

Abstract
One hypothesis for the etiology of neuropsychiatric disorders proposes that viral infection contributes to the induction of neuronal system dysfunction, resulting in a wide range of behavioral abnormalities. Recent research in molecular biology supports this hypothesis and refocuses on the role of viral infection in the development of psychiatric disorders. Viral infection can induce deleterious effects in the central nervous system by direct and/or indirect pathways. Understanding the mechanisms of glial cell dysfunction caused by persistent viral infection should lead to novel insights into the development of neurobehavioral disorders, including human mental illnesses, and to the possible development of treatments.
Neurobehavioral disorders, including most psychiatric diseases, can be multifactorial in origin, suggesting that both genetic and environmental factors play important roles in the manifestation of symptoms [1]. Each individual risk factor might exert a small effect on disease pathogenesis, but together their actions could result in more serious consequences, making it difficult to pinpoint the individual factors that contribute to these complex neurobehavioral disorders. Although recent approaches based on genetic research, as well as rapid progress in neurobiology, have led to the identification of several risk factors, including genetic variations or birth trauma, it is still difficult to determine the precise mechanisms and relative contributions of individual risk factors in the development of neuropsychiatric disorders.
Several working hypotheses propose that viral infection, as an environmental factor, contributes to the induction of neural cell dysfunction, resulting in a wide range of behavioral abnormalities, including cognitive, motor and social behavioral impairments. These hypotheses were initially based on observations from the 1918–1919 influenza virus epidemic, during which many infected patients developed psychiatric disorders similar to schizophrenia or depression, as well as on epidemiological studies of several viruses in psychiatric patients. An intriguing correlation between being born in winter and the development of schizophrenia in early adulthood has also reinforced the notion of a link between viral infection and increased risk for developing mental illness [2]. In spite of increased insight into the mechanisms of the neuropathogenesis of different viruses, a specific linkage between viral infection and the development of psychiatric disorders has not yet been identified.
Recent studies using molecular biology and genetic approaches revive the role of viral infection in the development of the behavioral disorders. It has recently been demonstrated that a virus product induces behavioral disorders in association with neurobiological abnormalities, and that impairment of glial cell function might exist as a common cause for the induction of neurobehavioral disorders [3]. A confirmed link between viral infection and neurobehavioral disorders would have a broad impact on diverse areas of biology and medicine, particularly the molecular pathogenesis of neurotropic viruses, neurobiology, biological psychiatry and public health.
1. Virus entry into the central nervous system
Many viruses can spread to the brain through the neural (peripheral or olfactory) or hematogenous route (figure 1). Poliovirus, rabies virus and α-herpesviruses, for example, invade at axon or sensory terminals and spread to the neurons. Many viruses able to spread via peripheral nerves can also enter the brain through the olfactory route. Neuronal spread and replication are characteristics of these viruses and define their pathogeneses. Other viruses, such as human immunodeficiency virus (HIV), measles virus, arenavirus and mumps virus, can invade the central nervous system (CNS) by the hematogenous route via the blood–brain barrier or microvascular endothelial cells. Such viruses can also replicate in resident cells within the brain. Some viruses that normally invade other tissues (e.g. influenza virus and reovirus) can enter the brain via the hematogenous route, although invasion of the CNS is a less frequent diversion from their normal mode of replication (in, for example, epithelial cells, lymphocytes or monocytes). Many viruses successfully infect brain cells and complete their life cycle in the CNS. Interestingly, in spite of significant differences among viruses in their modes of replication, as well as in the brain regions or neural cell types that they infect, most can establish a latent or persistent infection that could be essential for the induction of the unique pathogenesis of each virus [4].
Figure 1.
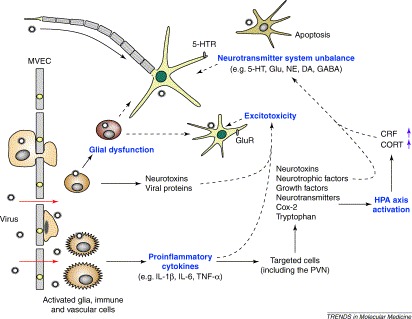
Possible mechanism of virus-induced neurological abnormalities linked to behavioral alterations. Unbroken arrows indicate stimulatory influences, broken arrows indicate negative effects and purple arrows indicate an increase in levels. Virus invades the CNS by the hematogenous route via the blood–brain barrier or microvascular endothelial cells (MVEC), or by a peripheral nerve route through the axon or sensory terminals. In this schema, initial infection of the CNS with a virus results in the production of proinflammatory cytokines by activated glia, neurons, vascular and immune cells. Proinflammatory cytokines activate the HPA axis by the production of Cox-2, tryptophan and norepinephrine from the targeted cells. Increased levels of TNF-α, neurotoxins and viral proteins can induce excitotoxicity via glutamate receptor pathways. Glial dysfunction by persistent viral infection might influence several neuronal functions, including synaptic formation and efficacy, as well as the maintenance of brain homeostasis. Abbreviations: CNS, central nervous system; CORT, cortisol; Cox-2, cyclooxygenase-2; CRF, corticotrophin-releasing factor; DA, dopamine; GABA, gamma-aminobutyric acid; Glu, glutamate; GluR, glutamate receptors; HPA, hypothalamic-pituitary-adrenal; 5-HT, serotonin; 5-HTR, serotonin receptors; IL, interleukin; NE, norepinephrine; PVN, paraventricular nucleus; TNF-α, tumor necrosis factor-α;.
2. Behavioral effects of neurotropic viral infection in animals
Behavioral effects of viral infection have been studied extensively in animal models. In experimental viral infection, behavioral alterations frequently dominate the clinical outcome. Virus-induced behavioral abnormality is predominantly manifested as a complex paradigm, formed by multiple elements of behavioral deficits induced by several different disabilities in the neuronal system. Viral infections are evaluated according to several behavioral phenotypes to determine whether correlations exist between neurophysiological and neuroanatomical disturbances, and ethological changes in infected hosts.
2.1. Sickness behaviors
Behavioral alterations following viral infection are often referred to as ‘sickness behavior’ 5, 6. Sickness behavior is recognized as part of the natural homeostatic reaction of the host during an infectious episode, and can include alterations in body weight, temperature, taste preference, food and water intake, and sleep patterns 5, 6. Although sickness behavior is generally believed to be triggered by proinflammatory cytokines, viruses might also play a direct role in the generation of sickness behavior. Borna disease virus (BDV) infection in neonatal rats induces body weight stunting and abnormal salt taste preference [7]. In addition, certain strains of mice infected with influenza virus show decreases in food and water intake, and body weight, as well as a reduced body temperature [8]. Although viral infection is normally associated with a reduction in body weight, a persistence of BDV or canine distemper virus is reported to induce an obesity syndrome. Of BDV-infected adult rats, 5–10% became obese, resulting in a body weight that is up to 300% that of normal rats [9]. Alteration of sleep pattern was also observed in adult mice and cats infected with influenza virus and feline immunodeficiency virus, respectively 10, 11.
2.2. Locomotor abnormalities
Locomotor activity, exploration and anxiety are frequently applied in the assessment of behavioral effects of CNS viral infection in animals. Locomotor activity tests have been used to find anxiety-like and exploratory responses in infected animals. Because rodents are neophobic and averse to bright light, the animals tend to avoid open spaces and stay close to the walls when they are placed in a novel, well-lit open field. The tests measure behavior induced by anxiety, and also exploratory processes that might be affected by multiple CNS dysfunctions. Locomotor abnormalities were reported in rodents inoculated with one of several viruses, including BDV, influenza virus, murine leukemia virus (MLV), herpes simplex virus (HSV), vesicular stomatitis virus (VSV), Venezuelan equine encephalomyelitis virus and lymphocytic choriomeningitis virus (LCMV), and also in non-human primates with BDV and simian immunodeficiency virus (SIV) 12, 13. Neonatally BDV-infected Lewis rats show an extreme locomotor hyperactivity [14]. The study of species-specific fear-related responses suggests that the locomotor hyperactivity in BDV-infected rats reflects increased, rather than decreased levels of anxiety, and is the result of increased escape responses to aversive stimuli [14].
2.3. Learning and memory deficits and abnormal social behaviors
Cognitive ability is a parameter that shows an integrated function of the neuronal system, because cognition involves many tasks, including learning, memory, motivational, and/or motor activities. Assessments using a Morris-type water maze or Y-maze are often performed to investigate dysfunctions or neuroanatomical abnormalities in specific brain regions, particularly in the hippocampus. Several viruses, including BDV, HSV, SIV, LCMV and MLV, induce learning and memory impairment in rodent or non-human primate models [12]. In many cases, the cognitive impairments are dependent upon the age of the animal at both inoculation and examination.
Social play behavior is well characterized in rats. Social behavior of BDV-infected neonatal Lewis rats has been tested using the ‘intruder–resident’ paradigm, in which an intruder was placed in the cage of socially isolated residents [15]. Although there was no difference in non-social exploratory activity (e.g. ambulation and rearing) between BDV- and uninfected residents, non-play social investigation (e.g. sniffing, approach and follow) was higher in BDV residents compared with uninfected residents on the first day of the testing. The disturbances in play behavior and other social interactions following neonatal BDV infection support the value of the neonatally BDV-infected rat as an animal model of autism; a disease that manifests as behavioral problems in social interaction and communication, and may be linked to developmental defects of higher brain function.
Aggression is also a complex social behavior with multiple causes 16, 17. In rodents, aggressive behavior is generally measured by offensive intermale fighting using a resident–intruder paradigm. Aggressive behavior in mice can be analyzed to elucidate the molecular mechanisms of violence in humans, because the molecular mechanisms in mice might be similar to those found in humans displaying aggression [e.g. serotonin (5-HT) system abnormalities] [16]. Aggressive behavior has been found in animals with rabies virus, BDV, HSV, SIV and tick-bone encephalitis viral infections 18, 19, 20, 21.
3. Virus-induced neuronal damages
3.1. Direct pathways
Brain damage as a result of viral infection defies a simple explanation, in which a single mechanism is involved in the devastation of neuronal systems. However, the mechanisms underlying these neuronal disturbances might fall into two general categories, involving direct and indirect pathways. Direct pathways include neural cell injury induced by the direct actions of virus replication or its components, as well as by the functional interplay between host and virus factors in an infected cell. Alternatively, indirect pathways involve a wide range of brain cell damage via neuroenvironmental alterations caused by viral infection. In indirect pathways, therefore, neurons and glial cells are widely injured, even if these cells are not actually infected.
Direct pathways are observed in the infections caused by HSV, rabies virus, reovirus and alphaviruses, where replication results in direct cell lysis or apoptosis [22]. Although HSV-1 can prevent apoptosis at the onset of infection, host cell death is induced subsequently as a result of the productive replication. It has been suggested that HSV-1 can induce host cell apoptosis before initiation of the host cellular immune response. Another well studied mechanism of the direct pathway involves BDV infection. In rodent models of BDV infection, the virus can cause neuronal cell dysfunctions in the absence of immune-mediated cell destruction or apoptosis, resulting in serious neurological disorders [23]. In addition, many viruses use one or more mechanisms to regulate the cell defense responses that are necessary for survival or functional stabilization of the neural cells being infected 24, 25. Such mechanisms include the inhibition or suppression of double-stranded RNA-dependent kinase, caspase activation, death receptors and proapoptotic activation of transcription factors, such as p53. Perturbation of such cellular factors could induce functional loss and/or abnormal activation of infected neural cells, leading to broad incoordination of the neural system.
3.2. Indirect pathways
Viral infection can alter the CNS environment as a result of host immune responses, as well as through the release of neurotoxins from macrophages, microglia and astrocytes. Indirect mechanisms involve the effects of such CNS alterations caused by host responses to infection. The host immune responses exist as a dominant indirect pathway to affect both infected and uninfected cells. Although T-cell responses elicited during infection can eliminate viruses efficiently without the destruction of infected neurons [26], infiltrated T cells can actually induce neuronal damage. In BDV-infected rodents, CD8+T cells are believed to play a major role in the immunopathogenesis of this virus. Administration of anti-CD8 antibodies to BDV-infected rats not only inhibits brain inflammation, but also prevents neuronal degeneration [27]. Proinflammatory cytokines, such as interleukin 1 (IL-1α and IL-1β), IL-6 and tumor necrosis factor (TNF)-α, in infected brain also have deleterious effects on neuronal functions. In addition, host autoimmune responses induced by viral infections are also present as an indirect pathway of virus-induced neuropathogenesis. It has been suggested that multiple sclerosis (MS), an autoimmune demyelinating disease, is initiated by persistent viral infection [28]. MS-like demyelination is induced in rodent brains infected with certain neurotropic viruses, such as Theiler's murine encephalomyelitis virus, murine coronavirus and Semliki Forest virus [28]. Perhaps the most thoroughly studied indirect mechanism is excitotoxicity, which involves abnormal stimulation or activation of neurons by the amino acid glutamate, and appears to play a role in HIV-associated neuronal injury [29]. HIV envelope protein gp120 or Tat, as well as the inflammatory mediator TNF-α, act via glutamate receptor pathways to increase oxidative stress in neurons and cause neuronal cell death [30]. Such indirect pathways might play a role in the development or aggravation of neurodegenerative disorders, such as Alzheimer's and Parkinson's diseases.
4. Molecular basis of virus-induced neurobehavioral disorders and a possible link to human mental disorders
4.1. Role of proinflammatory cytokines
Viral infection evokes cytokine upregulation, and persistent viral infection also seems to induce a chronic upregulation of cytokine levels in the brain (figure 1). Signaling molecules of the immune activation, particularly proinflammatory cytokines such as IL-1α, IL-1β, IL-6 and TNF-α, contribute to neuropsychiatric syndromes, especially major depression and chronic fatigue syndrome 31, 32. At the experimental level, proinflammatory cytokines induce alterations that are analogous to the behavioral and biological abnormalities occurring in depressed patients, including social withdrawal, anhedonia and cognitive impairment [5].
The role of proinflammatory cytokines in depressive behavior has been well documented in the findings of research on sickness behavior. IL-1 might be a dominant mediator for the induction of sickness behavior. In rats, peripheral and central administration of IL-1β induces conditioned taste aversions, as well as decreased food intake 33, 34. Furthermore, systemic injection of IL-1β to adult rats or mice decreases the time spent in their exploration of juveniles [35]. The involvement of brain IL-1 in depressive behavior has been confirmed by experiments using mice lacking IL-1β-converting enzyme or injected with IL-1 receptor antagonist 36, 37. IL-6 and TNF-α might not play a central role, but have been shown to act in conjunction with IL-1 in the induction of the symptoms.
The behavioral effects of proinflammatory cytokines are mediated by multiple pathways. The cytokines are known to activate rapidly the hypothalamic-pituitary-adrenal (HPA) axis, although IL-6 and TNF-α are less potent and effective than is IL-1. This activation might be mediated by the systematic regulation of cyclooxygenase, norepinephrine, 5-HT and brain tryptophan [38]. Hyperactivity of the HPA axis is a prominent biochemical change observed in patients with major depression [39]. That antidepressants directly regulate HPA axis function suggests that hyperactivity of the HPA axis is involved in the pathogenesis of depressive behaviors [40]. One current hypothesis proposes that activation of the HPA axis upregulates cortisol (a human glucocorticoid), the release of which is stimulated by adrenocorticotropin from the anterior pituitary, as well as by increased levels of corticotrophin-releasing factor (CRF) from the hypothalamus [39]. Both glucocorticoids and CRF are suggested to have multiple and complex effects on the circulating neurotransmitters, including 5-HT, dopamine and GABA (gamma-aminobutyric acid).
Proinflammatory cytokines might also affect, directly or indirectly, the levels of neurotrophins, growth factors, neurotransmitters and neurotoxins in the brain, via the activation of glia, neurons, vascular and immune cells (figure 1) [41]. Glial cells, particularly astrocytes, are a primary source of neurotrophins and growth factors, such as brain-derived neurotrophic factor (BDNF) and glial-derived neurotrophic factor. In addition, glial cells release neurotoxins following stimulation by IL-1 [42]. The contribution of proinflammatory cytokines to neuronal degeneration is believed to be dependent on the balance between neuroprotective and neurotoxic factors released from glial cells 41, 43. In virus-infected brains, a chronic upregulation of proinflammatory cytokines might tip the balance in favor of neurotoxic effects. Furthermore, proinflammatory cytokines might induce apoptosis in CNS cells. Although the role of IL-1 in the induction of apoptosis is less clear, it is well known that TNF-α can directly induce apoptosis via activation of Fas receptors in neuronal cells [44].
4.2. Role of 5-HT systems
Neuropsychiatric disorders are likely to be involved in the abnormalities of neurotransmitter systems, an assertion supported by the fact that drugs affecting neurotransmitter systems are potent antipsychotics [45]. 5-HT is the focus of intense investigation, as many experiments have revealed widespread distribution of 5-HT axons and multiple 5-HT receptor systems within the brain, as well as the importance of the 5-HT system in maintaining normal brain function [46]. Reduced levels of 5-HT in humans are associated with several mental health and behavioral problems, including aggression, violence, sexual dysfunctions, sleep and eating disorders. The reduction of 5-HT through tryptophan depletion induces depressive effects 39, 40. In rodent models, aggression increases after a reduction in the levels of brain 5-HT [16].
Viral infection can directly or indirectly alter 5-HT systems in both human and animal brains. The levels of cerebrospinal fluid 5-HT were significantly lower in HIV-1 patients compared with the control group [47]. Furthermore, several CNS infections, such as BDV, HSV, Japanese encephalitis virus, tick-bone encephalitis virus and VSV, are also reported to affect the level of 5-HT in rodent brains. As described above, 5-HT dysfunction is induced by the upregulation of proinflammatory cytokines and glucocorticoids, as well as by hyperactivity of the HPA axis (figure 1). The detailed mechanisms of 5-HT system disturbances by such indirect pathways have recently been reviewed 38, 48.
Recent studies have also focused on the alterations of 5-HT receptor expression. Genetic studies have shown that mice lacking 5-HT receptors display behavioral deficits, including aggression and anxiety 16, 49. Alteration of 5-HT receptors in the brain is observed in some viral infections. Semliki Forest viral infection increases the number of 5-HT1A receptors by up to 80% in the cortex but has no effect in the midbrain, pons and the hypothalamus, whereas there is no alteration in 5-HT2A receptor numbers in any of the brain regions [50]. Rabies viral infection in humans induces a series of clinical symptoms, suggesting involvement of the central 5-HT system. Studies using mouse models have suggested that 5-HT1D-like receptor subtypes are affected specifically and at an early stage following rabies viral infection [51]. Neonatal BDV infection induces upregulation of hippocampal 5-HT1A and cortical 5-HT2A receptors in Lewis rats and downregulation of cortical 5-HT2A receptors in Fisher344 rats [52]. It has recently been reported that transgenic mice synthesizing BDV phosphoprotein in glial cells exhibit an unbalanced expression of 5-HT receptors (1A, 1B and 2C) in the hippocampus at eight months of age [3]. Intriguingly, these mice exhibit intermale aggressiveness, as measured by offensive fighting attacks using a resident–intruder paradigm.
4.3. Effects of glial dysfunction
Current evidence indicates that psychiatric disorders, including schizophrenia, predominantly occur without neuronal degeneration. Neurodegenerative disorders are typically accompanied by glial proliferation, which involves the release of various neurotoxic factors. However, in major psychiatric disorders, the brain shows no signs of glial proliferation, and a spectrum of neurological abnormalities associated with glial cell dysfunction are frequently found 53, 54, 55. This observation gives rise to the hypothesis that neurological abnormalities associated with glial cell dysfunction contribute to the development of schizophrenia 54, 55. In fact, recent efforts to identify candidate genes that confer susceptibility to schizophrenia have revealed some genes whose functions might be linked to glial cell function or survival 56, 57.
It is well known that glial cells play an important role in generating the basic scaffolding of the brain during development. Furthermore, recent evidence has revealed that astrocytes might be required for synapse formation, maintenance and efficacy, as well as for the maintenance of brain homeostasis 58, 59. The long processes of astrocytes associate with the synaptic connections of neurons and participate in neuronal signaling, as a third element within the synapse. Astrocytes increase the number of structurally mature synapses in the CNS, as well as their function, and most synapses are generated synchronously with the development of glial cells [60]. It is most likely, therefore, that a reduction in astrocyte function causes a wide range of disturbances in brain development, as well as in synapses and neurons, leading to serious behavioral abnormalities (figure 2).
Figure 2.
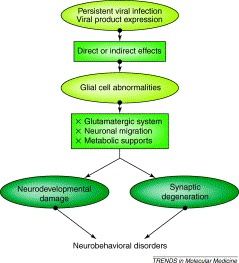
A possible hypothesis of glial cell dysfunction in the development of neurobehavioral disorders. Persistent viral infections or the expression of viral proteins may directly and/or indirectly affect glial cell functions in infected CNS. Glial dysfunctions lead to abnormalities in glutamatergic systems, neuronal migration and metabolic supports, resulting in neurodevelopmental damage or synaptic degeneration.
Many viruses can infect and subsequently activate glial cells. Several neurotoxic or immunomodulatory factors are released from activated glial cells, leading to altered neuronal function and eventual neuronal apoptosis, as described above. This process appears to be a common pathway leading to virus-induced neuronal injury, which is frequently accompanied by glial proliferation [61]. By contrast, this pathway seems to be unconnected to the neuropathology of major psychiatric disorders. Thus, the presence of virus that leads to a long-lasting persistent infection and induces glial cell dysfunctions without the destructions of neural cells might be key to understanding the linkage between CNS viral infection and neuropsychiatric disorders (figure 2). Recently, it has been demonstrated that glial expression of BDV phosphoprotein leads to behavioral and neurobiological abnormalities in transgenic mice without the loss of any types of glial cell [3]. The transgenic mice developed enhanced intermale aggressiveness, hyperactivity and spatial reference memory deficit. It was also shown that the brains of the transgenic mice exhibit a significant reduction in BDNF and 5-HT receptor expression, as well as a marked decrease in synaptic density [3]. Although the mechanism by which glial expression of BDV phosphoprotein induces neurological disturbances remains to be elucidated, the lack of reactive astrocytosis or neural cell loss in the transgenic brains suggests that phosphoprotein can directly affect astrocyte function. BDV phosphoprotein interferes with a multifunctional protein, amphoterin/HMGB1, and inhibits its function in cultured neuronal cells [62]. HMGB1 plays an important role not only in neurite outgrowth of neurons, but also in cell survival through the interaction with its cellular receptor, RAGE [63]. HMGB1 is crucial for neuron–glial cell interaction, which is correlated with diminished HMGB1 levels in glial cells [64]. These findings suggest that HMGB1 function is very important in the maintenance of astrocyte function, as well as of CNS homeostasis. BDV frequently establishes a persistent infection without immune-mediated brain damage in animal brains, and phosphoprotein is abundantly produced, even in the persistent state [23]. The possible relationship between glial cell dysfunction by persistent viral infection and virus-induced neurobehavioral disorders should be investigated further.
5. Concluding remarks
Viral infection frequently affects animal behavior, such as locomotor activity, learning, memory, aggression and social behavior, all of which are normally expressed as an integrated paradigm of different elements of CNS function. Numerous genetic and neurophysiological studies have demonstrated that abnormalities in neurotransmitter systems and glial cell function, as well as the release of proinflammatory cytokines, play crucial roles in the induction of behavioral disorders. Such abnormalities have also been observed in a wide variety of CNS viral infections. Does a specific virus cause psychiatric disorders in humans? Although frequently asked, there is currently no direct evidence that viral infection results in human mental illnesses. CNS viral infection in animals can induce neurochemical and neurophysiological abnormalities, as well as behavioral alterations, linked to human mental illnesses. Furthermore, epidemiological studies have demonstrated that people with behavioral diseases have a high prevalence for antibodies to certain viruses, such as BDV, herpesviruses and endogenous retrovirus, compared with controls 65, 66, 67, 68. However, such linkages might not be a direct answer. Given that most neuropsychiatric disorders are multifactorial, much more will be required to determine the role of viral infection among the many interlinked risk elements. A better understanding of the effects of chronic or persistent viral infection of CNS cells, particularly glial cells, should lead to novel insights into the molecular mechanisms underlying neurobehavioral disorders.
Acknowledgements
K.T is supported by Grants from the Ministry of Education, Culture, Sports, Science and Technology, a Grant-in-Aid from the Zoonosis Control Project of Ministry of Agriculture, Forestry and Fisheries and a Research Grant for Nervous and Mental Disorders from Ministry of Health, Labor and Welfare of Japan, and thanks Kazuyoshi Ikuta and Wataru Kamitani for stimulating discussions.
References
- 1.Cowan W.M. The human genome project and its impact on psychiatry. Annu. Rev. Neurosci. 2002;25:1–50. doi: 10.1146/annurev.neuro.25.112701.142853. [DOI] [PubMed] [Google Scholar]
- 2.Brown A.S., Susser E.S. In utero infection and adult schizophrenia. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:51–57. doi: 10.1002/mrdd.10004. [DOI] [PubMed] [Google Scholar]
- 3.Kamitani W. Glial expression of borna disease virus phosphoprotein induces behavioral and neurological abnormalities in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 2003;100:8969–8974. doi: 10.1073/pnas.1531155100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tyler K.L., Nathanson N. Pathogenesis of viral infections. In: Knipe D.M., Howley P.M., editors. Fields Virology. Lippincott Williams and Wilkins; 2001. pp. 199–243. [Google Scholar]
- 5.Konsman J.P. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002;25:154–159. doi: 10.1016/s0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- 6.Kelley K.W. Cytokine-induced sickness behavior. Brain Behav. Immun. 2003;17(Suppl 1):S112–S118. doi: 10.1016/s0889-1591(02)00077-6. [DOI] [PubMed] [Google Scholar]
- 7.Hornig M. Borna disease viral infection of adult and neonatal rats: models for neuropsychiatric disease. Curr. Top. Microbiol. Immunol. 2001;253:157–177. doi: 10.1007/978-3-662-10356-2_8. [DOI] [PubMed] [Google Scholar]
- 8.Conn C.A. Cytokines and the acute phase response to influenza virus in mice. Am. J. Physiol. 1995;268:R78–R84. doi: 10.1152/ajpregu.1995.268.1.R78. [DOI] [PubMed] [Google Scholar]
- 9.Herden C. Distribution of Borna disease virus in the brain of rats infected with an obesity-inducing virus strain. Brain Pathol. 2000;10:39–48. doi: 10.1111/j.1750-3639.2000.tb00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toth L.A. Strain differences in sleep and other pathophysiological sequelae of influenze virus infection in naı̈ve and immunized, ice. J. Immunol. 1995;58:89–99. doi: 10.1016/0165-5728(94)00193-r. [DOI] [PubMed] [Google Scholar]
- 11.Prospero-Garcia O. Sleep patterns are disturbed in cats infected with feline immunodeficiency virus. Proc. Natl. Acad. Sci. U. S. A. 1994;91:12947–12951. doi: 10.1073/pnas.91.26.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weed M.R., Gold L.H. Paradigms for behavioral assessment of viral pathogenesis. Adv. Virus Res. 2001;56:583–626. doi: 10.1016/s0065-3527(01)56039-x. [DOI] [PubMed] [Google Scholar]
- 13.Mohammed A.H. Viruses and behavioural changes: a review of clinical and experimental findings. Rev. Neurosci. 1993;4:267–286. doi: 10.1515/revneuro.1993.4.3.267. [DOI] [PubMed] [Google Scholar]
- 14.Pletnikov M.V. Persistent neonatal Borna disease virus (BDV) infection of the brain causes chronic emotional abnormalities in adult rats. Physiol. Behav. 1999;66:823–831. doi: 10.1016/s0031-9384(99)00021-9. [DOI] [PubMed] [Google Scholar]
- 15.Pletnikov M.V. Developmental brain injury associated with abnormal play behavior in neonatally Borna disease virus-infected Lewis rats: a model of autism. Behav. Brain Res. 1999;100:43–50. doi: 10.1016/s0166-4328(98)00111-9. [DOI] [PubMed] [Google Scholar]
- 16.Nelson R.J., Chiavegatto S. Molecular basis of aggression. Trends Neurosci. 2001;24:713–719. doi: 10.1016/s0166-2236(00)01996-2. [DOI] [PubMed] [Google Scholar]
- 17.Miczek K.A. Aggressive behavioral phenotypes in mice. Behav. Brain Res. 2001;125:167–181. doi: 10.1016/s0166-4328(01)00298-4. [DOI] [PubMed] [Google Scholar]
- 18.Charlton K.M. Experimental rabies in skunks: effects of immunosuppression induced by cyclophosphamide. Can. J. Comp. Med. 1984;48:72–77. [PMC free article] [PubMed] [Google Scholar]
- 19.Narayan O. Pathogenesis of Borna disease in rats: immune-mediated viral ophthalmoencephalopathy causing blindness and behavioral abnormalities. J. Infect. Dis. 1983;148:305–315. doi: 10.1093/infdis/148.2.305. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Hur T. A novel permissive role for glucocorticoids in induction of febrile and behavioral signs of experimental herpes simplex virus encephalitis. Neuroscience. 2001;108:119–127. doi: 10.1016/s0306-4522(01)00404-3. [DOI] [PubMed] [Google Scholar]
- 21.Moshkin M. Behavior, chemosignals and endocrine functions in male mice infected with tick-borne encephalitis virus. Psychoneuroendocinology. 2002;27:603–608. doi: 10.1016/s0306-4530(01)00096-8. [DOI] [PubMed] [Google Scholar]
- 22.Griffin D.E., Hardwick J.M. Perspective: viral infections and the death of neurons. Trends Microbiol. 1999;7:155–160. doi: 10.1016/s0966-842x(99)01470-5. [DOI] [PubMed] [Google Scholar]
- 23.Tomonaga K. Molecular and cellular biology of Borna disease viral infection. Microbes Infect. 2002;4:491–500. doi: 10.1016/s1286-4579(02)01564-2. [DOI] [PubMed] [Google Scholar]
- 24.Benedict C.A. To kill or be killed: viral evasion of apoptosis. Nat. Immunol. 2002;3:1013–1018. doi: 10.1038/ni1102-1013. [DOI] [PubMed] [Google Scholar]
- 25.Gale M., Jr, Katze M.G. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol. Ther. 1998;78:29–46. doi: 10.1016/s0163-7258(97)00165-4. [DOI] [PubMed] [Google Scholar]
- 26.Irani D.N., Griffin D.E. Regulation of T cell responses during central nervous system viral infection. Adv. Virus Res. 2001;56:175–197. doi: 10.1016/S0065-3527(01)56007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bilzer T., Stitz L. Immune-mediated brain atrophy. CD8+T cells contribute to tissue destruction during Borna disease. J. Immunol. 1994;153:818–823. [PubMed] [Google Scholar]
- 28.Fazakerley J.K., Walker R. Virus demyelination. J. Neurovirol. 2003;9:148–164. doi: 10.1080/13550280390194046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaul M. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 30.Ankarcrona M. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 31.Licinio J., Wong M.L. The role of inflammatory mediators in the biology of major depression: central nervous system cytokines modulate the biological substrate of depressive symptoms, regulate stress-responsive systems, and contribute to neurotoxicity and neuroprotection. Mol. Psychiatry. 1999;4:317–327. doi: 10.1038/sj.mp.4000586. [DOI] [PubMed] [Google Scholar]
- 32.Patarca R. Cytokines and chronic fatigue syndrome. Ann. N. Y. Acad. Sci. 2001;933:185–200. doi: 10.1111/j.1749-6632.2001.tb05824.x. [DOI] [PubMed] [Google Scholar]
- 33.Montkowski A. Central administration of IL-1 reduces anxiety and induces sickness behaviour in rats. Pharmacol. Biochem. Behav. 1997;58:329–336. doi: 10.1016/s0091-3057(97)00244-x. [DOI] [PubMed] [Google Scholar]
- 34.Kent S. Mechanisms of sickness-induced decreases in food-motivated behavior. Neurosci. Biobehav. Rev. 1996;20:171–175. doi: 10.1016/0149-7634(95)00037-f. [DOI] [PubMed] [Google Scholar]
- 35.Bluthe R.M. Mechanisms of the behavioral effects of interleukin 1. Role of prostaglandins and CRF. Ann. N. Y. Acad. Sci. 1992;650:268–275. doi: 10.1111/j.1749-6632.1992.tb49135.x. [DOI] [PubMed] [Google Scholar]
- 36.Burgess W. Interleukin-1β-converting enzyme-deficient mice resist central but not systemic endotoxin-induced anorexia. Am. J. Physiol. 1998;274:R1829–R1833. doi: 10.1152/ajpregu.1998.274.6.R1829. [DOI] [PubMed] [Google Scholar]
- 37.Dantzer R. Cytokine-induced sickness behavior: where do we stand? Brain Behav. Immun. 2001;15:7–24. doi: 10.1006/brbi.2000.0613. [DOI] [PubMed] [Google Scholar]
- 38.Dunn A.J. Cytokine activation of the HPA axis. Ann. N. Y. Acad. Sci. 2000;917:608–617. doi: 10.1111/j.1749-6632.2000.tb05426.x. [DOI] [PubMed] [Google Scholar]
- 39.Nestler E.J. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 40.Manji H.K. The cellular neurobiology of depression. Nat. Med. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- 41.Allan S.M., Rothwell N.J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001;2:724–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- 42.Giulian D. Brain glia release factors with opposing actions upon neuronal survival. J. Neurosci. 1993;13:29–37. doi: 10.1523/JNEUROSCI.13-01-00029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen M.D. Innate immunity: the missing link in neuroprotection and neurodegeneration. Nat. Rev. Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- 44.Park C. Expression of fas antigen in the normal mouse brain. Biochem. Biophys. Res. Commun. 1998;252:623–628. doi: 10.1006/bbrc.1998.9572. [DOI] [PubMed] [Google Scholar]
- 45.Sekine, Y. et al. Correlations between in vitro affinity of antipsychotics to various central neurotransmitter receptors and clinical incidence of their adverse drug reactions. Eur. J. Clin. Pharmacol. 55, 583–587. [DOI] [PubMed]
- 46.Azmitia E.C. Serotonin neurons, neuroplasticity, and homeostasis of neural tissue. Neuropsychopharmacology. 1999;21:33s–45s. doi: 10.1016/S0893-133X(99)00022-6. [DOI] [PubMed] [Google Scholar]
- 47.Kumar A.M. Cerebrospinal fluid 5-hydroxytryptamine and 5-hydroxyindoleacetic acid in HIV-1 infection. Neuropsychobiology. 2001;44:13–18. doi: 10.1159/000054908. [DOI] [PubMed] [Google Scholar]
- 48.Altar C.A. Neurotrophins and depression. Trends Pharmacol. Sci. 1999;20:59–61. doi: 10.1016/s0165-6147(99)01309-7. [DOI] [PubMed] [Google Scholar]
- 49.Overstreet D.H. Involvement of 5-HT1A receptors in animal tests of anxiety and depression: evidence from genetic models. Stress. 2003;6:101–110. doi: 10.1080/1025389031000111311. [DOI] [PubMed] [Google Scholar]
- 50.Mehta S., Kitchen I. Regional changes in 5-HT1A but not in 5-HT2A receptors in mouse brain after Semliki Forest viral infection: radioligand binding and autoradiographic studies. J. Neurovirol. 1998;4:606–618. doi: 10.3109/13550289809114227. [DOI] [PubMed] [Google Scholar]
- 51.Ceccaldi P.E. Rabies virus selectively alters 5-HT1 receptor subtypes in rat brain. Eur. J. Pharmacol. 1993;245:129–138. doi: 10.1016/0922-4106(93)90120-x. [DOI] [PubMed] [Google Scholar]
- 52.Pletnikov M.V. Effects of genetic background on neonatal Borna disease viral infection-induced neurodevelopmental damage. II. Neurochemical alterations and responses to pharmacological treatments. Brain Res. 2002;944:108–123. doi: 10.1016/s0006-8993(02)02724-5. [DOI] [PubMed] [Google Scholar]
- 53.Sawa A., Snyder S.H. Schizophrenia: diverse approaches to a complex disease. Science. 2002;296:692–695. doi: 10.1126/science.1070532. [DOI] [PubMed] [Google Scholar]
- 54.Cotter D.R. Glial cell abnormalities in major psychiatric disorders: the evidence and implications. Brain Res. Bull. 2001;55:585–595. doi: 10.1016/s0361-9230(01)00527-5. [DOI] [PubMed] [Google Scholar]
- 55.Moises H.W. The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. BMC Psychiatry. 2002;2:8. doi: 10.1186/1471-244X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tkachev D. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet. 2003;362:798–805. doi: 10.1016/S0140-6736(03)14289-4. [DOI] [PubMed] [Google Scholar]
- 57.Hakak Y. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 2001;98:4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pfrieger F.W., Barres B.A. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- 59.Fields R.D., Stevens-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ullian E.M. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- 61.Wyss-Coray T., Mucke L. Inflammation in neurodegenerative disease – a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 62.Kamitani W. Borna disease virus phosphoprotein binds a neurite outgrowth factor, amphoterin/HMG-1. J. Virol. 2001;75:8742–8751. doi: 10.1128/JVI.75.18.8742-8751.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rauvala H. Heparin-binding proteins HB-GAM (pleiotrophin) and amphoterin in the regulation of cell motility. Matrix Biol. 2000;19:377–387. doi: 10.1016/s0945-053x(00)00084-6. [DOI] [PubMed] [Google Scholar]
- 64.Daston M.M., Ratner N. Amphoterin (P30, HMG-1) and RIP are early markers of oligodendrocytes in the developing rat spinal cord. J. Neurocytol. 1994;23:323–332. doi: 10.1007/BF01188500. [DOI] [PubMed] [Google Scholar]
- 65.Carbone K.M. Borna disease virus and human disease. Clin. Microbiol. Rev. 2001;14:513–527. doi: 10.1128/CMR.14.3.513-527.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karlsson H. Retroviral RNA identified in the cerebrospinal fluids and brains of individuals with schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 2001;98:4634–4639. doi: 10.1073/pnas.061021998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ikuta K. Borna disease virus and infection in humans. Front. Biosci. 2002;7:D470–D495. doi: 10.2741/A789. [DOI] [PubMed] [Google Scholar]
- 68.Dickerson F.B. Association of serum antibodies to herpes simplex virus 1 with cognitive deficits in individuals with schizophrenia. Arch. Gen. Psychiatry. 2003;60:466–472. doi: 10.1001/archpsyc.60.5.466. [DOI] [PubMed] [Google Scholar]