

Abstract
Background: The antiviral effect of anti-influenza drugs such as zanamivir may be demonstrated in patients as an increased rate of decline in viral load over a time course of treatment as compared with placebo. Historically this was measured using plaque assays, or Culture Enhanced Enzyme Linked Immunosorbent Assay (CE-ELISA). Objectives: to develop and characterise real time quantitative PCR (qPCR) assays to measure influenza A and B viral load in clinical samples, that offer improvements over existing methods, in particular virus infectivity assays. Study design: The dynamic range and robustness were established for the real time qPCR assays along with stability of the assay components. Cross validation of the real time PCR assays with CE-ELISA was performed by parallel testing of both serial dilutions of three different subtypes of cultured virus and a panel of influenza positive throat swab specimens. Results: the assays were specific for influenza A and B and the dynamic ranges were at least seven logs. The assay variability was within acceptable limits but increased towards the lower limit of quantification, which was 3.33 log10 viral cDNA copies/ml of virus transport medium (ten viral RNA copies/PCR). The components of the assay were robust enough to withstand extended storage and several freeze–thaw cycles. For the real time PCR assays the limit of quantification was equivalent to the virus infectivity cut off, which equates to a 93-fold increase in sensitivity. Conclusion: Well characterised real time PCR assays offer significant improvements over the existing methods for measuring the viral load of strains of influenza A and B in clinical specimens.
Keywords: Influenza virus, Real-time PCR, Validation
1. Introduction
Influenza is an acute but normally self-limiting respiratory disease caused by influenza virus that results in considerable morbidity and lost working days. Patient groups such as the elderly, the immunocompromised and those with underlying chronic conditions such as asthma or chronic obstructive pulmonary disease (COPD) are vulnerable to complications that can result in mortality. Influenza virus is an enveloped, single stranded RNA virus with a segmented genome. The three genera known as A, B and C are grouped by differences in their core proteins; influenza A and B are the most common and are strongly associated with epidemics. Historically, the only therapies for influenza A infection were amantidine and rimantidine that inhibit viral entry into cells by targeting the M2 protein of influenza A virus. More recently treatment options for influenza A and B have expanded to include the neuraminidase inhibitors, zanamivir (RELENZA™) and oseltamivir (TAMIFLU™). Neuraminidase promotes influenza virus release from infected cells and facilitates virus spread within the respiratory tract. These inhibitors block replication of both influenza A and B viruses, cause fewer side effects and have less potential to select resistant variants than the M2 inhibitors. Early treatment with either drug reduces the severity and duration of influenza symptoms and associated complications (Hayden et al., 1996, Hayden et al., 1997, Treanor et al., 2000, Makela et al., 2000, Monto et al., 2000, Kashiwagi et al., 2000).
The antiviral efficacy of zanamivir was monitored by analysis of the rate of decline in upper airway viral load determined during a time course of treatment. The viral titres present in patient samples were historically determined by a culture-enhanced enzyme-linked immunosorbent assay (CE-ELISA), or plaque assay as described by Barnett et al. (2000). Since these methods rely on the ability of clinical isolates to replicate in cell culture, it may not be possible to quantify viruses that replicate inefficiently, or fail to produce cytopathic effects in cell culture. A fluorogenic real-time PCR-based technique that detects and quantifies influenza A and B virus RNA has been developed to serve as an alternative assay that is not dependent upon the replicative efficiency of the virus in cell culture, the capacity of the virus to form plaques, or the sensitivity and specificity of monoclonal antibodies. Real time qPCR is a modified form of PCR that measures an increase in PCR product over time. Reactions that exploit the 5′–3′ nuclease activity of Taq polymerase to cleave a sequence specific fluorescent labelled probe are sometimes known as TaqMan® PCR (Holland et al., 1991, Lee et al., 1993, Livak et al., 1995).
The purpose of the study was to characterise the real time qPCR assays and to cross-validate them with CE-ELISA methods, using laboratory strains of virus and two panels of clinical isolates. The CE-ELISA detects nucleoprotein from infectious virus particles and is expressed as TCID50/ml, whereas real time qPCR assays detect the total number of matrix protein gene viral nucleic acids (infectious and non-infectious). Given the differences between the two methods, the relationship between viral load determined by the CE-ELISA method and the real time qPCR assay and the relative sensitivity of both assays were determined.
2. Materials and methods
2.1. Virology
2.1.1. Laboratory strains
Laboratory grown stocks of A/Texas/1/77 (H1N1) or B/Victoria/102/85 with titres of 8.1×107 and 5.3×107 pfu/ml, respectively, were diluted 103, 104 and 106 fold in 10–100 fold steps in pooled virus transport medium to give validation control samples at nominal concentrations of Influenza A and Influenza B at high, medium and low titre. The approximate concentrations of these validation control samples were 8.1×104, 8.1×102 and 8 pfu/ml for influenza A and 5.3×104, 5.3×102 and 5 pfu/ml, respectively. Virus transport medium was obtained from Virocult® swabs (Medical Wire and Equipment Co., Corsham, Wiltshire, England). Serial 10 fold dilutions of A/Shangdong/3/93 (H3N2), A/Taiwan/1/86 (H1N1) and B/Lisbon/3/96 were prepared in phosphate-buffered saline (PBS).
2.1.2. Clinical specimens
Panel A consisted of 233 throat swab samples taken within 1, 3 and/or 6 days of the symptoms of influenza onset from 91 patients. Influenza was originally diagnosed in all of these patients from the testing of a nasal swab taken from each patient on Day 1. A positive result from diagnostic multiplex PCR as described by Stockton et al. (1998) and/or virus culture was obtained for each Day 1 sample.
Panel B consisted of 63 throat swabs from patients with symptoms of upper airway infection, only 40 of whom were diagnosed as positive for influenza. Throat swabs were taken 1, 3 and 6 days after the onset of influenza-like symptoms. Informed consent was obtained for the collection of all samples after the nature and possible consequence of the studies was fully explained.
2.2. Virus detection by cell culture and culture enhanced ELISA
Virus detection by cell culture and CE-ELISA was performed as described by Barnett et al. (2000).
2.3. Real time qPCR
2.3.1. RNA isolation and cDNA preparation
Viral RNA was isolated from 280 μl of sample using a QIAamp® Viral RNA Kit and the cDNA synthesis was carried using an Omniscript™ kit according to the manufacturer's instructions (Qiagen, Hilden, Germany). Reactions were primed with a mixture of 1 μM random hexamers and 1 μM each of primers specific to a highly conserved region of the matrix protein gene (5′TCT AAC CGA GGT CGA AAC GTA 3′ influenza A, 5′TCA TGG CCT TCT GCT ATT TC 3′ influenza B), which were incubated at 42 °C for 60 min, heated to 95 °C for 5 min, then cooled to 4 °C in a 9600 thermal cycler (Applied Biosystems, Foster City, CA, USA).
2.3.2. Assay design
Twenty temporally and spatially divergent influenza A (ten H1N1 and ten H3N2) and 20 influenza B matrix protein gene sequences were retrieved from public databases. Since there was insufficient homology between the matrix protein gene sequences of the two genera, each was aligned separately using megalign v4.05, within the lasergene software package (DNAStar). Regions of homology were identified and primer/probe sets along with primers for reverse transcription were designed in these regions; primer express ® Software v1.0 was used to verify the selected primer and probe sequences (Applied Biosystems). The primers (senseA) 5′AAG ACC AAT CCT GTC ACC TCT GA 3′ and (antisenseA) 5′CAA AGC GTC TAC GCT GCA GTC C 3′ amplify a 104-base pair fragment in the M1 gene of influenza A. The influenza A specific probe FAM (6-carboxyfluorescein)-5′ TTT GTG TTC ACG CTC ACC GT 3′-TAMRA (6-carboxytetramethylrhodamine) annealed to part of the sequence amplified by the two primers. The primers (senseB) 5′GAG ACA CAA TTG CCT ACC TGC TT 3′ and (antisenseB) 5′TTC TTT CCC ACC GAA CCA AC 3′ amplify a 92-base pair fragment in the M gene of influenza B. The probe specific for influenza B VIC-5′AGA AGA TGG AGA AGG CAA AGC AGA ACT AGC 3′-TAMRA similarly annealed to part of the sequence amplified by the two primers. Probes and primers were obtained from Applied Biosystems.
2.3.3. Real-time qPCR protocol
The PCR consisted of a final concentration of 1× Universal Master Mix (Applied Biosystems), 900 nM each primer; 225 nM of the Influenza A probe and 100 nM of the influenza B probe), plus 2 μl of target cDNA and was made up to a volume of 25 μl with nuclease free water (Promega Corp. Madison, USA). After UNG treatment at 50 °C for 2 min and UNG inactivation/Amplitaq Gold activation at 95 °C for 10 min, the cDNA was amplified by 40 two step cycles (15 s at 95 °C for denaturation of the DNA, 1 min at 60 °C for primer annealing and extension). The qPCR reactions were carried out in a 96 well microtitre plate. The real time quantitative PCR amplifications were measured in real time mode using the ABI7700 (Applied Biosystems). Data was gathered, analysed and viral load calculated using sequence detection systems (v1.6.3), and microsoft excel 97 (Microsoft Corp. Redmond, Washington) was used to export and manipulate viral load data. The copy number of viral cDNA in copies/ml virus transport medium was determined for influenza A and B by comparison with a serially diluted plasmid standard of known concentration included on each 96 well plate. At least four calibration standards containing a known copy number of virus were included on each plate to indicate any changes in the efficiency of the viral RNA extraction and RT reaction.
Separate plasmids containing Influenza A and B M1 derived inserts that included the real time qPCR assay amplicons were constructed by ligation of a PCR amplified matrix gene fragment in pCRII according to the instructions of the T/A Cloning® Kit Dual Promotor (Invitrogen, Groningen, The Netherlands). The cloned influenza A fragment comprised the entire M1 protein gene and was obtained by RT-PCR from stocks of A/Texas/1/77 (H1N1). The cloned influenza B fragment was a 371 bp region of the influenza B isolate B/Victoria/102/85, amplified by primers 5′ AGG AAC GCT CTG TGC TTT GTG 3′ and 5′ TCT TTG GCT TGG ATT TCT 3′. The plasmid DNA was amplified in E. coli strain TOP10 according to the manufacturer's protocol and purified using a Qiagen Plasmid Midi kit (Qiagen). Plasmid insert DNA sequences were verified by sequencing in both directions using dye-labelled dideoxy-terminator cycle sequencing. Sequences were analysed using an ABI Model 377 (Applied Biosystems and data were assembled with seqman v4.05 (DNAStar), manually proof read and aligned with representative published sequences (GenBank Accession no. U52940 for A/Texas/1/77 (H1N1) and AF100376 for B/Victoria/102/85). The concentration and purity of the plasmid DNA was calculated by measuring the OD260/280 of a 1:100 and 1:1000 dilution in TE buffer, pH 8.0. Plasmid DNA was then serially diluted tenfold in TE buffer, pH 8.0, from 5×105 to 5 plasmid copies/μl for use in real time PCR.
2.3.4. Assay specificity
The degree of homology between publicly available sequences and the primer and probe sequences were compared using Basic Local Alignment Search Tool (BLASTn; Basic BLAST n (Altschul et al., 1999)). The plasmid vector pCRII containing parts of the genomes of other respiratory viruses (respiratory syncytial virus, parainfluenza I and III, human rhinovirus 16, coronavirus 229E and OC43, and Adenovirus 5) were cloned according to manufacturers instructions (Invitrogen). Plasmids were purified using a Plasmid Mini Kit (Qiagen) and diluted to 1×107 copies/μl. cDNA generated from stocks of the same viruses and human RNA using random hexamers were prepared using an Omniscript™ kit (Qiagen). SYBR® Green assays were performed by replacing 2× Universal Master Mix with 2× SYBR® Green PCR Master Mix containing SYBR® Green dye (Applied Biosystems). The PCR mix was consisted of a final concentration of 1× SYBR® Green Master Mix (Applied Biosystems), primers as described above optimised to 50 nM, 2 μl of target cDNA and the PCR reaction volume was made up 25 μl with nuclease free water (Promega Corp.). Cycling conditions were identical to those described above.
3. Results
3.1. Overall agreement of real time PCR assays with culture enhanced-ELISA and virus culture using laboratory grown strains of virus
Serial 10 fold dilutions of A/Shangdong/3/93 (H3N2), A/Taiwan/1/86 (H1N1) and B/Lisbon/3/96 were used to establish the relationship between influenza viral load determined by the CE-ELISA method and the real time PCR assays. (i.e. between TCID50/ml and viral cDNA copies) and the relative sensitivity of both assays (Fig. 1 ). This confirmed that real time qPCR was consistently more sensitive and showed a broader dynamic range. Three log10 copies/ml corresponded to 1 TCID50/ml or 1 pfu/ml as defined in the assay system.
Fig. 1.
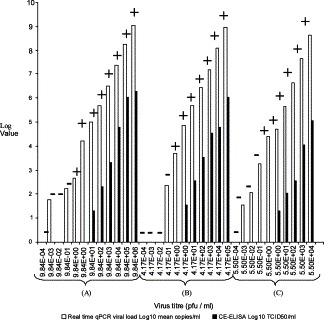
Comparison of real time qPCR, CE-ELISA and virus culture over a range of 10-fold serial dilutions of (A) A/Shangdong/3/93 (H3N2), (B) A/Taiwan/1/86 (H1N1) and (C) B/Lisbon/3/96. The virus titre in pfu/ml for the input virus dilutions is estimated from the stock concentration. log values (Y-axis) are given as mean vRNA copies/ml for real time qPCR and TCID50/ml for CE-ELISA. +/− Refers to the presence/absence of haemagglutination activity in virus culture.
Since stocks of viral isolates are required for subsequent drug-susceptibility analyses, we determined the minimum number of influenza virus genome copies in a sample (as determined by real time qPCR) required to generate a stock of infectious virus. There was little difference between the different viral strains tested in terms of the minimum viral loads, expressed as log10 viral RNA copies/ml, that were detectable in the CE-ELISA assay. The viral loads measured were 5.01 log10 copies/ml (1.3 log10 TCID50/ml) for A/Shangdong (H3N2), 4.89 log10 copies/ml (1.55 log10 TCID50/ml) for A/Taiwan (H1N1 subtype) and 4.69 vRNA log10 copies/ml (1.3 log10 TCID50/ml) for B/Lisbon. However, there was a difference in the minimum number of copies of influenza virus genomes required to generate a stock of infectious virus. The minimum genome copy number of A/Shandong/3/93 (H3N2), A/Taiwan/1/86 (H1N1) and B/Lisbon/3/96 viruses required to generate virus stocks when 150 μl of virus dilution was used were 71, 735 and 3768 genome copies, respectively.
3.2. Overall agreement and sensitivity of real time PCR assays and culture enhanced ELISA using clinical specimens
The sensitivity and agreement between real time PCR and CE-ELISA was assessed by blinded testing of throat swab samples from panel A, which included only patients with confirmed influenza. Table 1 depicts contingency tables of qualitative and quantitative results measured by real time PCR in comparison with CE-ELISA. There were 50 positive samples detected by both CE-ELISA and real time PCR and 87 negative samples by both methods. Real time PCR detected 90 additional positives and failed to detect six positives that were previously detected by CE-ELISA. Fisher's Exact Test was applied to the contingency table to check whether the proportions of positives and negatives are the same for each method. The CE-ELISA results differed significantly to real time qPCR (P<0.0001). Those samples that were positive by both CE-ELISA and real time PCR had a mean viral load of 6.17 log10 copies/ml, as compared with 4.97 log10 copies/ml for those positive by real time PCR alone (not significant). Those that were positive by both real time PCR and CE-ELISA had a mean TCID50/ml of 2.24, as compared with 1.88 TCID50/ml for those positive by CE-ELISA alone (not significant). There were 22 additional samples that were positive for influenza by real time qPCR only but were below the limit of quantification so could not be included in the analysis, as compared with no additional positives below the limit of quantification in the group positive by both tests. In common with the data generated with laboratory strains of virus, this suggested that the real time PCR assays were more sensitive than the CE-ELISA method.
Table 1.
The qualitative and quantitative results obtained by testing throat swabs with real time qPCR were compared with the results generated previously by CE-ELISA
| Real time qPCR |
|||||
| + |
− |
||||
| Influenza A | Influenza B | Total | Total | ||
| CE-ELISA n=233 | |||||
| +n=56 | n | 31 | 19 | 50 | 6 |
| Viral load (qPCR) | 6.25 | 6.04 | 6.17 | ||
| TCID50 (CE-ELISA) | 2.33 | 2.07 | 2.24 | 1.88 | |
| −n=177 | n | 50+11 BLQ | 18+11 BLQ | 68+22 BLQ | 87 |
| Viral load (qPCR) | 5.02 | 4.76 | 4.97 | ||
The mean viral load generated by real time qPCR is expressed as log10 vRNA copies/ml of virus transport medium. BLQ, below limit of quantification for real time qPCR. CE-ELISA results are given as mean TCID50/ml.
Fig. 2 depicts a comparison of influenza quantification from throat swabs by real time qPCR and CE-ELISA using a subset of 40 patients from panel B that were diagnosed influenza A (n=26) or influenza B (n=14) positive. Drops in viral load over time were demonstrated using both methods, but the decreases in viral load were generally less rapid when measured by real time qPCR. The ability of real time qPCR assays to differentiate influenza A and B infections was investigated by comparing the influenza virus type obtained with those suggested by diagnostic multiplex PCR, virus culture and CE-ELISA. There was 100% concordance of influenza virus type between the three assays.
Fig. 2.
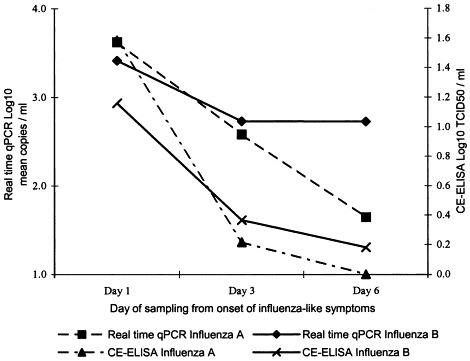
Comparison of influenza quantification by real time qPCR and CE-ELISA. A subset of 40 patients that were diagnosed influenza A (n=26) or influenza B (n=14) positive and had a complete set of throat swab samples taken on days 1, 3 and 6 were included.
3.3. Real time qPCR assay sensitivity, specificity, dynamic range and variability
BLASTn searching of the primer and probe sequences suggested that the primer and probe sequences were genus specific. The oligonucleotides designed to anneal with influenza A were 100% homologous in sequence to a broad range of virus subtypes including H1N1 and H3N2 isolates reported over several decades. Similarly, the oligonucleotides designed to anneal with influenza B were also 100% homologous in sequence to a broad range of influenza B isolates reported over a number of years.
The specificity of each real time qPCR assay was determined experimentally by spiking influenza A and B real time PCR reactions individually with 1×107 copies of pCRII plasmid (Invitrogen) containing parts of the genomes of other respiratory viruses. In addition, stocks of cDNA generated from stocks of the same viruses and human RNA using random hexamers were added to individual PCR reactions. The specificity of the method was also assessed by the inclusion of blank samples in each assay to check for non-specific reactions between the components. The specificity of the real time PCR assays were confirmed by both the negative readings given by the ABI7700 for blank samples and inappropriate targets that included human and viral cDNA and plasmids containing sequences from other respiratory viruses, and the positive readings generated for five laboratory grown strains of influenza. Furthermore, the assays differentiated influenza A and B with no cross-reactivity, allowing multiplexing of the two assays.
The specificity of the primers was tested by comparing the performance of the probe-based assay with a non-specific real time reporter system. This involved substituting the influenza A virus specific labelled hydrolysis probe for SYBR® Green dye that fluoresces when bound non specifically to double stranded DNA, and repeating the assays using the same PCR primers in real time mode. Throat swabs from panel B were tested using both probe based and SYBR® Green assays. Twenty-four samples were influenza A positive and 33 samples were negative by both systems. Four influenza positive samples previously detected by the probe-based assay were not detected by the SYBR Green assay and two positives detected by the SYBR® Green assay were not detected by the previous probe assay. Fig. 3 depicts a comparison of viral load measurements obtained using the probe-based assay with those obtained using a SYBR® Green assay. Although a relationship between the two assays was present, the SYBR® Green assay consistently reported higher copy numbers and there was less agreement between the two assays at low copy numbers.
Fig. 3.
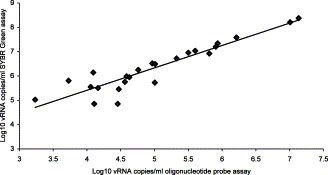
Comparison of influenza A vRNA copies/ml in influenza A positive throat swabs taken from subjects with evidence of an upper airway infection, measured by oligonucleotide probe and SYBR Green based real time qPCR assays.
Dilutions of validation control virus stocks were used to determine the variability, dynamic ranges and limits of quantification of the real time qPCR assays. The real time qPCR assay variability expressed as mean coefficient of variation was determined by assessing the agreement between the measured and nominal concentrations of the serially diluted laboratory strains of influenza A and B viruses designated as validation control samples that were analysed in replicates of three, on six separate occasions (Fig. 4 ). The precision of the method was determined by assessing the agreement between replicate measurements of validation control samples by transforming the raw data to their common logarithms and performing analysis of variance (ANOVA). The lower and upper limits of quantification were defined by the validation control sample concentrations possessing acceptable precision, which was the lowest amount of influenza in vRNA copies/ml present in a sample for which the coefficient of variation does not exceed 100%. The lower limit of quantitation was 3.33 log10 copies/ml (ten copies/PCR reaction). To obtain the linear range of the assays, the threshold cycle of each laboratory strain dilution was plotted against the corresponding initial template concentration. The coefficient of regression of the graph was consistently greater than 0.997 for all the experiments carried out. The results indicate that there is a good correlation, in the range of 10–107 copies/PCR reaction (3.33–8.33 log10 copies/ml), and that the linear range of the assay is at least seven orders of magnitude. It was not possible to verify the upper end of the dynamic range any further since samples with such a high viral load are rare. When the real time qPCR assays were used to measure the influenza viral load in clinical samples, more than 99% of samples containing influenza RNA were quantifiable without the need for further sample dilution or concentration.
Fig. 4.
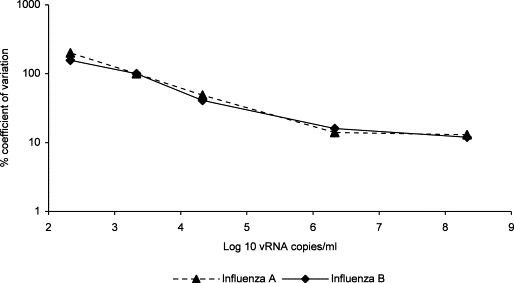
Percentage coefficient of variation over the dynamic range of the real time qPCR assays. * Denotes the limit of detection and ** the limit of quantification.
The inter-assay coefficient of variation was in the range of 10–25% for most of the dynamic range, but increased to 49% at viral loads of 4.33 log10 vRNA copies/ml. Inter-assay coefficient of variation expresses the variation within different experiments, e.g. carried out on different days; using different equipment and different batches of reagents. The intra-assay coefficient of variation ranged from approximately 12 to 50% over the measured range of concentrations. Overall, the method has a% CV of the order of 30–40%, but the individual %CV was greater with low concentrations of virus.
The variability introduced by using different batches of reagents, or ABI7700 instruments used to cycle and monitor real time qPCR reactions, or thermal cyclers used to make cDNA (for example, GeneAmp® 9600 and 9700; Applied Biosystems) was also investigated using the validation control samples. There was no difference in viral load results generated as a result of using different ABI7700 instruments to perform and monitor the real time qPCR, different thermal cyclers to prepare cDNA (GeneAmp® 9600 or 9700), or between different batches of any of the reagents.
3.4. Stability of virus and real time qPCR assay processing intermediates
Assay performance can also be affected by the quality of the sample and processing intermediates. To determine the length of time for which the quantity of virus in a sample stored in virus transport medium might still be meaningfully interpreted, the stability of the validation control stock viruses diluted 1:10 in transport medium (in replicates of six) stored at −80 °C, was assessed by monitoring the virus copy number measured by real time qPCR over a 6 month period. The viral RNA within influenza A and influenza B particles stored at −80 °C in virus transport medium did not deteriorate significantly as measured by no significant deviation from the copy number measured initially, over almost a 2 month period. The viral RNA associated with virus particles at the concentrations tested was stable in virus transport medium stored at −80 °C for at least 59 days. Further unpublished observation indicates that virus stored in this way may be stable for much longer periods than this. However, the effects on the viability of the virus are not known.
The effect of long term storage on the quality of cDNA generated from clinical samples was determined by real time qPCR testing of cDNA that was prepared from throat swab samples from Panel B. The results obtained using freshly prepared cDNA were compared with results obtained from the same cDNA after storage at −20 °C for 16 months. There was no significant difference between the results obtained using freshly made influenza B cDNA and those that were tested after storage at −20 °C for 16 months. The 11 samples that tested positive for influenza B re-tested positive and the 39 samples that were originally negative remained negative. Of the influenza A cDNA tested, of the 24 that were originally positive, 22 remained quantifiable and the 26 that were originally negative remained negative. Two samples were below the limit of quantitation. Quantitative differences were within the %CV limits described above for the method.
The effect of freezing and thawing influenza virus in virus transport medium, viral RNA, or cDNA was determined by using the real time qPCR assays to measure the copy number of virus in the validation control samples at medium concentration. The validation control samples were tested in replicates of three, over four freeze–thaw cycles from −80 °C to ambient temperature. Influenza A and influenza B concentrations after five freeze–thaw cycles were not significantly different to the concentrations measured prior to freezing the samples. Variation in the viral load was within the %CV limits described for the real time qPCR assays that were used to measure the viral load. Influenza A and B viral RNA was found to give a trend of decreased viral load as the number of freeze–thaw cycles which became a significant decline in viral load from that measured initially after four freeze–thaw cycles (P<0.001). Viral cDNA returned similar viral load values after four freeze–thaw cycles to those of freshly prepared cDNA.
To establish the shelf life of working stocks of plasmid used as an assay standard, stability at 4 °C was assessed by comparing the threshold cycle (Ct) values generated by real time qPCR assay, of duplicate 10 fold dilutions spanning 1×107–10 copies/PCR reaction, against those of freshly prepared solutions at 1 and 6 month intervals. The threshold cycle or Ct value is a value generated in real time PCR that is the PCR cycle at which a statistically significantly increase in normalised fluorescence intensity is first detected. Similarly, the stability of high concentration (>1×1010 plasmid copies/μl) stocks stored at −80 °C was assessed over a 24 month period by monitoring the Ct values obtained from diluting to 5×105 copies/μl for testing by real time qPCR assay. Analytical solutions of plasmid containing inserts of influenza A and influenza B genome used as a standard were relatively stable in well sealed tubes when stored at 4 °C for at least 59 days.
The results obtained from real time PCR reactions performed using the validation control samples in triplicate, prepared 24 h in advance of PCR and stored at 4 °C were compared with freshly prepared reactions to determine if it was possible to prepare and store PCR reactions in advance. The results obtained from real time qPCR of the validation control samples when the reactions were prepared 24 h in advance of PCR and stored at 4 °C were not significantly different to freshly assembled reactions, suggesting that it is possible to set up these reactions in advance of performing the qPCR.
4. Discussion
A sensitive quantitative assay as described is critical for the high throughput monitoring of patient samples, since one way to monitor the antiviral efficacy of neuraminidase inhibitors is to analyse the rate of viral load decline in the upper airway during a time course of treatment. The real time quantitative PCR assays described have been applied to determine the antiviral activity of inhaled zanamivir for the treatment of naturally acquired influenza in military recruits living in residential units. A significant increase in the rate of viral load reduction was shown once zanamivir therapy had commenced as compared with placebo (Puhakka et al., 2003). Assays used historically were semi-quantitative and dependent upon the growth of infectious virus, and for plaque assays, upon the capacity of the virus to form plaques. The variation in the minimum number of influenza virus copies of different virus subtypes required to generate a stock of infectious virus may be due to differences in adaptation to cell culture or variation in the levels of non-infectious virus particles in these laboratory cultured virus strains. The success of assays such as the CE-ELISA is also dependent on the availability of monoclonal antibodies of high sensitivity and specificity to detect viral nucleoprotein. There is evidence to suggest that the results of diagnostic tests dependent upon the detection of nucleoprotein by an enzyme conjugated monoclonal antibody, such as Directigen™ FLU-A test (Beckton Dickinson, Cockeysville, Maryland, US), are affected by freezing and thawing of specimens prior to testing (Waner et al., 1991). The nucleic acid based real time PCR assays were not affected by up to five sample freeze–thaw events, suggesting that even though influenza is an RNA virus, methods based upon the amplification of short segments of viral RNA are robust. Decreases in viral load were generally less rapid when measured by real time qPCR than when measured by CE-ELISA. This was probably due to the slower decay rate of the viral nucleic acid measured by PCR relative to infectious virus.
Employing real time PCR technology for the purpose of screening patients participating in clinical trials of neuraminidase inhibitors has several further advantages. The inclusion of an external standard curve and the reliable quantification of influenza cDNA in assays which are linear over seven orders of magnitude eliminates the need for sample dilution, which minimises sample handling and associated increases in variability. Limited data (not shown) suggests that for samples with higher viral loads an accurate quantitative result can be obtained by extrapolation of the standard curve but the reporting of low viral load quantities should be interpreted with caution, particularly small magnitude viral load changes at low viral loads. The chance of experiencing cross-contamination and hence false positive results is low as the need for post PCR sample handling is removed, since the detection of the PCR products occurs online during real-time PCR amplification. Real time qPCR reactions can be set up in advance of cycling, allowing processing in batches. Additionally, appropriately archived samples or cDNA primed with random hexamers or appropriate specific primers can be re-tested using these real time qPCR assays as they are not dependent on the infectivity of the virus.
Since no international standard is available for quantitation, results were expressed as viral RNA copies/ml of swab transport medium. The lowest amount of influenza virus cDNA that can be quantified by this assay is 3.33 log10 copies/ml. The assays were set up with speed and throughput as the priority, but it is likely that a more sensitive limit of quantification could be achieved by increasing the quantity of material extracted. At concentrations below the limit of quantification the coefficient of variation between repeated tests becomes unacceptably large and the actual quantity difficult to interpret, as is the case for all PCR based assays. Probability rather than sample quality variation is the predominant cause of variability at low copy numbers (De Vries et al., 1999). The theoretical uncertainty of measurements made by quantitative PCR has been modelled. The theoretical variability of viral loads of below ten copies/PCR reaction (3.33 log10 copies/ml) was calculated to be greater than 30%, even assuming optimal PCR efficiency and excluding other variables (Peccoud and Jacob, 1996). The results for intra- and inter-assay precision indicate that the assay is reproducible, even between different batches of reagents or different thermal cycler instruments used.
Additional factors that can affect real time qPCR assay reproducibility are the susceptibility of virus and processing intermediates of the assay such as RNA and cDNA to degradation over time in storage and by repeated freezing and thawing. Influenza virus stored at −80 °C and cDNA stocks made from virus kept at −20 °C were stable for long periods and over several freeze–thaw cycles, suggesting that it is possible to apply real time qPCR assays to quantify influenza in appropriately stored archive samples and appropriately primed cDNA stocks. However, samples with viral loads below the limit of quantitation may be vulnerable to degradation. Our data suggests that at the viral loads tested it is feasible to thaw and re-test throat swab samples stored in virus transport medium up to five times. Samples intended to be used for real time qPCR should be stored as collected in virus transport medium, or processed to make cDNA for long-term storage and future analysis, if it is likely that the samples will be revisited for further molecular studies, but not stored as naked RNA. Although the viral RNA at the concentrations tested is relatively stable stored at −80 °C, it is affected by freezing and thawing events.
When three influenza strains were tested in the real time PCR assays and compared with CE-ELISA and virus culture, the results were in good agreement and it is concluded that the assay is comparable with the results obtained in other laboratories. However, the real time PCR assays were 93 fold more sensitive than the virus infectivity assay (CE-ELISA), so potentially more useful where maximal sensitivity was required for the analysis of influenza viral load as is required for clinical trials. Similarly, the real time qPCR assays appeared to be more sensitive when applied to clinical samples. For clinical trials of zanamivir there was a requirement to quantify virus in the throat, which is the main site of action of the drug, as well as the nose where titres may be higher. Therefore, use of the real time PCR assays allows quantitation of viral load at lower concentrations of virus, which may be negative by the CE-ELISA method. However, a further distinction between the two assays is that the CE-ELISA detects only infectious virus, whereas PCR assays including these real time qPCR assays detect both infectious and non-infectious virus. Therefore, detection and quantitation of viral nucleic acid by the real time PCRs may reflect the presence of some virus, which is unable to produce an infection. The production of non-infectious virus particles, known as the Von Magnus effect, is particularly common in laboratory strains of virus. Conversely, it is possible that virus at a concentration below the limit of detection of the real time qPCR would be able to infect cells and produce a positive result in a culture based system.
Given the high degree of sequence conservation in the sites of binding for the primers and probes, it is likely that these assays could be applied to detect a wide variety of different human and animal strains of influenza. Three different haemaglutinin and two neuraminidase subtypes account for virtually all human infections (Hayden and Palese, 1997). All of the M1 sequences from these subtypes found in humans including the H5N1 strain recovered from an outbreak in Hong Kong (Claas et al., 1998), aligned with the probe and primer sequences for influenza A. Unlike the assay described here, other published assays (van Elden et al., 2001, Schweiger et al., 2000) are unlikely to quantify accurately or sensitively detect H5N1 isolates due to misalignment of at least one and up to four nucleotide changes in the probe sequence. This highlights the need for regular reviews of any primer or probe sequences used diagnostically or for quantification of viruses that are subject to genetic drift. The robustness of the assay primers was confirmed by the similar qualitative results obtained by SYBR Green assays, although the SYBR Green assay tended to report slightly higher viral load values.
Using the real time qPCR assays described above in a routine setting for throat swab screening confirms that it allows, rapid throughput of a high number of samples generating reliable quantitative results and with greater sensitivity than has been achieved by previous commonly used influenza assays. Large-scale screening and identification of influenza virus using real time qPCR has been carried out as part of the development of zanamivir. This new approach should also help to further knowledge of the dynamics of influenza virus infection and the effect of novel therapies on influenza viral load.
References
- Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1999;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett J.M., Cadman A., Gor D., Dempsey M., Walters M., Candlin A., Tisdale M., Morley P.J., Owens I.J., Fenton R.J., Lewis A.P., Claas E.C.J., Rimmelzwaan G.F., De Groot R., Osterhaus A.D.M.E. Zanamivir susceptibility monitoring and characterisation of influenza virus clinical isolates obtained during phase II clinical efficacy studies. Antimicrob. Agents Chemother. 2000;44:78–87. doi: 10.1128/aac.44.1.78-87.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claas E.C., Osterhaus A.D., van Beek R., De Jong J.C., Rimmelzwaan G.F., Senne D.A., Krauss S., Shortridge K.F., Webster R.G. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet. 1998;351:472–477. doi: 10.1016/S0140-6736(97)11212-0. [DOI] [PubMed] [Google Scholar]
- De Vries T.J., Fourkour A., Punt C.J.A., van de Locht L.T.F., Wobbes T., van den Bosch S., de Rooij M.J.M., Mensink E.J.B.M., Ruiter D.J., van Muijen G.N.P. Reproducibility of detection of tyrosine and MART-1 transcripts in the peripheral blood of melanoma patients: a quality control study using real-time quantitative RT-PCR. Br. J. Cancer. 1999;80:883–891. doi: 10.1038/sj.bjc.6690436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden F.G., Palese P. Influenza virus. In: Rickmann D.D., Whittey R.J., Hayden F.G., editors. Clinical Virology. Churchill Livingstone; New York: 1997. [Google Scholar]
- Hayden F.G., Treanor J.J., Betts R.F., Lobo M., Esinhart J.D., Hussey E.K. Safety and efficacy of the neuraminidase inhibitor GG167 in experimental human influenza. J. Am. Med. Assoc. 1996;275:295–299. [PubMed] [Google Scholar]
- Hayden F.G., Osterhaus A.D., Treanor J.J., Fleming D.M., Aoki F.Y., Nicholson K.G., Bohnen A.M., Hirst H.M., Keene O., Wightman K. Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatment of influenza virus infections. New Engl. J. Med. 1997;337:874–880. doi: 10.1056/NEJM199709253371302. [DOI] [PubMed] [Google Scholar]
- Holland P.M., Abramson R.D., Watson R., Gelfand D.H. Detection of specific polymerase chain reaction product by utilizing the 5′ to 3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA. 1991;88:7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi S., Kudoh S., Watanabe A., Yoshimura I. Clinical efficacy and safety of the selective oral neuraminidase inhibitor oseltamivir in treating acute influenza—placebo-controlled double-blind multicenter phase III trial. Kansenshogaku Zasshi—J. Jpn. Assoc. Infect. Dis. 2000;74:1044–1061. doi: 10.11150/kansenshogakuzasshi1970.74.1044. [DOI] [PubMed] [Google Scholar]
- Lee L.G., Connell C.R., Bloch W. Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucleic Acids Res. 1993;21:3761–3766. doi: 10.1093/nar/21.16.3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Flood S.J., Marmaro J., Giusti W., Deetz K. Oligonucleotides with fluorescent dyes at the opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridisation. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- Makela M.J., Pauksens K., Rostila T., Fleming D.M., Man C.Y., Keene O.N., Webster A. Clinical efficacy and safety of the orally inhaled neuraminidase inhibitor zanamivir in the treatment of influenza: a randomised, double-blind, placebo-controlled European study. J. Infect. 2000;40:42–48. doi: 10.1053/jinf.1999.0602. [DOI] [PubMed] [Google Scholar]
- Monto A.S., Moult A.B., Sharp S.J. Effect of zanamivir on duration and resolution of influenza symptoms. Clin. Ther. 2000;22:1294–1305. doi: 10.1016/s0149-2918(00)83026-x. [DOI] [PubMed] [Google Scholar]
- Peccoud J., Jacob C. Theoretical uncertainty of measurements using quantitative polymerase chain reaction. Biophys. J. 1996;71:101–108. doi: 10.1016/S0006-3495(96)79205-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhakka T., Lehti H., Vainionpaa R., Jormanainen V., Pulkkinen M., Sharp S., Kerr C., Dempsey M., Ring C.J., Ward C., Tisdale M. Zanamivir: a significant reduction in viral load during treatment in military conscripts with influenza. Scand. J. Infect. Dis. 2003;35:52–58. doi: 10.1080/0036554021000026981. [DOI] [PubMed] [Google Scholar]
- Schweiger B., Zadow L., Heckler R., Timm H., Pauli G. Application of a fluorogenic PCR assay for typing and subtyping of influenza viruses in respiratory samples. J. Clin. Microbiol. 2000;38:1552–1558. doi: 10.1128/jcm.38.4.1552-1558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockton J., Ellis J.S., Saville M., Clewley J.P., Zambon M.C. Multiplex PCR for typing and subtyping influenza and respiratory syncytial viruses. J. Clin. Microbiol. 1998;36:2990–2995. doi: 10.1128/jcm.36.10.2990-2995.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treanor J.J., Hayden F.G., Vrooman P.S., Barbarash R., Bettis R., Riff D., Singh S., Kinnersley N., Ward P., Mills R.G. Efficacy and safety of the oral neuraminidase inhibitor oseltamivir in treating acute influenza: a randomised controlled trial. US Oral Neuraminidase Study Group. J. Am. Med. Assoc. 2000;283:1016–1024. doi: 10.1001/jama.283.8.1016. [DOI] [PubMed] [Google Scholar]
- van Elden L.J., Nijhuis M., Schipper P., Schuurman R., van Loon A.M. Simultaneous detection of influenza viruses A and B using real-time quantitative PCR. J. Clin. Microbiol. 2001;39:196–200. doi: 10.1128/JCM.39.1.196-200.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waner J.L., Todd S.J., Shalaby H., Murphy P., Wall L.V. Comparison of directigen FLU-A with viral isolation and direct immunofluorescence for the rapid detection and identification of influenza virus. J. Clin. Microbiol. 1991;29:479–482. doi: 10.1128/jcm.29.3.479-482.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]