1080/10715760903045296. [DOI] [PubMed] [Google Scholar]
- 24.Flohé L., Günzler W. A. Assays of glutathione peroxidase. Methods in Enzymology. 1984;105:114–121. doi: 10.1016/s0076-6879(84)05015-1. [DOI] [PubMed] [Google Scholar]
- 25.Beutler E. The relationship of red cell enzymes to red cell life-span. Blood Cells. 1988;14(1):69–91. [PubMed] [Google Scholar]
- 26.Holmgren A., Björnstedt M. Thioredoxin and thioredoxin reductase. Methods in Enzymology. 1995;252:199–208. doi: 10.1016/0076-6879(95)52023-6. [DOI] [PubMed] [Google Scholar]
- 27.Pech L. G. M., del Carmen Caballero-Chacón S., Guarner-Lans V., Díaz-Díaz E., Gómez A. M., Pérez-Torres I. Effect of oophorosalpingo-hysterectomy on serum antioxidant enzymes in female dogs. Scientific Reports. 2019;9(1) doi: 10.1038/s41598-019-46204-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benzie I. F. F., Strain J. J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of "Antioxidant Power": The FRAP Assay. Analytical Biochemistry. 1996;239(1):70–76. doi: 10.1006/abio.1996.0292. [DOI] [PubMed] [Google Scholar]
- 29.Ellman G. L. Tissue sulfhydryl groups. Archives of Biochemistry and Biophysics. 1959;82(1):70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 30.Jagota S. K., Dani H. M. A new colorimetric technique for the estimation of vitamin C using Folin phenol reagent. Analytical Biochemistry. 1982;127(1):178–182. doi: 10.1016/0003-2697(82)90162-2. [DOI] [PubMed] [Google Scholar]
- 31.Pérez Ruiz T., Martínez Lozano M. D. C., Hernández Lozano M. Determinación de selénio por su efecto catalítico en la reducción del azul de toluidina por el ion sulfuro. Anales de la Universidad de Murcia Ciencias. 1983;39 [Google Scholar]
- 32.Rodríguez A. C., Huel G., Saucedo B. L., et al. Determinación del contenido de selenio en suero sanguíneo de embarazadas normotensivas y preeclámpticas y en el cordón umbilical de sus bebés. Revista CENIC Ciencias Químicas. 2010;41(1):3–8. [Google Scholar]
- 33.Takeda N., Hara H., Fujiwara T., Kanaya T., Maemura S., Komuro I. TGF-β signaling-related genes and thoracic aortic aneurysms and dissections. International Journal of Molecular Sciences. 2018;19(7):p. 2125. doi: 10.3390/ijms19072125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh K. K., Rommel K., Mishra A., et al. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Human Mutation. 2006;27(8):770–777. doi: 10.1002/humu.20354. [DOI] [PubMed] [Google Scholar]
- 35.Dietz H. C., Cutting C. R., Pyeritz R. E., et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 36.Meienberg J., Rohrbach M., Neuenschwander S., et al. Hemizygous deletion of COL3A1, COL5A2, and MSTN causes a complex phenotype with aortic dissection: a lesson for and from true haploinsufficiency. European Journal of Human Genetics. 2010;18(12):1315–1321. doi: 10.1038/ejhg.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsubouchi K., Araya J., Yoshida M., et al. Involvement of GPx4-regulated lipid peroxidation in idiopathic pulmonary fibrosis pathogenesis. The Journal of Immunology. 2019;203(8):2076–2087. doi: 10.4049/jimmunol.1801232. [DOI] [PubMed] [Google Scholar]
- 38.Isekame Y., Gati S., Aragon-Martin J. A., Bastiaenen R., Kondapally Seshasai S. R., Child A. Cardiovascular management of adults with Marfan syndrome. European Cardiology Review. 2016;11(2):102–110. doi: 10.15420/ecr/2016:19:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L., Cheng X., Li H., et al. Quercetin reduces oxidative stress and inhibits activation of c-Jun N-terminal kinase/activator protein-1 signaling in an experimental mouse model of abdominal aortic aneurysm. Molecular Medicine Reports. 2014;9(2):435–442. doi: 10.3892/mmr.2013.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong K. W., Zhu G. Y., Wang L. H., Tang C. S. Effect of active oxygen species on intimal proliferation in rat aorta after arterial injury. Journal of Vascular Research. 1996;33(1):42–46. doi: 10.1159/000159130. [DOI] [PubMed] [Google Scholar]
- 41.Yamakura F., Kawasaki H. Post-translational modifications of superoxide dismutase. Biochimica et Biophysica Acta. 2010;1804(2):318–325. doi: 10.1016/j.bbapap.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 42.Rocha S., Gomes D., Lima M., Bronze-da-Rocha E., Santos-Silva A. Peroxiredoxin 2, glutathione peroxidase, and catalase in the cytosol and membrane of erythrocytes under H2O2-induced oxidative stress. Free Radical Research. 2015;49(8):990–1003. doi: 10.3109/10715762.2015.1028402. [DOI] [PubMed] [Google Scholar]
- 43.Hussain R. M., Abdullah N. F., Amom Z. Killing of Staphylococcus aureus by allylpyrocatechol is potentiated by induction of intracellular oxidative stress and inhibition of catalase activity. Journal of Integrative Medicine. 2016;14(6):456–464. doi: 10.1016/S2095-4964(16)60279-0. [DOI] [PubMed] [Google Scholar]
- 44.Hecker P. A., Leopold J. A., Gupte S. A., Recchia F. A., Stanley W. C. Impact of glucose-6-phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. American Journal of Physiology Heart and Circulatory Physiology. 2013;304(4):H491–H500. doi: 10.1152/ajpheart.00721.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deponte M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochimica et Biophysica Acta. 2013;1830(5):3217–3266. doi: 10.1016/j.bbagen.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 46.Rayman M. P. Selenium and human health. Lancet. 2012;379(9822):1256–1268. doi: 10.1016/S0140-6736(11)61452-9. [DOI] [PubMed] [Google Scholar]
- 47.Natasha M. S., Niazi N. K., Khalid S., Murtaza B., Bibi I., Rashid M. I. A critical review of selenium biogeochemical behavior in soil-plant system with an inference to human health. Environmental Pollution. 2018;234(1):915–934. doi: 10.1016/j.envpol.2017.12.019. [DOI] [PubMed] [Google Scholar]
- 48.Zwolak I., Zaporowska H. Selenium interactions and toxicity: a review. Selenium interactions and toxicity. Cell Biology and Toxicology. 2012;28(1):31–46. doi: 10.1007/s10565-011-9203-9. [DOI] [PubMed] [Google Scholar]
- 49.Lei C., Niu X., Wei J., Zhu J., Zhu Y. Interaction of glutathione peroxidase-1 and selenium in endemic dilated cardiomyopathy. Clinica Chimica Acta. 2009;399(1-2):102–108. doi: 10.1016/j.cca.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 50.Brigelius-Flohé R., Flohé L. Selenium and redox signaling. Archives of Biochemistry and Biophysics. 2017;617:48–59. doi: 10.1016/j.abb.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Kieliszek M. Selenium fascinating microelement, properties and sources in food. Molecules. 2019;24(7):p. 1298. doi: 10.3390/molecules24071298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayes J. D., Flanagan J. U., Jowsey I. R. Glutathione transferases. Annual Review of Pharmacology and Toxicology. 2005;45(1):51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 53.Lu J., Holmgren A. The thioredoxin antioxidant system. Free Radical Biology and Medicine. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 54.Tinkov A. A., Bjørklund G., Skalny A. V., et al. The role of the thioredoxin/thioredoxin reductase system in the metabolic syndrome: towards a possible prognostic marker? Cellular and Molecular Life Sciences. 2018;75(9):1567–1586. doi: 10.1007/s00018-018-2745-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rahal A., Kumar A., Singh V., et al. Oxidative stress, prooxidants, and antioxidants: the interplay. BioMed Research International. 2014;2014:19. doi: 10.1155/2014/761264.761264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu W., Fang Y. Z., Yang S., Lupton J. R., Turner N. D. Glutathione metabolism and its implications for health. The Journal of Nutrition. 2004;134(3):489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 57.Couto N., Wood J., Barber J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radical Biology and Medicine. 2016;95:27–42. doi: 10.1016/j.freeradbiomed.2016.02.028. [DOI] [PubMed] [Google Scholar]
- 58.Anand P., Rajakumar D., Jeraud M., Felix A. J., Balasubramanian T. Effects of taurine on glutathione peroxidase, glutathione reductase and reduced glutathione levels in rats. Pakistan Journal of Biological Sciences. 2011;14(3):219–225. doi: 10.3923/pjbs.2011.219.225. [DOI] [PubMed] [Google Scholar]
- 59.Paşaoğlu H., Muhtaroğlu S., Güneş M., Utaş C. The role of the oxidative state of glutathione and glutathione-related enzymes in anemia of hemodialysis patients. Clinical Biochemistry. 1996;29(6):567–572. doi: 10.1016/s0009-9120(96)00097-5. [DOI] [PubMed] [Google Scholar]
- 60.Shen K., Ji L., Chen Y., Yu Q., Wang Z. Influence of glutathione levels and activity of glutathione-related enzymes in the brains of tumor-bearing mice. Bioscience Trends. 2011;5(1):30–37. doi: 10.5582/bst.2011.v5.1.30. [DOI] [PubMed] [Google Scholar]
- 61.Soto M. E., Guarner-Lans V., Herrera-Morales K. Y., Pérez-Torres I. Participation of arachidonic acid metabolism in the aortic aneurysm formation in patients with Marfan syndrome. Frontiers in Physiology. 2018;9 doi: 10.3389/fphys.2018.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Satta S., Mahmoud A. M., Wilkinson F. L., Yvonne Alexander M., White S. J. The role of Nrf2 in cardiovascular function and disease. Oxidative Medicine and Cellular Longevity. 2017;2017:18. doi: 10.1155/2017/9237263.9237263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soto M. E., Soria-Castro E., Guarner-Lans V., et al. Preliminary analysis of the association of TRPV1 to the formation of Marfan syndrome aneurysms. Histology and Histopathology. 2019;34(12):1329–1343. doi: 10.14670/HH-18-127. [DOI] [PubMed] [Google Scholar]
- 64.Cui N., Hu M., Khalil R. A. Biochemical and biological attributes of matrix metalloproteinases. Progress in Molecular Biology and Translational Science. 2017;147:1–73. doi: 10.1016/bs.pmbts.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nissinen L., Kähäri V. M. Matrix metalloproteinases in inflammation. Biochimica et Biophysica Acta (BBA) - General Subjects. 2014;1840(8):2571–2580. doi: 10.1016/j.bbagen.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 66.Wheeler J. B., Ikonomidis J. S., Jones J. A. Connective tissue disorders and cardiovascular complications: the indomitable role of transforming growth factor-beta signaling. Advances in Experimental Medicine and Biology. 2014;802(1):107–127. doi: 10.1007/978-94-007-7893-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Habashi J. P., Judge D. P., Holm T. M., et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shores J., Berger K. R., Murphy E. A., Pyeritz R. E. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan's syndrome. New England Journal of Medicine. 1994;330(19):1335–1341. doi: 10.1056/NEJM199405123301902. [DOI] [PubMed] [Google Scholar]
- 69.Gersony D. R., McClaughlin M. A., Jin Z., Gersony W. M. The effect of beta-blocker therapy on clinical outcome in patients with Marfan's syndrome: a meta-analysis. International Journal of Cardiology. 2007;114(3):303–308. doi: 10.1016/j.ijcard.2005.11.116. [DOI] [PubMed] [Google Scholar]
- 70.McLoughlin D., McGuinness J., Byrne J., et al. Pravastatin reduces Marfan aortic dilation. Circulation. 2011;124:S168–S173. doi: 10.1161/CIRCULATIONAHA.110.012187. [DOI] [PubMed] [Google Scholar]
- 71.Ramirez F., Dietz H. C. Fibrillin-rich microfibrils: structural determinants of morphogenetic and homeostatic events. Journal of Cellular Physiology. 2007;213(2):326–330. doi: 10.1002/jcp.21189. [DOI] [PubMed] [Google Scholar]
- 72.Neptune E. R., Frischmeyer P. A., Arking D. E., et al. Dysregulation of TGF-β activation contributes to pathogenesis in Marfan syndrome. Nature Genetics. 2003;33(3):407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 73.Maciejczyk M., Zalewska A., Ładny J. R. Salivary antioxidant barrier, redox status, and oxidative damage to proteins and lipids in healthy children, adults, and the elderly. Oxidative Medicine and Cellular Longevity. 2019;2019:12. doi: 10.1155/2019/4393460.4393460 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.