

The severe acute respiratory syndrome epidemic brought into the spotlight the need for rapid development of effective antiviral drugs against newly emerging viruses.
Keywords: HIV, Entry inhibitors, RNA polymerase, Membrane fusion
Abstract
The severe acute respiratory syndrome (SARS) epidemic brought into the spotlight the need for rapid development of effective anti-viral drugs against newly emerging viruses. Researchers have leveraged the 20-year battle against AIDS into a variety of possible treatments for SARS. Most prominently, based solely on viral genome information, silencers of viral genes, viral-enzyme blockers and viral-entry inhibitors were suggested as potential therapeutic agents for SARS. In particular, inhibitors of viral entry, comprising therapeutic peptides, were based on the recently launched anti-HIV drug enfuvirtide. This could represent one of the most direct routes from genome sequencing to the discovery of antiviral drugs.
Severe acute respiratory syndrome (SARS) is a pulmonary infection that has been identified in multiple outbreaks around the world, emerging initially in Guangdong Province, China, in November 2002. The number of reported cases increased exponentially and reached 8422, which resulted in 916 deaths by August 2003 (WHO website). The syndrome is caused by a previously unknown virus - SARS-associated coronavirus (SARS-CoV) [1, 2, 3]. The global SARS outbreak has been contained, mainly owing to strict patient isolation and aggressive containment of infected regions, but the virus itself has potential to reappear. This concern is supported by studies reporting a cyclic pattern for other human-infective coronaviruses that attack mainly in the winter, sometimes skipping years, for example, the related human coronavirus, HCoV-OC43, which breaks out every two to four years [4].
To date, there are ∼36 antiviral drugs, half of which were developed in the past 15 years to treat a single virus, HIV-1. Development of these antiviral drugs gained from the advances in molecular and structural biology coupled with advances in medicinal chemistry and in the industrialization of the drug discovery process. The SARS epidemic has emphasized the need to develop drugs against emerging viral infections quickly, and demonstrates the usage of genomic technologies in antiviral research. In the past two decades, biology has become an information-driven science as a result of the emergence of genomic technologies and the expansion of the Internet that allows analysis of genomic databases at every researcher's desktop. These advanced genomic technologies led to rapid sequencing of SARS-CoV [5, 6] and, for the first time in history, the sequencing of a genome of an infective agent preceded the understanding of its basic biology and etiology. Armed with this genomic information, research groups around the world suggested multi-disciplinary approaches to attain anti-SARS drugs. In general, physicians tried to relieve the symptoms mainly by modulating parts of the immune system, while vaccinologists began the long process of developing a vaccine against the virus. Molecular and structural biologists suggested ways to interfere with the viral life cycle, and these are the focus of this review (Figure 1). The strategy starts from virus identification, goes through genome sequencing and, hopefully, ends with an antiviral drug. Drug development remains a challenge, but the acceleration in drug discovery offered by genome technologies will hopefully enable significant timetable cuts in achieving antiviral medicine. Other, more classical anti-viral strategies used to treat SARS patients, like the use of interferon, will not be discussed here. The interested reader is referred to [7, 8]. This review includes a retrospective summary of the development of anti-HIV drugs, followed by an appraisal of anti-SARS strategies and their applicability for rapid development of antivirals against SARS-CoV, if it does resurface, and against the next, probably inevitable, viral threat.
FIGURE 1.
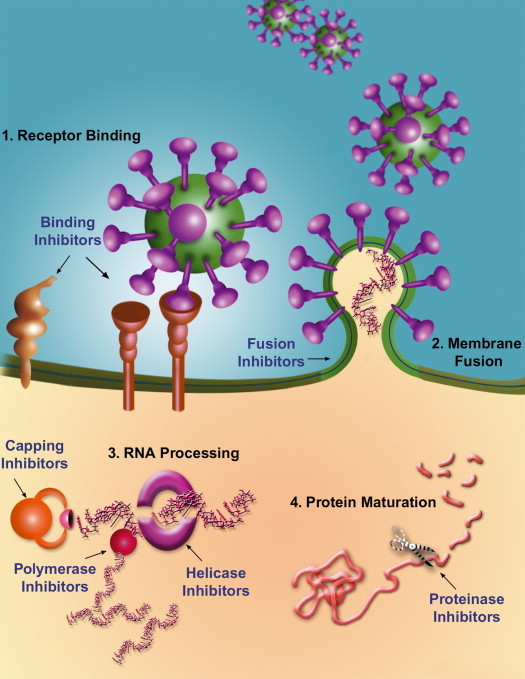
The SARS-CoV life cycle is vulnerable to therapeutic intervention in several places. (1) Virus binding to cellular receptors. Outside the cell, blocking the interaction of SARS-CoV with the cellular receptor will prevent the virus from attaching to host cells. Co-receptor antagonists will prevent the initiation of the next step. (2) Membrane fusion of the virion with the host. Fusion inhibitors will block merging of the viral membrane with the host cell membrane. (3) Viral RNA processing. Within the cell, transcription and multiplication of the viral RNA can be blocked by polymerase and helicase inhibitors. Translation of the viral proteins might be inhibited by blocking mRNA capping. (4) Protein maturation. Viral proteins will not mature in the presence of protease inhibitors, rendering them useless.
History lessons: HIV and AIDS
In the early 1980s, a sudden increase in life-threatening opportunistic infections and Kaposi's sarcoma, considered rare until then, was observed among homosexuals and injecting drug users. These infections were attributed to an acquired immunodeficiency syndrome (AIDS). About two years of extensive studies passed until the human immunodeficiency virus (HIV) was identified as the etiological agent [9, 10]. An additional two years passed before completing the sequencing of its genome [11]. The huge efforts invested in developing the first drug to treat AIDS patients bore fruit in record speed when the FDA approved azidothymidine (AZT) in 1987. AZT, a nucleoside analog, inhibits the viral reverse transcriptase - an enzyme that is essential for HIV replication. Unfortunately, isolates from these patients showed decreased sensitivity after six months of AZT administration. The finding that some of these isolates also exhibited cross-resistance to other nucleoside analogs [12] raised further concern. AZT and other nucleoside reverse transcriptase inhibitors were still the sole treatment available for almost a decade. The next important milestones were the development of inhibitors against the protease, another essential viral enzyme, and non-nucleoside reverse transcriptase inhibitors, first approved in 1995 and 1997, respectively. While the nucleoside reverse transcriptase inhibitors were discovered by cell culture screening directed towards a specific class of chemical agent, the non-nucleoside reverse transcriptase inhibitors were discovered by HTS of large compound libraries. The discovery of HIV protease inhibitors represents one of the best examples of the application of protein structural knowledge to rational drug design. Since then, AIDS patients have been treated with a cocktail of inhibitors against HIV reverse transcriptase and protease.
However, despite the unprecedented successes in the therapy of HIV infection, HIV continues to spread, causing more than 14,000 new infections every day, 95% of these in the developing world (WHO website). A major problem with current AIDS treatment is the high frequency of HIV mutations, resulting in drug resistance. Thus, efforts are being made to develop agents addressing yet untargeted steps in the HIV life cycle. Viral-induced fusion between viral and host cell membranes was acknowledged as a useful drug target with the approval of the HIV fusion inhibitor, enfuvirtide (Fuzeon), in 2003 [13]. Unlike other HIV drugs that are small molecules developed against viral enzymes, enfuvirtide is a peptide that corresponds to a specific segment of the viral envelope protein. Importantly, this segment can be directly pinpointed by computational sequence analysis [14, 15]. This strategy seems promising in developing anti-viral therapeutic peptides to other viruses that possess type 1 viral fusion proteins [e.g. measles virus and respiratory syncytial virus (RSV)], which share some structural motifs with HIV. Little is known about viral-induced membrane fusion of other viruses that do not share these motifs.
Entry inhibitors
Viruses can be divided into two groups based on the composition of their outer surface: (a) non-enveloped viruses are enclosed by a protein shell called a capsid; (b) enveloped viruses are surrounded by a membrane ‘stolen’ from their last host. In order to infect host cells, that is, to inject their genetic material into the cell, enveloped viruses need to overcome both viral and cellular membrane barriers. Viral entry of many enveloped viruses, including SARS-CoV, involves two major steps. First, the virion binds to receptor(s) localized on the surface of its host cell, and second, the viral membrane fuses with the host cell membrane. SARS-CoV spike glycoprotein, responsible for these two steps, is translated as a large polypeptide that is subsequently cleaved to produce two functional subunits, S1 and S2. S1 is the peripheral protein, which binds to cellular receptor(s), whereas S2 is a type I transmembrane protein that catalyzes the membrane fusion reaction. Both steps are crucial for viral infection, and therefore were suggested as targets for antivirals.
Blocking the interaction between SARS-CoV and its cellular receptors
Since the identification of CD4 as the cellular receptor for HIV in 1984 [16, 17], several therapeutic agents aiming to inhibit the binding of HIV to CD4 were suggested. Unfortunately, these efforts have yet to bear fruit. Major difficulties that slow down the development of inhibitors for the binding of CD4 to gp120 include: (i) the gp120-binding site for CD4 consists largely of a recessed pocket; (ii) antibodies that bind to CD4 antigen are likely to block virus attachment but can be immunosuppressive because they will lead to depletion of CD4 cells. Currently, both a recombinant CD4-IgG2 fusion protein (PRO-542) and a small-molecule (BMS-488043) aiming to prevent HIV from attaching to CD4, are in clinical trials. These efforts reflect the motivation of inhibiting the first step in the viral life cycle, that is, the binding of the virus to its host cell. This approach was strengthened when CXCR4 and CCR5 were identified as additional essential cellular receptors for HIV [18, 19], and with the discovery that CCR5-deficient people are resistant to infection by HIV [20].
Similar to HIV, binding of the viral spike glycoprotein to some receptor(s) on host cells is the first step in SARS-CoV infection. Blocking the interaction between these receptors and the virus could prevent infection, thus inspiring the search for SARS-CoV cellular receptors. Recently, human angiotensin converting enzyme-related carboxypeptidase (ACE2), a type I integral membrane metalloprotease, was identified as a receptor for SARS-CoV [21]. A soluble form of ACE2 and an antibody recognizing SARS-CoV S1 efficiently neutralized SARS-CoV in vitro, supporting the speculation that the ACE2-binding site of the spike glycoprotein is an attractive target for vaccine and drug development [21, 22]. This is further supported by an ACE2 inhibitor, which also inhibits SARS-CoV infection in vitro [23]. Notably, a 193-amino acid fragment of SARS-CoV S1, which efficiently bound ACE2, blocked spike glycoprotein-mediated infection with an IC50 of less than 10 nM [24]. More recently, a human lung cDNA library was screened to identify receptors for SARS-CoV, revealing that human CD209L can also mediate infection by SARS-CoV, although it is a much less efficient receptor than ACE2. Interestingly, CD209L is expressed in human lung in type II alveolar cells, which are an important target for SARS-CoV infection [25]. It is still not known whether interactions between ACE2 and CD209L play a role in SARS-CoV infection and pathogenesis.
Fusion inhibitors
HIV entry involves the binding of the viral envelope glycoproteins (comprising gp120 and gp41, which are the homologous of SARS-CoV S1 and S2, respectively) to CD4 on the host cell plasma membrane. This induces conformational changes, enabling the N-terminal heptad repeat region (N-HR) of gp41 to be exposed. At this stage, enfuvirtide binds to the N-HR of gp41, hence blocking further conformational changes required for membrane fusion. Enfuvirtide is a synthetic peptide inhibitor corresponding to a segment of gp41, known as the C-terminal heptad repeat (C-HR). Following the CD4-induced conformational change of gp41, plasma membrane CCR5 (or CXCR4) molecules are recruited to the binding site, and bind to the CD4-envelope complexes. This triggers a highly stable interaction between the C-HR and the N-HR regions of gp41, which drives the membrane fusion reaction to completion. Thus, enfuvirtide can no longer inhibit the fusion process [26]. Slower engagement of the co-receptor with the CD4-envelope complexes, results in a stronger inhibition by C-HR-derived peptides [27, 28]. Furthermore, reduction in CCR5 binding efficiency resulted in slower fusion kinetics and increased sensitivity to enfuvirtide [28, 29]. Further support for this model is provided by the finding that CCR5 and CXCR4 antagonists showed strong anti-HIV synergy with enfuvirtide against CCR5-dependent and CXCR4-dependent HIV isolates, respectively [30, 31]. In addition, PRO-542 acts in concert with enfuvirtide in virus-cell and cell-cell fusion assay, by triggering formation of gp41 fusion intermediates, enabling enfuvirtide to act on free HIV-1 virions [32].
There are no peptide fusion inhibitors for influenza virus. It is noteworthy that influenza virus uses a different mechanism to enter its host cells: it is first endocytosed into the cell, followed by a pH-dependent fusion between the viral and the endosome membranes. Strikingly, it takes only few milliseconds from the time the pH drops in the endosomes until the fusion process is completed [33, 34, 35, 36]. In contrast, the time scale of HIV infection is about 20 minutes, allowing ample time for binding of entry inhibitors [26, 27, 37]. SARS-CoV entry kinetics resembles that of HIV. At 5 minutes after exposure, the SARS-CoV lined the plasma membrane of Vero cells [38]. Fusion and entry of the viral load into the cytoplasm was observed mainly between 15 and 20 minutes [38]. The [39],(http://www.virology.net/Articles/sars/s2model.html). Indeed, preliminary reports revealed anti-SARS activity for peptides corresponding to the C-HR of SARS-CoV S2 protein [40, 41, 42], and indicated a mode of action similar to that of enfuvirtide [43, 44, 45, 46].
The kinetic similarity of SARS-CoV and HIV entries suggests a synergism between SARS-CoV spike glycoprotein inhibitors and agents that block some of its receptors. The role of different cellular receptors in SARS-CoV entry should be characterized to discover the receptor(s) that trigger conformational changes and transform the spike protein into the stable ‘fusogenic’ form. Antagonists for these receptor(s) could synergize with fusion inhibitors. The synergy between SARS fusion inhibitors and ACE2 or CD209L antagonists has not yet been investigated. The first step is to determine optimal fusion inhibitors. Intriguingly, whereas polar residues disrupt the heptad repeat in the C-HR of HIV-1 gp41, the C-HR of SARS-CoV S2 has a perfect leucine/isoleucine heptad repeat (Figure 2d). This could explain why the exact sequence boundaries of the C-HR-derived peptides are crucial for efficient inhibition [41, 42, 44, 47], as aggregation of the peptides in solution could abolish anti-viral activity. Interestingly, two reports demonstrate that N-HR-derived peptides are also active [40, 41], while others found that only C-HR-derived peptides have anti-SARS activity [42, 47]. It is noteworthy that the reason for the poor inhibitory activities of N-HR-derived peptides in other viruses is contributed to their tendency to aggregate in solution, suggesting that, similar to the C-HR-derived peptides, the exact sequence boundaries of the N-HR-derived peptides are important.
FIGURE 2.
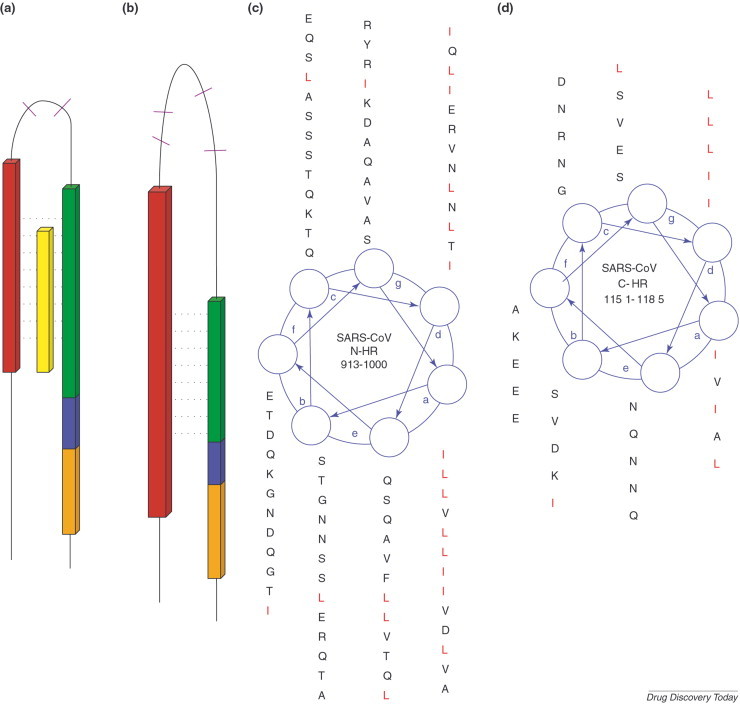
Similarity between the fusion proteins of HIV-1 and SARS-CoV. Schematic illustration of (a) HIV-1 gp41 and (b) the equivalent S2 protein from the SARS-CoV. A Leucine/Isoleucine heptad repeat adjacent to the N-terminus of both proteins appears in red. The C-HR is in green. Cysteine residues (purple) confining a loop structure are located between the two heptad repeats. An aromatic residues-rich motif is marked blue, and the transmembrane segment is in orange. A peptide corresponding to the C-HR, which acts as potent inhibitor of HIV-1 entry into the cell, appears in yellow. The helical wheel is a top view of a single strand of a coiled coil. In the wheel projection of the N-HR (c) and C-HR (d) of SARS-CoV S2 protein, each of the seven positions (a-g) corresponds to the location of an amino acid residue that makes up the coiled coil. The arrows between the seven positions indicate the relative locations of adjacent residues in an amino acid subsequence. The helical wheel demonstrates how a potential antiviral drug can be discovered based solely on sequence information.
The main advantage of fusion inhibitors is their immediate discovery as they are simply the corresponding fragments of a known protein. However, their drawbacks as therapeutic peptides are lack of oral bioavailability and high production costs. Auspiciously, SARS is a respiratory syndrome, thus, peptidic fusion inhibitors could be given by inhalation. This approach was applied successfully in RSV-infected mice [48].
SARS-CoV enzymes as targets for antivirals
To serve as drug targets, viral proteins should fulfill two criteria: (i) they should be essential for the viral life cycle; and (ii) they should exhibit low similarity to host proteins. SARS-CoV genome analysis was performed to predict its proteome [49], and three viral enzymes were suggested as targets for drug discovery: the helicase, the RNA-dependent RNA polymerase and the main protease. These enzymes are crucial for replication, transcription, translation and post-translational polyprotein processing (Box 1). Assay development based on these three SARS-CoV target enzymes was initiated [50, 51, 52], thus paving the way for high-throughput in vitro screening approaches to identify candidate inhibitors in compound libraries.
BOX 1. SARS-CoV enzymes as targets for antiviral agents.
Currently, inhibitors target three enzymes, crucial for SARS-CoV life cycle:
Helicase: Protein-fold recognition methods followed by a biochemical study suggested a dual use of SARS-CoV helicase in both RNA synthesis and cap formation, suggesting new avenues to treat the virus [65, 66]. Screening of a compound library by plaque reduction assay resulted in a helicase inhibitor at the low microM range. In vitro assay confirmed SARS-CoV helicase as a validated drug target [67].
RNA-dependent RNA polymerase: Molecular modeling revealed that SARS-CoV RNA-dependent RNA polymerase does not contain a hydrophobic pocket for non-nucleoside inhibitors. This is in contrast with the non-nucleoside inhibitors activity against HIV-1 reverse transcriptase [68]. Of the many nucleoside analogues screened, SARS-CoV most selective nucleoside analogue inhibitor is Beta-D-N4-hydroxycytidine, albeit at low efficacy (EC90 of 6 microM by virus yield reduction assay) [69]. Ribavirin, a broad-spectrum nucleoside analogue efficacious in the treatment of several viral infections, was used in various countries against SARS-CoV. While ribavirin was promising in vitro [70], recent reports revealed that ribavirin did not appear to confer any benefit for patients with SARS [71, 72].
Main protease: Sequence similarity was found between the substrate-binding sites of SARS-CoV main protease and the main protease of related viruses. Remarkably, the SARS-CoV main protease cleaved the porcine coronavirus transmissible gastroenteritis coronavirus (TGEV) main protease substrate [73]. This rationalizes screening of known protease inhibitors for anti-SARS activity. This approach was reinforced by the crystallization of the SARS-CoV main protease together with a TGEV inhibitor [74]. The findings revealed that homology modeling is often inadequate for the prediction of the mutual orientation of domains in multidomain proteins. However, the TGEV-based homology model also shows that a reasonable model of a substrate-binding site can serve to develop useful ideas for inhibitor design that can inspire medicinal chemists to start a synthesis program long before the 3D structure of the target enzyme is experimentally determined (reviewed in [75]). In parallel, HTS of compound libraries identified inhibitors of the SARS-CoV main protease in the low μM range [67, 76, 77]. There are a few studies demonstrating that inhibitors of the HIV protease can also inhibit SARS-CoV, albeit with much lower efficiency [52, 78].
Other approaches
The traditional and, in many cases, the most cost-efficient way of dealing with viruses has been through vaccines. The logic of vaccine development against SARS-CoV emerges from the combination of several findings: (i) re-infection with SARS-CoV causes only mild illness; (ii) SARS is fatal mainly to old people who have difficulty in producing good humoral and cellular immune responses; and (iii) the case fatality ratio of SARS ranges from 0–50% depending on the age group affected, with an overall estimate of case fatality of 14–15% (WHO website). Thus, most infected individuals recover from SARS. Furthermore, the success of a vaccine against other mammal-infective coronaviruses is encouraging [53, 54]. Modern antiviral vaccine development depends heavily on the viral genome. The availability of the human genome, together with the recent sequencing of the SARS-CoV genome, largely increases the probability of success of vaccine development. The full-length spike glycoprotein of SARS-CoV, expressed by vaccinia virus, induces binding and neutralizing antibody and protectively immunizes mice against a subsequent infection with SARS-CoV [55]. In addition, DNA vaccine encoding the spike glycoprotein of the SARS-CoV induces T-cell and neutralizing antibody responses, as well as protective immunity, in a mouse model [56].
The discovery of RNAi raises many hopes regarding antiviral strategies and carries the promise of a shortcut in the drug discovery process. Usually, target discovery is followed by exhaustive HTS and/or structure-based screening of many thousands of compounds in the hope that some of them will efficiently bind to the target. Theoretically, with small-interfering RNA (siRNA) as a drug, the course from target to drug is much shorter. Encouraging results in mice were obtained using an RNAi-based therapy against hepatitis B virus (HBV): transfection with plasmids expressing short hairpin RNAs (shRNAs) homologous to HBV mRNAs effectively inhibited replication initiation in cultured cells and mice liver, showing that such an approach could be useful in the treatment of viral diseases [57]. Currently, there are attempts to use siRNA as anti-SARS drugs, but they are still in preliminary in vitro stages [58, 59, 60]. The application of this relatively new technology to therapeutics faces several safety and technical issues, including delivery of the RNA molecule into the virus-infected cells and the activation of interferon system [61, 62].
The challenges ahead…
SARS-CoV reminds us that viral infections are a global threat. It is vital that the scientific community acquire the ability to develop anti-viral therapy promptly. We can be encouraged by the remarkable speed with which the global community acted in a coordinated research effort to investigate SARS-CoV. Immediately after the last nucleotide of the SARS-CoV genome was verified, the sequence was distributed through the internet to the worldwide scientific community. Among the genomic-based approaches that followed, inhibitors of the viral-induced membrane fusion seem the most promising.
The mutation rate of SARS-CoV is much slower than that of HIV-1 and is among the lowest of RNA viruses [63, 64]. However, viral resistance will be an obstacle. The solution could lie in the use of a drug cocktail, combining antiviral drugs with different modes of action (e.g. protease and polymerase inhibitors), to lower the chances for drug-resistant viruses to arise. In addition, drug cocktails are beneficial when the optimal dose of a drug, given as a mono-therapy, is toxic - then, combining drugs with distinct modes of action, in sub-optimal doses, might alleviate toxicity issues. Moreover, the recent advancement in the understanding of HIV entry into its host cell revealed an opportunity for synergism, based on the molecular mechanism of viral entry. Drugs that inhibit the interaction between CCR5 and the CD4-envelope complexes enhance the efficiency of HIV fusion inhibitors by elongating the exposure time of their target site. The timescale similarity of the SARS-CoV fusion process to that of HIV is encouraging. Hopefully, future identification and characterization of SARS-CoV receptors will open a way for an efficient antiviral strategy, by synergistically combining viral entry inhibitors.
Within a few months, scientists have managed to leverage the technological advances of the past 20 years of anti-AIDS research into an unprecedented antiviral campaign against SARS (Figure 3). SARS served as a test tube for novel approaches developed following the AIDS epidemic. The current SARS epidemic was finally contained, yet quick development of antivirals is still high priority. Today, we are closer than ever to achieving therapeutic solutions for a viral epidemic shortly after viral outbreak.
FIGURE 3.
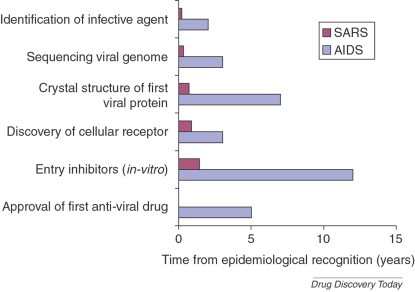
A time line comparing key achievements in AIDS and SARS research. Effective international collaborations and technological advances greatly accelerated the understanding of viral diseases. It is anticipated that these research achievements will also lead to faster drug discovery and development.
Acknowledgements
We thank L. Rychlewski, A. Wool, E. Eisenberg, N. Rabbie, A. Toporik, M. Olshansky and S.G. Peisajovich for critical reading of the manuscript. We are also grateful to artist R. Lieber for capturing the central idea of the article with her illustration.
References
- 1.Ksiazek T.G. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.Drosten C. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Peiris J.S. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vabret A. An outbreak of coronavirus OC43 respiratory infection in Normandy, France. Clin. Infect. Dis. 2003;36:985–989. doi: 10.1086/374222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marra M.A. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 6.Rota P.A. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 7.Sainz B., Jr Interferon-beta and interferon-gamma synergistically inhibit the replication of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) Virology. 2004;329:11–17. doi: 10.1016/j.virol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujii T. Current concepts in SARS treatment. J. Infect. Chemother. 2004;10:1–7. doi: 10.1007/s10156-003-0296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barre-Sinoussi F. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 10.Gallo R.C. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science. 1984;224:500–503. doi: 10.1126/science.6200936. [DOI] [PubMed] [Google Scholar]
- 11.Muesing M.A. Nucleic acid structure and expression of the human AIDS/lymphadenopathy retrovirus. Nature. 1985;313:450–458. doi: 10.1038/313450a0. [DOI] [PubMed] [Google Scholar]
- 12.Richman D.D. Zidovudine resistance of human immunodeficiency virus. Rev. Infect. Dis. 1990;12(Suppl. 5):507. doi: 10.1093/clinids/12.supplement_5.s507. [DOI] [PubMed] [Google Scholar]
- 13.Matthews T. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004;3:215–225. doi: 10.1038/nrd1331. [DOI] [PubMed] [Google Scholar]
- 14.Berger B., Singh M. An iterative method for improved protein structural motif recognition. J. Comput. Biol. 1997;4:261–273. doi: 10.1089/cmb.1997.4.261. [DOI] [PubMed] [Google Scholar]
- 15.Singh M. LearnCoil-VMF: computational evidence for coiled-coil-like motifs in many viral membrane-fusion proteins. J. Mol. Biol. 1999;290:1031–1041. doi: 10.1006/jmbi.1999.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klatzmann D. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 1984;312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 17.Dalgleish A.G. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 18.Feng Y. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 19.Alkhatib G. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 20.Samson M. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 21.Li W. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sui J. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huentelman M.J. Structure-based discovery of a novel angiotensin-converting enzyme 2 inhibitor. Hypertension. 2004;44:903–906. doi: 10.1161/01.HYP.0000146120.29648.36. [DOI] [PubMed] [Google Scholar]
- 24.Wong S.K. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeffers S.A. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U. S. A. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallo S.A. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta. 2003;1614:36–50. doi: 10.1016/s0005-2736(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 27.Gallo S.A. HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry. 2001;40:12231–12236. doi: 10.1021/bi0155596. [DOI] [PubMed] [Google Scholar]
- 28.Reeves J.D. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16249–16254. doi: 10.1073/pnas.252469399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reeves J.D. Impact of mutations in the coreceptor binding site on human immunodeficiency virus type 1 fusion, infection, and entry inhibitor sensitivity. J. Virol. 2004;78:5476–5485. doi: 10.1128/JVI.78.10.5476-5485.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tremblay C.L. Anti-human immunodeficiency virus interactions of SCH-C (SCH 351125), a CCR5 antagonist, with other antiretroviral agents in vitro. Antimicrob. Agents Chemother. 2002;46:1336–1339. doi: 10.1128/AAC.46.5.1336-1339.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tremblay C.L. Strong in vitro synergy between the fusion inhibitor T-20 and the CXCR4 blocker AMD-3100. J. Acquir. Immune Defic. Syndr. 2000;25:99–102. doi: 10.1097/00042560-200010010-00001. [DOI] [PubMed] [Google Scholar]
- 32.Nagashima K.A. Human immunodeficiency virus type 1 entry inhibitors PRO 542 and T-20 are potently synergistic in blocking virus-cell and cell-cell fusion. J. Infect. Dis. 2001;183:1121–1125. doi: 10.1086/319284. [DOI] [PubMed] [Google Scholar]
- 33.Blumenthal R. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell-cell fusion events. J. Cell Biol. 1996;135:63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mittal A. Kinetics of influenza hemagglutinin-mediated membrane fusion as a function of technique. Anal. Biochem. 2002;303:145–152. doi: 10.1006/abio.2002.5590. [DOI] [PubMed] [Google Scholar]
- 35.Mittal A. Kinetically differentiating influenza hemagglutinin fusion and hemifusion machines. Biophys. J. 2003;85:1713–1724. doi: 10.1016/S0006-3495(03)74601-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Markosyan R.M. Evolution of intermediates of influenza virus hemagglutinin-mediated fusion revealed by kinetic measurements of pore formation. Biophys. J. 2001;80:812–821. doi: 10.1016/S0006-3495(01)76060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markosyan R.M. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol. Biol. Cell. 2003;14:926–938. doi: 10.1091/mbc.E02-09-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng M.L. Early events of SARS coronavirus infection in vero cells. J. Med. Virol. 2003;71:323–331. doi: 10.1002/jmv.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kliger Y., Levanon E.Y. Cloaked similarity between HIV-1 and SARS-CoV suggests an anti-SARS strategy. BMC Microbiol. 2003;3:20. doi: 10.1186/1471-2180-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu S. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan K. Suppression of SARS-CoV entry by peptides corresponding to heptad regions on spike glycoprotein. Biochem. Biophys. Res. Commun. 2004;319:746–752. doi: 10.1016/j.bbrc.2004.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu J. Following the rule: formation of the 6-helix bundle of the fusion core from severe acute respiratory syndrome coronavirus spike protein and identification of potent peptide inhibitors. Biochem. Biophys. Res. Commun. 2004;319:283–288. doi: 10.1016/j.bbrc.2004.04.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simmons G. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ingallinella P. Structural characterization of the fusion-active complex of severe acute respiratory syndrome (SARS) coronavirus. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8709–8714. doi: 10.1073/pnas.0402753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Y. Crystal structure of SARS-CoV spike protein fusion core. J. Biol. Chem. 2004;279:49414–49419. doi: 10.1074/jbc.M408782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tripet B. Structural characterization of the SARS-coronavirus spike S fusion protein core. J. Biol. Chem. 2004;279:20836–20849. doi: 10.1074/jbc.M400759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bosch B.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pastey M.K. A RhoA-derived peptide inhibits syncytium formation induced by respiratory syncytial virus and parainfluenza virus type 3. Nat. Med. 2000;6:35–40. doi: 10.1038/71503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snijder E.J. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thiel V. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 51.Tanner J.A. The severe acute respiratory syndrome (SARS) coronavirus NTPase/helicase belongs to a distinct class of 5′ to 3′ viral helicases. J. Biol. Chem. 2003;278:39578–39582. doi: 10.1074/jbc.C300328200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu C.Y. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koolen M.J. Immunogenic peptide comprising a mouse hepatitis virus A59 B-cell epitope and an influenza virus T-cell epitope protects against lethal infection. J. Virol. 1990;64:6270–6273. doi: 10.1128/jvi.64.12.6270-6273.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takamura K. Field study of bovine coronavirus vaccine enriched with hemagglutinating antigen for winter dysentery in dairy cows. Can. J. Vet. Res. 2002;66:278–281. [PMC free article] [PubMed] [Google Scholar]
- 55.Bisht H. Severe acute respiratory syndrome coronavirus spike protein expressed by attenuated vaccinia virus protectively immunizes mice. Proc. Natl. Acad. Sci. U. S. A. 2004;101:6641–6646. doi: 10.1073/pnas.0401939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Z.Y. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature. 2004;428:561–564. doi: 10.1038/nature02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCaffrey A.P. Inhibition of hepatitis B virus in mice by RNA interference. Nat. Biotechnol. 2003;21:639–644. doi: 10.1038/nbt824. [DOI] [PubMed] [Google Scholar]
- 58.Zhang R. Inhibiting severe acute respiratory syndrome-associated coronavirus by small interfering RNA. Chin Med J (Engl) 2003;116:1262–1264. [PubMed] [Google Scholar]
- 59.Zhang X.W., Yap Y.L. Old drugs as lead compounds for a new disease? Binding analysis of SARS coronavirus main proteinase with HIV, psychotic and parasite drugs. Bioorg. Med. Chem. 2004;12:2517–2521. doi: 10.1016/j.bmc.2004.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z. Inhibition of severe acute respiratory syndrome virus replication by small interfering RNAs in mammalian cells. J. Virol. 2004;78:7523–7527. doi: 10.1128/JVI.78.14.7523-7527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sledz C.A. Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 2003;5:834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- 62.Opalinska J.B., Gewirtz A.M. Nucleic-acid therapeutics: basic principles and recent applications. Nat. Rev. Drug Discov. 2002;1:503–514. doi: 10.1038/nrd837. [DOI] [PubMed] [Google Scholar]
- 63.Yeh S.H. Characterization of severe acute respiratory syndrome coronavirus genomes in Taiwan: Molecular epidemiology and genome evolution. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2542–2547. doi: 10.1073/pnas.0307904100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chinese S.M.E.C. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–1669. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- 65.von Grotthuss M. mRNA cap-1 methyltransferase in the SARS genome. Cell. 2003;113:701–702. doi: 10.1016/S0092-8674(03)00424-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ivanov K.A. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 2004;78:5619–5632. doi: 10.1128/JVI.78.11.5619-5632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kao R.Y. Identification of novel small-molecule inhibitors of severe acute respiratory syndrome-associated coronavirus by chemical genetics. Chem. Biol. 2004;11:1293–1299. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu X. Molecular model of SARS coronavirus polymerase: implications for biochemical functions and drug design. Nucleic Acids Res. 2003;31:7117–7130. doi: 10.1093/nar/gkg916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barnard D.L. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARSCoV) by calpain inhibitors and beta-D-N4-hydroxycytidine. Antivir. Chem. Chemother. 2004;15:15–22. doi: 10.1177/095632020401500102. [DOI] [PubMed] [Google Scholar]
- 70.Chen F. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J. Clin. Virol. 2004;31:69–75. doi: 10.1016/j.jcv.2004.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leong H.N. Investigational use of ribavirin in the treatment of severe acute respiratory syndrome, Singapore, 2003. Trop. Med. Int. Health. 2004;9:923–927. doi: 10.1111/j.1365-3156.2004.01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang W.K. Temporal relationship of viral load, ribavirin, interleukin (IL)-6, IL-8, and clinical progression in patients with severe acute respiratory syndrome. Clin. Infect. Dis. 2004;39:1071–1075. doi: 10.1086/423808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anand K. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 74.Yang H. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hillisch A. Utility of homology models in the drug discovery process. Drug Discov. Today. 2004;9:659–669. doi: 10.1016/S1359-6446(04)03196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kao R.Y. Characterization of SARS-CoV main protease and identification of biologically active small molecule inhibitors using a continuous fluorescence-based assay. FEBS Lett. 2004;576:325–330. doi: 10.1016/j.febslet.2004.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blanchard J.E. High-Throughput Screening Identifies Inhibitors of the SARS Coronavirus Main Proteinase. Chem. Biol. 2004;11:1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamamoto N. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]